Bright fluorescent nanoparticles for developing potential optical imaging contrast agents†
Guorong
Sun
ab,
Mikhail Y.
Berezin
*b,
Jinda
Fan
b,
Hyeran
Lee
b,
Jun
Ma
ab,
Ke
Zhang
ab,
Karen L.
Wooley
*abc and
Samuel
Achilefu
*b
aDepartment of Chemistry, Washington University in Saint Louis, Saint Louis, MO 63130, USA
bDepartment of Radiology, Washington University School of Medicine, Saint Louis, MO 63110, USA. E-mail: berezinm@mir.wustl.edu; achilefu@wustl.edu
cDepartment of Chemistry and Department of Chemical Engineering, Texas A&M University, College Station, TX 77842, USA. E-mail: wooley@mail.chem.tamu.edu
First published on 8th February 2010
Abstract
Fluorescent cross-linked nanoparticles with variable fluorophore loading amounts, locations, and particle sizes were synthesized from sequential one-pot functionalization/cross-linking of block copolymer micelles with amine-terminated dye and cross-linker molecules, via reductive amination and amidation. The fluorescence quantum yield and brightness of these nanoparticles were evaluated by steady-state and dynamic fluorescence methods. The results demonstrate that the quantum yield and brightness of the fluorescent nanoparticles correlated directly with the number of dyes/nanoparticle and the nanoparticle size. A strategy to increase the fluorescence brightness of nanoparticles with fluorescein and near-infrared dyes is proposed.
Introduction
During the past decades, in vivo fluorescence imaging has experienced a substantial growth with the “opening” of the near-infrared (NIR) window (wavelengths between 650 and 900 nm) of the electromagnetic spectrum.1–7 By utilizing fluorescent probes, such as quantum dots,8–10 fluorescent proteins,11,12 and organic NIR dyes,13–20 tissue autofluorescence and light attenuation associated with deep-tissue imaging are minimized, facilitating depth profiling of tissues to within a few centimetres by in vivo optical imaging.4,5Among the organic NIR fluorophores (NIRFs), carbocyanine-based dyes are of particular interest, due to their high molar extinction coefficients (usually on the order of 105 M−1 cm−1) and the tunability of their photo-physical properties.19 The ease of functionalizing these NIRFs with bioactive ligands enables targeted imaging.15,21–23 However, the in vivo application of cyanine dyes generally suffers from short blood circulation times and non-specific tissue/organ accumulations.24–27 One approach to overcome these drawbacks is to encapsulate/couple cyanine dyes within/onto nanoscale platforms, which can provide protection and also impart “stealth” properties from the blood clearance systems.14,28–45 The nanoscale platform can further be conjugated with multiple homo- or heterogeneous targeting agents to improve specificity by taking advantage of the interactions with cell-surface receptors. Furthermore, this approach may allow for “hybridization” of different probes within a single nanoparticle to provide multi-modal imaging, thereby improving the diagnostic accuracy.42,46–50
Nanoparticles (NPs) have been widely studied in molecular imaging. To date, cyanine NIRF-functionalized dendrimers,37,48 silica NPs,30,34,35,39,44,51,52 and natural/engineered viruses29,33 have been reported. However, the design of fluorescent nanoparticles of small diameters is challenging because multiple fluorophores on the surface of a nanoparticle quench each other if they are located within the energy-transfer distance. Since the Förster radius for common fluorophores (1–10 nm)53 is on the order of the nanoparticle diameter, care must be taken in synthesizing particles with desirable fluorescence properties. Herein, we investigated the optical properties of nanoparticles labelled with different numbers of common fluorescent dyes based upon fluorescein and cypate (a NIRF).21 A synthetic procedure was developed for conjugating block copolymer-based nanoscale micellar assemblies with fluorescent molecules, followed sequentially in situ by covalent cross-linking of the polymers to afford robust nanoparticles having differential placement, number, and type of fluorescent chromophores. Investigation of their steady-state and dynamic optical properties, together with the synthetic efforts, allowed for optimization of these unique fluorescent nanoparticles to achieve a high level of fluorescence intensity.
Experimental
Materials
Cypate,14 HL-800 (LS-277),17S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (DDMAT),54 4-(dimethy1amino)pyridinium 4-toluenesulfonate (DPTS),55 4-vinylbenzaldehyde (VBA),56 mPEG 2 kDa macro-chain transfer agent (macro-CTA),57 and poly(ethylene oxide)-block-poly(N-acryloxysuccinimide)-block-polystyrene (PEO45-b-PNAS95-b-PS60)58 were synthesized according to literature reports. Mono-methoxy-terminated mono-hydroxy poly(ethylene glycol) (mPEG 2 kDa and mPEG 5k Da, Mw = 2000 and 5000 Da, respectively, PDI = 1.06 and 1.07, respectively) were purchased from Intezyne Technologies (Tampa, FL) and used without purification. Other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO) and Acros Organics (Thermo Fisher Sci.) and were used without purification unless otherwise noted. Methylene chloride (CH2Cl2) was distilled from calcium hydride and stored under N2. Nano-pure water was 18.0 MΩcm.Measurements
1H NMR spectra were recorded on a Varian 500 MHz spectrometer with CD2Cl2 and DMSO-d6 as solvents and internal standards. Infrared spectra were obtained on a Perkin-Elmer Spectrum BX FT-IR system using diffuse reflectance sampling accessories with FT-IR Spectrum v2.00 software.Absolute molecular weight and molecular weight distribution were determined by gel permeation chromatography (GPC). GPC was performed on a Waters 1515 HPLC system (Water Chromatography, Inc.), equipped with a Waters 2414 differential refractometer, a PD2020 dual-angle (15 and 90) light scattering detector (Precision Detectors, Inc.), and a three-column series PL gel 5 μm Mixed columns (Polymer Laboratories, Inc.). The eluent was anhydrous tetrahydrofuran (THF) with a flow rate of 1 mL min−1. All instrumental calibrations were conducted using a series of nearly monodisperse poly(styrene) standards. Data were collected after an injection of 200 μL of polymer solution in THF (ca. 5 mg mL−1) and then analyzed using the Discovery 32 software (Precision Detectors, Inc.).
The GPC with N,N-dimethylformamide (DMF) as a mobile phase was conducted on a Waters Chromatography, Inc. (Milford, MA) system equipped with an isocratic pump model 1515, a differential refractometer model 2414, and a two-column set of Styragel HR 4 and HR 4E 5 μm DMF 7.8 × 300 mm columns. The system was equilibrated at 70 °C in pre-filtered DMF containing 0.05 M LiBr, which served as polymer solvent and eluent (flow rate set to 1.00 mL min−1). Polymer solutions were prepared at a concentration of ca. 3 mg mL−1 and an injection volume of 200 μL was used. Data collection and analysis was performed with Empower Pro software (Waters, Inc.). The system was calibrated with poly(ethylene glycol) standards (Polymer Laboratories, Amherst, MA) ranging from 615 to 442![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 800 Da.
800 Da.
Samples for transmission electron microscopy (TEM) measurements were diluted with a 1% phosphotungstic acid (PTA) stain (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Carbon grids were exposed to oxygen plasma treatment to increase the surface hydrophilicity. Micrographs were collected at 100000× magnification and calibrated using a 41 nm polyacrylamide bead from NIST. The number average particle diameters (Dav) and standard deviations were generated from the analysis of a minimum of 150 particles from at least three different micrographs. The aggregation number (Naggr) was calculated based upon the diameter measured from TEM by using the following equation:
:1). Carbon grids were exposed to oxygen plasma treatment to increase the surface hydrophilicity. Micrographs were collected at 100000× magnification and calibrated using a 41 nm polyacrylamide bead from NIST. The number average particle diameters (Dav) and standard deviations were generated from the analysis of a minimum of 150 particles from at least three different micrographs. The aggregation number (Naggr) was calculated based upon the diameter measured from TEM by using the following equation:
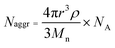 | (1) |
Hydrodynamic diameters (Dh) and size distributions for the nanoparticles in aqueous solutions were determined by dynamic light scattering (DLS). The DLS instrument consisted of a Brookhaven Instruments Ltd (Worcestershire, U.K.) system equipped with a model BI-200SM goniometer, a model BI-9000AT digital correlator, a model EMI-9865 photomultiplier, and a model 95-2 Ar ion laser (Lexel Corp.) operated at 514.5 nm. Measurements were made at 25 ± 1 °C. Scattered light was collected at a fixed angle of 90°. The digital correlator was operated with 522 ratio spaced channels, and initial delay of 5 μs, a final delay of 100 ms, and a duration of 8 min. A photomulitplier aperture of 400 μm was used and the incident laser intensity was adjusted to obtain a photon counting of between 150 and 200 kcps. Only measurements in which the measured and calculated baselines of the intensity autocorrelation function agreed to within 0.1% were used to calculate particle size. The calculations of the particle size distributions and distribution averages were performed with the ISDA software package (Brookhaven Instruments Co.), which employed single-exponential fitting, cumulants analysis, and CONTIN particle size distribution analysis routines. All experiments were repeated 5 times and the results are represented as intensity-averaged mean values with standard deviations between runs.
The fluorescence spectra of NPs were obtained at room temperature using a Varian Cary Eclipse and Fluorolog III fluorescence spectrophotometer. Fluorescence lifetime was measured using TCSPS method as described previously.59 Briefly, the fluorescence decays were measured in aqueous media using λex 460/λem 570 nm for fluorescein-based NPs or λex 773/λem 820 nm for cypate-based NPs with bandpass 20 nm. In both measurements, a two-exponential decay analysis was applied.
For optical measurements, the fluorescein-based NPs were diluted with 0.1 M NaOH solution. NIR-based NPs were measured in PBS buffer at pH 7.2 (5 mM with 5 mM of NaCl). Samples were diluted to give the absorbance maximum value of 0.1 to avoid photon re-absorption.
The UV–vis absorption spectra of NPs were collected at room temperature using a Varian Cary 500 Bio UV–visible spectrophotometer in a quartz cuvette with a 10 mm light path.
The Förster radius of a homoFRET pair (R0) was calculated using previously developed FRET calculator37 from steady-state absorption and emission spectra using known equations:
| R0 = 0.211(k2n − 4ϕDJ(λ))1/6 Å | (2) |
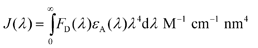 | (3) |
Synthesis
:5:5:5, v/v) was added and stirred for 1 h. The solvent was removed under reduced pressure and the crude product was washed with diethyl ether (3 × 5 mL) and purified by medium pressure chromatography with a C-18 reverse phase column (acetonitrile–water as eluent). A green solid was obtained as pure product (10.9 mg, 60.6% yield). MS (EI): 709.0 (M + 1)+.
:5:5:5, v/v)) for 1 h. Diethyl ether was then added to the crude product to precipitate solid, washed with diethyl ether (3 × 5 mL) and further purified by medium pressure chromatography with a C-18 reverse phase column (acetonitrile–water as eluent). The solvents were evaporated to give HL-800-amine (3.18mg, 29.2% yield) as a green solid. MS (EI): 955.2 (M + 1)+.
The solid was precipitated by addition of anhydrous ethyl ether (150 mL) at 0 °C twice and the crude product obtained was further purified by flash column chromatography (2–3% MeOH–CH2Cl2, v/v) to afford mPEG 5 kDa macro-CTA as a yellow solid (1.2 g, 60% yield). 1H NMR (500 MHz, CD2Cl2, δ): 0.88 (t, J = 6.5 Hz, 3H), 1.26 (m, 16H), 1.38 (t, J = 6.5 Hz, 2H), 1.66 (t, J = 7.5 Hz, 2H), 1.68 (s, 6H), 3.27 (t, J = 7.2 Hz, 2H), 3.33 (s, 3H), 3.40–3.80 (m, 440H), 4.21 (t, J = 5.0 Hz, 2H).
General procedure for construction of PEO-b-PVBA micelles
To a solution of PEO-b-PVBA block copolymer in DMF (ca. 1.0 mg mL−1) was added an equal volume of water dropwise via a syringe pump at a rate of 15.0 mL h−1, and the mixture was further stirred for 16 h at rt. The solution was then transferred to pre-soaked dialysis tubing (MWCO ca. 3500 Da) and dialyzed against water for 4 days to afford a solution of micelles.General procedure for one-pot functionalization and cross-linking of PEO-b-PVBA micelles with fluorescein-5-thiosemicarbazide to construct core cross-linked fluorescent nanoparticles (CCFNP1–4)
To a solution of PEO-b-PVBA micelles in 10.0 mL of water, was added a solution of fluorescein-5-thiosemicarbazide (20 mol%, 2 mol%, 1 mol%, and 0.5 mol%, relative to the aldehyde residues, respectively) in DMF. The reaction mixture was allowed to stir for 2 h at rt in the dark. To this reaction mixture, was added a solution of 2,2′-(ethylenedioxy)-bis(ethylamine) (150 mol%, relative to the aldehyde residues) in water dropwise over 10 min and further stirred for 48 h at rt in the dark. NaBH3CN (200 mol%, relative to the aldehyde residues) in water was then added to the reaction solution and further stirred for 16 h at rt in the dark. Finally, the mixture was transferred to pre-soaked dialysis tubing (MWCO ca. 3500 Da) and dialyzed against 5.0 mM PBS (pH 7.2, with 5.0 mM NaCl) for 7 days to remove the small molecule byproducts and afford an aqueous solution of fluorescein-functionalized cross-linked nanoparticles.General procedure for one-pot functionalization and cross-linking of PEO113-b-PVBA46 micelles with cypate-diamine to prepare core cross-linked fluorescent nanoparticles (CCFNP5–7)
To a solution of PEO113-b-PVBA46 micelles (2.7 mg of polymer, 11 μmol of aldehyde residues) in 10.0 mL of water, was added a solution of cypate-diamine (1 mol%, 0.5 mol%, and 0.2 mol%, relative to the aldehyde residues, respectively) in DMF. The reaction mixture was allowed to stir for 2 h at rt in the dark. To this reaction mixture was added a solution of 2,2′-(ethylenedioxy)-bis(ethylamine) (2.4 mg, 16 μmol) in water dropwise over 10 min and further stirred for 48 h at rt in the dark. NaBH3CN (1.4 mg, 22 μmol) in water was then added to the reaction solution and further stirred for 16 h at rt in the absence of light. Finally, the mixture was transferred to pre-soaked dialysis tubing (MWCO ca. 6000–8000 Da) and dialyzed against 5.0 mM PBS (pH 7.2, with 5.0 mM NaCl) for 7 days to remove the small molecule byproducts to afford an aqueous solution of functionalized and cross-linked nanoparticles.Micellization of PEO45-b-PNAS95-b-PS60
To a solution of PEO45-b-PNAS95-b-PS60 block copolymer in DMF (ca. 1.0 mg mL−1), was added dropwise an equal volume of water via a syringe pump at a rate of 15.0 mL h−1, and the mixture was further stirred for 1 h at rt before use for the following reactions.Functionalization and cross-linking of PEO45-b-PNAS95-b-PS60 micelles with cypate-diamine or HL-800-amine to prepare shell cross-linked fluorescent Knedel-like nanoparticles (SCFK1–2)
To a solution of PEO45-b-PNAS95-b-PS60 micelles (4.8 mg of block copolymer precursor, 27 μmol of NAS residues) in 10 mL of DMF–H2O (v:v = 1:1) at rt, was added the solution of cypate-diamine (38.3 μg, 0.054 μmol) or HL-800-amine (51.6 μg, 0.054 μmol) in 50 μL of DMF. The reaction mixture was allowed to stir for 2 h at rt in the dark. To this reaction mixture, was added a solution of 2,2′-(ethylenedioxy)-bis(ethylamine) (1.0 mg, 6.8 μmol) in water dropwise over 10 min and further stirred for 24 h at rt in the dark. The mixture was transferred to pre-soaked dialysis tubing (MWCO 6000–8000 Da) and dialyzed against 5.0 mM PBS (pH 7.2, with 5.0 mM NaCl) for 7 d to remove DMF, un-reacted crosslinker, and the small molecule byproducts to afford an aqueous solution of cross-linked nanoparticles.
One-pot functionalization and cross-linking of PEO113-b-PVBA46 micelle with HL-800-amine (CCFNP8)
To a solution of PEO113-b-PVBA46 micelles (2.7 mg of polymer, 11 μmol of aldehyde residues) in 10.0 mL of water, was added a solution of HL-800-amine (20.7 μg, 0.022 μmol) in DMF. The reaction mixture was allowed to stir for 2 h at rt in the absence of light. To this reaction mixture, was added a solution of 2,2′-(ethylenedioxy)-bis(ethylamine) (2.4 mg, 16 μmol) in water dropwise over 10 min and further stirred for 48 h at rt in the dark. NaBH3CN (1.4 mg, 22 μmol) in water was then added to the reaction solution and further stirred for 16 h at rt in the absence of light. Finally, the mixture was transferred to pre-soaked dialysis tubing (MWCO ca. 6000–8000 Da) and dialyzed against 5.0 mM PBS (pH 7.2, with 5.0 mM NaCl) for 7 days to remove the small molecule by-products and to afford an aqueous solution of functionalized and cross-linked nanoparticles.Results and discussion
To construct fluorescent NPs, diblock copolymer (poly(ethylene oxide)-block-poly(4-vinyl benzaldehyde), PEO-b-PVBA) and triblock copolymer (poly(ethylene oxide)-block-poly(N-acryloxysuccinimide)-block-polystyrene, PEO-b-PNAS-b-PS) micelles were functionalized with fluorescein-5-thiosemicarbazide or amine-functionalized carbocyanine dyes through either reductive amination or amidation, respectively (Scheme 1). The PEO-b-PVBA diblock copolymers I (PEO45-b-PVBA18, Mn, NMR = 4,700 Da, Mn, GPC = 3,900 Da, PDI = 1.2, ESI† Fig. S1A) and II (PEO113-b-PVBA46, Mn, NMR = 12,600 Da, Mn, GPC = 12,400 Da, PDI = 1.4, ESI† Fig. S1B)60 were used for construction of core functionalizable cross-linked nanoparticles (CCFNPs) while the PEO-b-PNAS-b-PS triblock copolymer III (PEO45-b-PNAS95-b-PS60, Mn, NMR = 18,400 Da, PDI = 1.2, ESI† Fig. S1C) was utilized as the precursor for shell cross-linked and shell fluorescently-labelled Knedel-like nanoparticles (SCFKs). All block copolymers were prepared by reversible addition-fragmentation chain transfer (RAFT) polymerization61–64 based upon our previous reports.57,58 PEO (Mw = 2000 and 5000 Da, respectively) was selected as the hydrophilic segment because of its good water solubility, well-known biocompatibility, and low immunogenic response,65 and it was designed to occupy the corona of each of the nanostructures. PVBA was used as the hydrophobic and reactive segment for the core-cross-linked and fluorescently-labelled nanostructures, due to the broad reaction scope and the selectively-high reactivity of aldehydes under mild aqueous conditions. PNAS segments were utilized as pre-installed active esters along the central block segment of triblock copolymers for conjugation with amine-functionalized fluorophores within the shell layer of the shell cross-linked nanoparticles.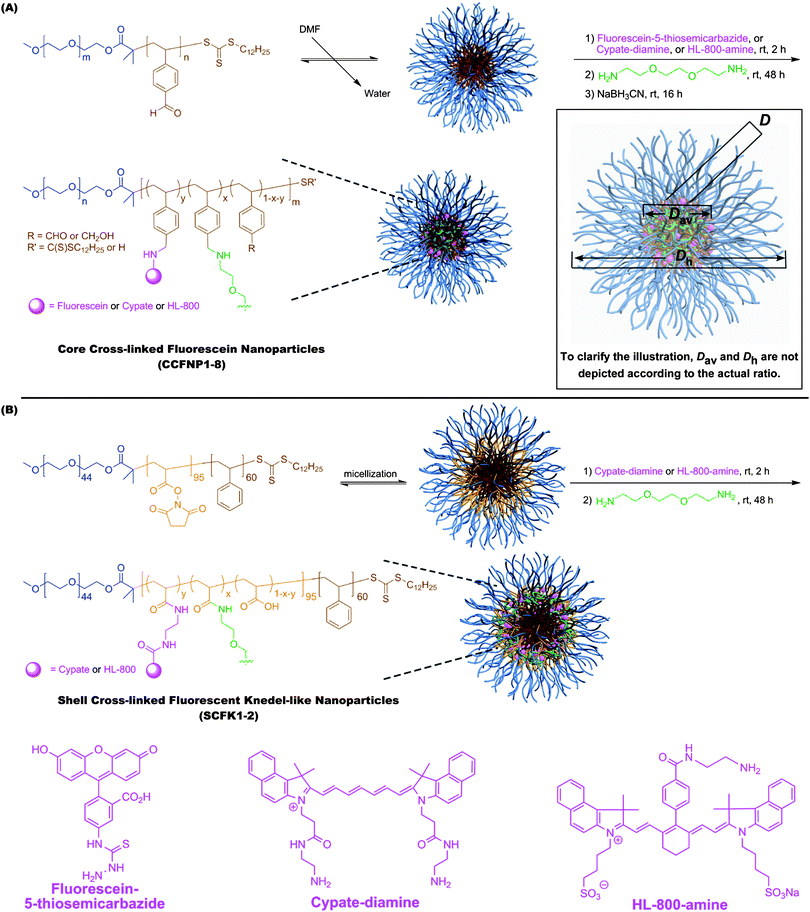 | ||
| Scheme 1 Construction of fluorescent nanoparticles through reductive amination to afford core-crosslinked and core fluorescently-labelled nanoparticles (CCFNPs) (A) and through amidation to afford shell-crosslinked and shell fluorescently-labelled knedel-like nanoparticles (SCFKs) (B). | ||
Aqueous assembly of block copolymers I and II followed conventional methods for micellization of amphiphilic block copolymers. A PEO selective solvent, i.e. water, was added to the solution of PEO-b-PVBA in organic solvent (DMF, ca. 1.0 mg mL−1) at a rate of ca. 15 mL h−1 to induce the micellization and further stabilize the formed nanoscale-aggregates. The organic solvent was then removed by dialysis to afford PEO-b-PVBA micelles with PEO shells and PVBA core domains. The uniformity and narrow size distribution of the assembled micelles were demonstrated through a combination of DLS and TEM (Fig. 1). The DLS measurements showed that these micelles had an intensity-averaged hydrodynamic diameter (Dh, intensity) of 23 ± 2 nm (from PEO45-b-PVBA18) and 26 ± 2 nm (from PEO113-b-PVBA46), respectively. TEM micrographs revealed their globular shapes with an average core domain diameter (Dav) of 13 ± 1 nm and 19 ± 1 nm, respectively, depending upon the length of PVBA block segment.
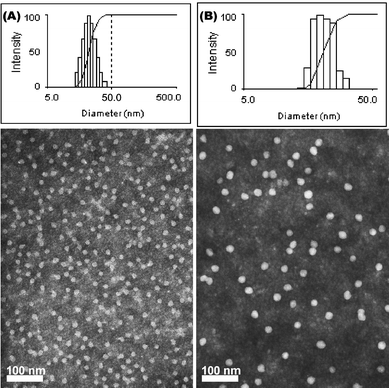 | ||
| Fig. 1 Characterization of PEO-b-PVBA micelles. Intensity-average weighted hydrodynamic diameter distribution histograms by DLS (top) and TEM micrograph (bottom, stained with PTA): (A) PEO45-b-PVBA18 micelles; (B) PEO113-b-PVBA46 micelles. | ||
One-pot chemical functionalization, with fluorescein-5-thiosemicarbazide, and sequential cross-linking, with diamine cross-linker, of the PEO45-b-PVBA18 micelles were achieved by our previously published procedure (Scheme 1A).57 Similar to our previous findings,57 buffer solutions with certain ionic strength (pH 7.2, 5 mM PBS with 5 mM of NaCl in this study) were found to be required to prevent the precipitation of the cross-linked nanoparticles. For CCFNP1 (prepared with a feeding ratio of fluoresceins/aldehydes = 0.2:1), the DLS (Dh, intensity = 21 ± 1 nm) and TEM (Dav = 13 ± 1 nm) characterizations of the functionalized cross-linked NPs showed no obvious morphology, size, and size distribution changes (Fig. 2A), compared with the micelle precursors (Fig. 1A). UV–vis measurement of the fluorescein-NP solutions revealed that a slight red-shift of ca. 8 nm (Fig. 2E left) occurred for the maximum absorption peak (λmax, 496 nm for the fluorescein-functionalized NPs vs. 488 nm for the unconjugated fluorescein), which might be related to the change of fluorescein local environment before and after incorporation into the nanostructure. Although the thiosemicarbazide is a strong nucleophile, the coupling efficiency of fluorescein into the NPs was only ca. 12%. This might be associated with the low accessibility of hydrophilic fluorescein to the hydrophobic benzaldehyde functionalities (packed inside the hydrophobic core domain of the micelles).
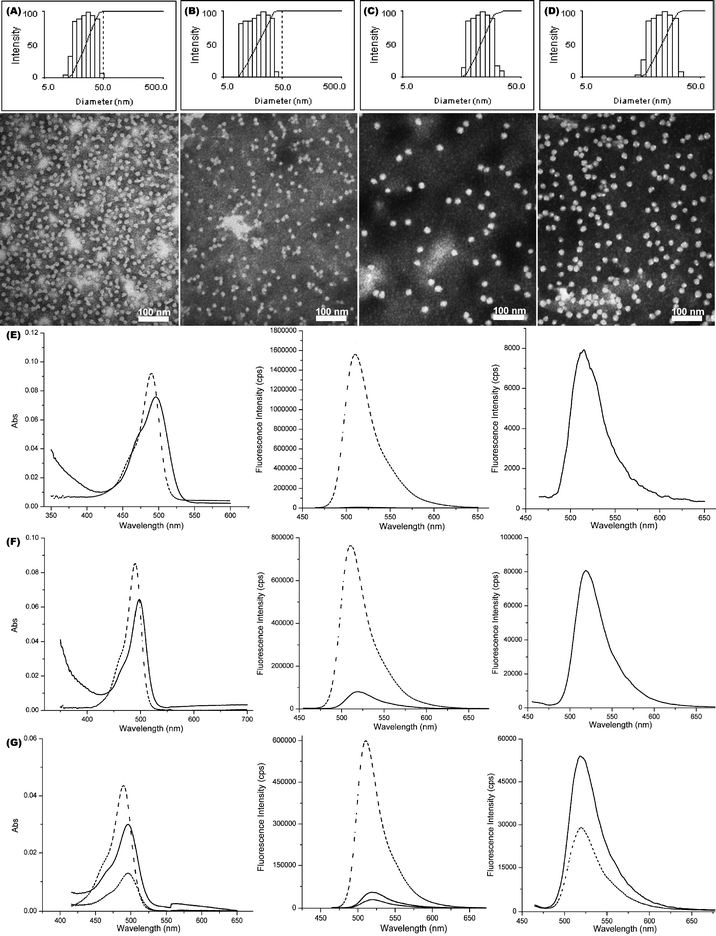 | ||
| Fig. 2 Characterizations of CCFNP1–4. (A–D) Intensity-average weighted hydrodynamic diameter distribution by DLS (top) and TEM micrograph (bottom, stained with PTA) of CCFNP1–4, respectively. (E) UV–vis (left) and fluorescence emission (middle and right) spectra of fluorescein (dashed) and CCFNP1 (solid). (F) UV–vis (left) and fluorescence emission (middle and right) spectra of fluorescein (dashed) and CCFNP2 (solid). (G) UV–vis (left) and fluorescence emission (middle and right) spectra of fluorescein (dashed), CCFNP3 (short dashed) and CCFNP4 (solid). | ||
A steady-state fluorescence spectrum of the diluted NP solution was recorded at the excitation wavelength of 450 nm over the range of 465–650 nm (Fig. 2E). Compared with the spectrum of the fluorescein small molecule, much lower fluorescence emission intensity of the CCFNP1 sample (200-fold drop at the same concentration magnitude) was observed. The decrease in fluorescence of fluorescein attached to nanoparticles could be attributed to self-quenching via non-radiative energy transfer mechanism (homoFRET) between fluorophores in close proximity. We hypothesized that incorporating ca. 150 dye molecules into a confined space with ca. 13 nm diameter placed the fluorophores within distances much shorter than the Förster radius R0 – the distance at which 50% of energy transfers to another fluorophore (vide infra).
The R0 measurement for fluorescein using steady-state spectra (see the Experimental section) provided a homoFRET distance of 4.7 nm, similar to the literature value (4.4 nm).66 The average distance between two fluorescein moieties on the NPs (r) was calculated from both steady-state and dynamic emission spectra by using the following equations:
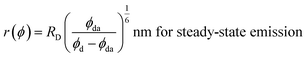 | (4) |
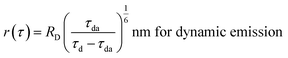 | (5) |
Ideally, both methods provide the same results, however, the distances obtained from the lifetime measurements were consistently higher than those from quantum yield calculations (Table 1). Such discrepancy in the results suggests the presence of mechanisms other than FRET for quenching. For example, at high fluorophore concentrations, photon re-absorption and subsequent photon re-emission can increase the fluorescence lifetime values significantly. Accordingly, the local concentration of fluorescein within the nanoparticles is rather high, which might create an environment favourable for re-absorption. This condition is supported by the fact that at lower conjugation number, r(ϕ) and r(τ) values become closer. Another possible explanation for the discrepancy in results may be attributed to the fact that the relative fluorescence quantum yield used in this work did not take into account that the molar absorptivity of fluorescein is highly sensitive to a substitution pattern as well as to a polarity of the solvent. However, the measurement of molar absorptivity in systems like nanoparticles is challenging because it requires the knowledge of exact nanoparticle compositions. Finally, in addition to the homoFRET, other non-energy transfer quenching mechanisms, such as dynamic and static quenching have to be taken into account.
| Sample | D h, intensity /nm | D av /nm | N agg | N | D /nm | τ /ns | r(τ)g/nm | ϕ | r(ϕ)i/nm | B × 10−3 M−1 cm−1 |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Intensity-averaged hydrodynamic diameter.
b Average core domain diameter.
c Nanoparticle aggregation number.
d Dyes per nanoparticle, calculated based upon ε = 76000 M−1 cm−1.
e Calculated distance between two adjacent fluorophores within nanoparticle.
f Fluorescence average lifetime. Complex multiexponential decay of nanoparticles (see the beginning of the decay shown in ESI1 Fig. S2) suggested the presence of many distances between fluorescein moieties.
g Distance between two fluorophores based on lifetime measurement [eqn (5)].
h Fluorescence quantum yield.
i Distance between two fluorophores based on steady-state data [eqn (4)].
j Fluorescence brightness [see eqn (8)].
|
||||||||||
| Fluorescein | — | — | — | — | — | 3.92 | — | 0.79 | — | 62.4 |
| CCFNP1 | 21 ± 1 | 13 ± 1 | 350 | 150 | 1.88 | 2.01 | 4.76 | 0.005 | 2.01 | 59.3 |
| CCFNP2 | 22 ± 2 | 13 ± 1 | 350 | 15 | 5.95 | 2.97 | 5.65 | 0.088 | 3.30 | 104.3 |
| CCFNP3 | 24 ± 2 | 19 ± 1 | 430 | 15 | 8.69 | 3.33 | 6.23 | 0.233 | 4.04 | 276.1 |
| CCFNP4 | 24 ± 2 | 19 ± 1 | 430 | 30 | 6.15 | 3.11 | 5.84 | 0.215 | 3.96 | 509.6 |
We have previously established a semi-quantitative model for evaluation of the PEG surface coverage density across a nanoparticle.67 Because only ∼2.5% of the VBA units were actually “decorated” with fluorescein, the fluorescein molecules should be attached to the most accessible VBA units, i.e. benzaldehydes around the periphery of the core domain. Therefore, the previous model could be easily revised to estimate the distance between adjacent fluorescein molecules (depicted as D in the Scheme 1 insertion) through the following equation:
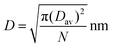 | (6) |
According to a known Förster equation [eqn (7)],66 no energy transfer and therefore no quenching would occur if the distance between two fluoresceins (r) exceeds 9 nm (ET = 0.02%).
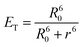 | (7) |
For CCFNP1, the calculated distances between two fluorescein molecules, both from eqn (6) (1.88 nm) and from the experimental calculations (2.00 nm from steady-state [eqn (4)] and 4.75 nm from dynamic [eqn (5)] measurements, respectively), were far below 9 nm. This condition would result in substantial self-quenching and low brightness of the nanoparticles (B = 59.3 × 103 M−1 cm−1 for CCFNP1). Thus, 150 fluorescein moieties had the brightness of a single fluorescein molecule, which is certainly not satisfactory.
| B = εϕN, M−1 cm−1 | (8) |
000 M−1 cm−1 and ∼200000 M−1 cm−1 for, respectively, fluorescein at 488 nm, cypate at 780 nm, and HR-800 at 800 nm, actual molar absorptivity was not determined), ϕ is the fluorescence quantum yield, and N represents the number of fluorophores per nanoparticle.
To improve the brightness of fluorescein-incorporated nanoparticles, the number of fluorophores per nanoparticle should be decreased to overcome the homoFRET quenching. Accordingly, CCFNP2 with ca. 15 fluoresceins per NP was prepared from the same block copolymer micelle as CCFNP1, maintaining similar size (Dh, intensity = 22 ± 2 nm and Dav = 13 ± 1 nm) and size distribution (Fig. 2B).68 The quantum yield was significantly improved by a factor of 18 (Table 1) due to the increase of the distance between fluorescein moieties, as calculated from the experimental data [eqns (4) and (5)] and from eqn (6) (Table 1). However, the overall improvement of fluorescence brightness was only marginal (1.043 × 105 M−1 cm−1).
A simple calculation (not shown here) estimates a maximum number of 5 fluoresceins per nanoparticle for the PEO45-b-PVBA18 system (13 nm diameter core) to allow at least 9 nm distance between fluoresceins and to further eliminate possible homoFRET quenching. With such a small loading, even in the absence of any other quenching mechanisms the fluorescence brightness cannot exceed a theoretical value of 3 × 105 M−1 cm−1. To improve the overall brightness of fluorescent nanoparticles without inducing homoFRET quenching, it is advantageous to use nanoparticles with larger cores to allow an increased number of fluorophores per nanoparticle as indicated in eqn (6). Accordingly, CCFNP3 and CCFNP4 with larger core sizes (19 vs. 13 nm, Fig. 2C and 2D, respectively) were prepared from PEO113-b-PVBA46 block copolymer micelles.69
With a core size of 19 nm and 15 fluorescein molecules/NP, the calculated D for CCFNP3 was 8.69 nm, which is close to the target 9 nm cut-off range. In fact, only 15.7% of the absorbed energy was non-radiatively transferred between fluorescein moieties (based on lifetime measurement). As a result, the quantum yield of CCFNP3 increased to 0.233, a 2.5-fold enhancement compared with CCFNP2 with brightness of 276.1 x103 M−1 cm−1. For CCFNP4, which bears 30 fluoresceins per particle, a small drop of quantum yield (0.215) was observed. This is likely due to the relative proximity between fluorophores (D = 6.15 nm). However, the slightly decreased quantum yield was compensated by the higher number of fluorophores, rendering its overall fluorescence intensity 190% as large as that of CCFNP3 (Fig. 2G, right). The calculated brightness of 509.6 × 103 M−1 cm−1 is nearly an order of magnitude higher than that for a single fluorescein molecule.
The PEO113-b-PVBA46 micelle system and the optimized stoichiometry of fluorescein loading (less than 1 mol%, relative to the aldehydes) were then extended to construct NIR fluorescent nanoparticles using cypate (Scheme 1A). CCFNP5–7 with ca. 70, 45, and 10 cypates per particle, respectively, were synthesized and evaluated by photophysical methods (Fig. 3). Coupling yields using cypate improved to ∼36% to 47% compared with the fluoresceins at ∼12% to 15%. This improvement could be attributed to the fact that cypate is more hydrophobic than fluorescein and interacts favourably with the hydrophobic PVBA core.
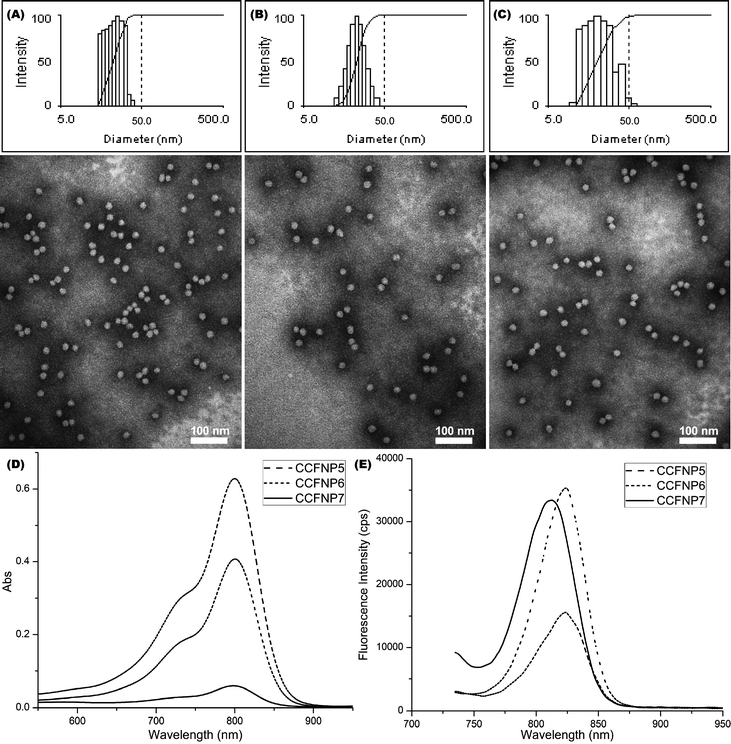 | ||
| Fig. 3 (A–C) Intensity-average weighted hydrodynamic diameter distribution histogram by DLS (top) and TEM micrograph (bottom, stained with PTA) of CCFNP5, CCFNP6, and CCFNP7, respectively. (D–E) UV–vis and fluorescence profiles of CCFNP5 (dashed), CCFNP6 (short dashed), and CCFNP7 (solid), respectively. | ||
The calculated distances derived from eqn (6) between neighbouring cypate molecules for CCFNP5–7 were 4.02 nm, 5.02 nm, and 10.65 nm, respectively (Table 2). The brightness values significantly improved from 4.2 × 103 M−1 cm−1 for a single cypate molecule to 3.6 × 104 M−1 cm−1 for CCFNP6. However, a low quantum yield of 0.004 was obtained. For CCFNP7, with a higher D value of over 10 nm, an enhanced quantum yield value of 0.019 was observed, which is very close to the free cypate in water (0.021), as expected. However, due to the lower number of fluorophores on the nanoparticles, the overall brightness (3.8 × 104 M−1 cm−1) remained similar to CCFNP6.
| Sample | D h, intensity /nm | D av /nm | N agg | N | D /nm | ϕ | B × 10−3 M−1 cm−1 |
|---|---|---|---|---|---|---|---|
|
a Intensity-averaged hydrodynamic diameter.
b Average core domain diameter.
c Nanoparticle aggregation number.
d Dyes per nanoparticle, calculated based upon ε = 200000 M−1 cm−1.
e Calculated distance between two adjacent cypates within nanoparticle.
f Fluorescence quantum yield, relative to indocyanine green (ICG) in methanol (quantum yield = 0.09).
g Fluorescence brightness.
|
|||||||
| Cypate | — | — | — | — | — | 0.021 | 4.2 |
| CCFNP5 | 23 ± 1 | 19 ± 1 | 430 | 70 | 4.02 | 0.001 | 14.0 |
| CCFNP6 | 23 ± 1 | 19 ± 1 | 430 | 45 | 5.02 | 0.004 | 36.0 |
| CCFNP7 | 22 ± 2 | 19 ± 1 | 430 | 10 | 10.65 | 0.019 | 38.0 |
Previously, we have shown that the fluorescence lifetime of cyanine dyes, such as cypate, is significantly affected by the polarity of the media.59 To further explore the relationship between the brightness of the fluorescent nanoparticles with the locations of the fluorophores, NIRF shell cross-linked fluorescent Knedel-like nanoparticles (NIR-SCFKs, SCFK1–2) were prepared, in which carbocyanine molecules having differing hydrophobicities/hydrophilicities (Cypate for SCFK1 and HL-800 for SCFK2, respectively) were distributed throughout the hydrophilic shell domain (Scheme 1B). These particles showed relatively narrow size distributions and globular shape, as observed through DLS and TEM (Fig. 4A–B). The photophysical characteristics of SCFK1 and SCFK2 are summarized in Table 3.
| Sample | D h, intensity /nm | D av /nm | N agg | N | ϕ | B × 10−3 M−1 cm−1 |
|---|---|---|---|---|---|---|
|
a Intensity-averaged hydrodynamic diameter.
b Average core domain diameter.
c Nanoparticle aggregation number.
d Dyes per nanoparticle, calculated based upon ε = 200000 M−1 cm−1.
e Fluorescence quantum yield.
f Relative to ICG in methanol (quantum yield = 0.09).
g Fluorescence brightness.
|
||||||
| SCFK1 | 59 ± 3 | 30 ± 1 | 1450 | 40 | 0.005 | 40.0 |
| HL-800 | — | — | — | — | 0.088f | 17.6 |
| SCFK2 | 56 ± 2 | 30 ± 1 | 1450 | 20 | 0.036 | 144.0 |
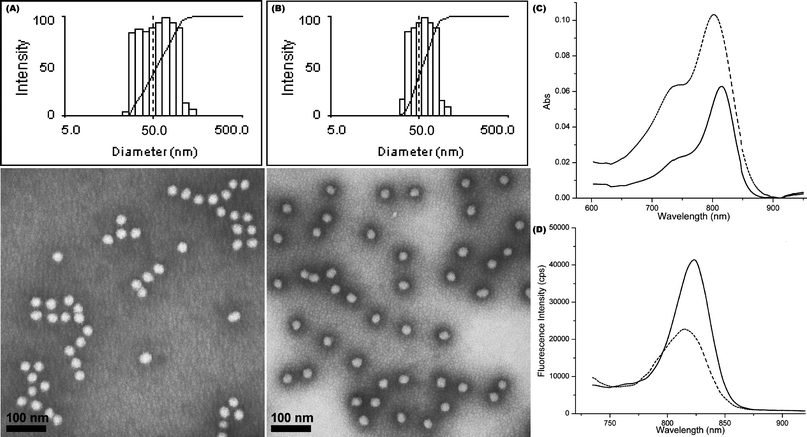 | ||
| Fig. 4 Physical and photo-physical properties of shell cross-linked NIR fluorescent nanoparticles. (A–B) Intensity-average weighted hydrodynamic diameter distribution histograms by DLS (top) and TEM micrographs (bottom, stained with PTA) of SCFK1 and SCFK2, respectively. (C–D) UV–vis and fluorescence profiles of SCFK1 (dashed) and SCFK2 (solid), respectively. | ||
At a similar cypate loading capacity as CCFNP6, SCFK1 showed similar quantum yield and brightness. NIR-SCFKs with improved optical characteristics were achieved when cypate was replaced with HL-800 at a loading capacity of 20 HL-800s/SCFK (SCFK2). The D value for SCFK2 was between the range of 12 nm to 22 nm. The quantum yield of SCFK2 (0.036, Table 3) was enhanced by a factor of 7 and the overall fluorescence brightness gained a 3.6-fold increase relative to SCFK1. Encouraged by the high brightness, HL–800 was further introduced into the PEO113-b-PVBA46 system to afford CCFNP8 (Scheme 1A) bearing 5 dyes in the core of each NP while maintaining a similar size as those of CCFNP5–7 (ESI† Fig. S3A, Dh, intensity = 22 ± 1 nm and Dav = 19 ± 1 nm) and comparable inter-dye spacing, similar to SCFK2 (D = 15 nm). Unfortunately, a dramatic decrease in quantum yield from 0.036 to 0.002 resulted in very low (2 × 103 M−1 cm−1) brightness of these core-cross-linked and HL-800-labelled nanoparticles. The mechanism for such lower fluorescence emission is unclear.
Conclusions
In summary, we have prepared unique core or shell-cross-linked and fluorescently-labelled nanomaterials and studied their photo-physical properties. Fluoresceins or NIR carbocyanine (cypate and HL-800) dyes were incorporated at different stoichiometric loadings and within different regions of the nanostructures to modify the local environment and inter-dye spacings. The brightness of the fluorescent nanoparticles was evaluated by varying the fluorophore loading capacity and location within nanoparticles of different sizes. As a result of optimization, we successfully prepared nanoscale agents with fluorescent brightness values that are ∼10-fold higher than those of free fluorescein or NIR dye molecules. Such bright fluorescent nanoparticles could be utilized in a variety of biological applications, particularly in fluorescence microscopy and in vivo imaging for significant contrast enhancement.Acknowledgements
This material is based upon work supported partially by the National Heart, Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HL080729), and by the National Institutes of Health grant number 1R01EB008111. The authors thank Mr. G. M. Veith for his kind assistance with TEM imaging; and Dr J. Kao for assistance with NMR measurements.Notes and references
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–805 CrossRef.
- V. Ntziachristos, J. Ripoll, L. V. Wang and R. Weissleder, Nat. Biotechnol., 2005, 23, 313–320 CrossRef CAS.
- K. Licha and C. Olbrich, Adv. Drug Delivery Rev., 2005, 57, 1087–1108 CrossRef CAS.
- V. Ntziachristos, Annu. Rev. Biomed. Eng., 2006, 8, 1–33 CrossRef CAS.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS.
- V. Ntziachristos, Genomic and Personalized Medicine, 2009, 524–531 Search PubMed.
- B. M. W. Tsui and D. L. Kraitchman, J. Nucl. Med., 2009, 50, 667–670 CrossRef.
- X. Gao, Y. Cui, R. M. Levenson, L. W. K. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- M. K. So, C. Xu, A. M. Leoning, S. S. Gambhir and J. Rao, Nat. Biotechnol., 2006, 24, 339–343 CrossRef CAS.
- R. M. Hoffman, Nat. Rev. Cancer, 2005, 5, 796–806 CrossRef CAS.
- N. Shaner, P. A. Steinbach and Y. T. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS.
- S. Achilefu, Technol. Cancer Res. Treat., 2004, 3, 393–410 Search PubMed.
- Y. Ye, S. Bloch and S. Achilefu, J. Am. Chem. Soc., 2004, 126, 7740–7741 CrossRef CAS.
- Z. Cheng, Y. Wu, Z. Xiong, S. S. Gambhir and X. Chen, Bioconjugate Chem., 2005, 16, 1433–1441 CrossRef CAS.
- W. Pham, Z. Medarova and A. Moore, Bioconjugate Chem., 2005, 16, 735–740 CrossRef CAS.
- H. Lee, J. C. Mason and S. Achilefu, J. Org. Chem., 2006, 71, 7862–7865 CrossRef CAS.
- A. Masotti, P. Vicennati, F. Boschi, L. Calderan, A. Sbarbati and G. Ortaggi, Bioconjugate Chem., 2008, 19, 983–987 CrossRef CAS.
- L. Strekowski, Heterocyclic Polymethine Dyes: Synthesis, Properties and Applications, Springer,Berlin/Heidelberg, 2008 Search PubMed.
- Z. Zhang, M. Y. Berezin, J. L. F. Kao, A. d'Avignon, M. Bai and S. Achilefu, Angew. Chem., Int. Ed., 2008, 47, 3584–3587 CrossRef CAS.
- S. Achilefu, R. B. Dorshow, J. E. Bugal and R. Rajagopalan, Invest. Radiol., 2000, 35, 479–485 CrossRef CAS.
- S. Achilefu, S. Bloch, M. A. Markiewicz, T. Zhong, Y. Ye, R. B. Dorshow, B. Chance and K. Liang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7976–7981 CrossRef CAS.
- Y. Ye, S. Bloch, B. Xu and S. Achilefu, J. Med. Chem., 2006, 49, 2268–2275 CrossRef CAS.
- W. West and S. Pearce, J. Phys. Chem., 1965, 69, 1894–1903 CrossRef CAS.
- P. Ott, Pharmacol. Toxicol., 1998, 83, 1–48 CrossRef.
- V. Saxena, M. Sadoqi and J. Shao, J. Pharm. Sci., 2003, 92, 2090–2097 CrossRef CAS.
- J. A. Cardillo, R. Jorge, R. A. Costa, S. M. Nunes, D. Lavinsky, B. D. Kuppermann, A. C. Tedesco and M. E. Farah, Br. J. Ophthalmol., 2008, 92, 276–280 CrossRef CAS.
- V. Saxena, M. Sadoqi and J. Shao, J. Photochem. Photobiol., B, 2004, 74, 29–38 CrossRef CAS.
- C. Wu, H. Barnhill, X. Liang, Q. Wang and H. Jiang, Opt. Commun., 2005, 255, 366–374 CrossRef CAS.
- T. Deng, J.-S. Li, J.-H. Jiang, G.-L. Shen and R.-Q. Yu, Adv. Funct. Mater., 2006, 16, 2147–2155 CrossRef CAS.
- K. Kim, M. Lee, H. Park, J.-H. Kim, S. Kim, H. Chung, K. Choi, I.-S. Kim, B. L. Seong and I. C. Kwon, J. Am. Chem. Soc., 2006, 128, 3490–3491 CrossRef CAS.
- P. Sharma, S. Brown, G. Walter, S. Santra and B. Moudgil, Adv. Colloid Interface Sci., 2006, 123–126, 471–485 CrossRef CAS.
- C. M. Soto, A. S. Blum, G. J. Vora, N. Lebedev, C. E. meador, A. P. Won, A. Chatterji, J. E. Johnson and B. R. Ratna, J. Am. Chem. Soc., 2006, 128, 5184–5189 CrossRef CAS.
- X. He, J. Chen, K. Wang, D. Qin and W. Tan, Talanta, 2007, 72, 1519–1526 CrossRef CAS.
- Z. Yang, S. Zheng, W. J. Harrison, J. Harder, X. Wen, J. G. Gelovani, A. Qiao and C. Li, Biomacromolecules, 2007, 8, 3422–3428 CrossRef CAS.
- J. Yu, M. A. Yaseen, B. Anvari and M. S. Wong, Chem. Mater., 2007, 19, 1277–1284 CrossRef CAS.
- A. Almutairi, W. J. Akers, M. Y. Berezin, S. Achilefu and J. M. J. Fréchet, Mol. Pharmacol., 2008, 5, 1103–1110 CrossRef CAS.
- E. I. Altınoǧlu, T. J. Russin, J. M. Kaiser, B. M. Barth, P. C. Eklund, M. Kester and J. H. Adair, ACS Nano, 2008, 2, 2075–2084 CrossRef CAS.
- J. F. Bringley, T. L. Penner, R. Wang, J. F. Harder, W. J. Harrison and L. Buonemani, J. Colloid Interface Sci., 2008, 320, 132–139 CrossRef CAS.
- W. Chen, J. Nanosci. Nanotechnol., 2008, 8, 1019–1051 CrossRef CAS.
- S. Lee, E.-J. Cha, K. Park, S.-Y. Lee, J.-K. Hong, I.-C. Sun, S. Y. Kim, K. Choi, I. K. Kwon, K. Kim and C.-H. Ahn, Angew. Chem., Int. Ed., 2008, 47, 2804–2807 CrossRef CAS.
- Y. T. Lim, Y.-W. Noh, J. H. Han, Q.-Y. Cai, K.-H. Yoon and B. H. Chung, Small, 2008, 4, 1640–1645 CrossRef CAS.
- V. B. Rodriguez, S. M. Henry, A. S. Hoffman, P. S. Stayton, X. Li and S. H. Pun, J. Biomed. Opt., 2008, 13, 014025–25 CrossRef.
- A. A. Burns, J. Vider, H. Ow, E. Herz, O. Penate-Medina, M. Baumgart, S. M. Larson, U. Wiesner and M. Bradbury, Nano Lett., 2009, 9, 442–448 CrossRef CAS.
- A.-K. Kirchherr, A. Briel and K. Maeder, Mol. Pharmacol., 2009, 6, 480–491 CrossRef CAS.
- L. Josephson, M. F. Kircher, U. Mahmood, Y. Tang and R. Weissleder, Bioconjugate Chem., 2002, 13, 554–560 CrossRef CAS.
- M. F. Kircher, U. Mahmood, R. S. King, R. Weissleder and L. Josephson, Cancer Res., 2003, 63, 8122–8125 CAS.
- O. Veiseh, C. Sun, J. Gunn, N. Kohler, P. Gabikian, D. Lee, N. Bhattarai, R. Ellenbogen, R. Sze, A. Hallahan, J. Olson and M. Zhang, Nano Lett., 2005, 5, 1003–1008 CrossRef CAS.
- J. H. Choi, F. T. Nguyen, P. W. Barone, D. A. Heller, A. E. Moll, D. Patel, S. A. Boppart and M. S. Strano, Nano Lett., 2007, 7, 861–867 CrossRef.
- W. B. Edwards, W. J. Akers, Y. Ye, P. P. Cheney, S. Bloch, B. Xu, R. Laforest and S. Achilefu, Mol. Imaging, 2009, 8, 101–110 CAS.
- L. Wang, V. Reipa and J. Blasic, Bioconjugate Chem., 2004, 15, 409–412 CrossRef.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy 2nd edn, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- J. S. Moore and S. I. Stupp, Macromolecules, 1990, 23, 65–70 CrossRef CAS.
- G. Sun, C. Cheng and K. L. Wooley, Macromolecules, 2007, 40, 793–795 CrossRef CAS.
- G. Sun, H. Fang, C. Cheng, P. Lu, K. Zhang, A. V. Walker, J. Taylor and K. L. Wooley, ACS Nano, 2009, 3, 673–681 CrossRef CAS.
- G. Sun, N. S. Lee, W. L. Neumann, J. N. Freskos, J. J. Shieh, R. B. Dorshow and K. L. Wooley, Soft Matter, 2009, 5, 3422–3429 RSC.
- M. Y. Berezin, H. Lee, A. Walter and S. Achilefu, Biophys. J., 2007, 93, 2892–2899 CrossRef CAS.
- Unexpected shoulders were observed in the GPC profile of II (ESI† Fig. S1B), at both high and low molecular weights. This observation indicated lower chain transfer efficiency of the mPEG5k macro-CTA, compared with the mPEG2k macro-CTA used for the chain extension to block copolymer I (smaller low-molecular-weight shoulder). However, the relatively larger polydispersity of II did not affect its assembly behavior and uniform core–shell micelles with narrow size distributions were still obtained.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- J. M. Harris, ed., Poly(ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications, Plenum Press, New York, 1992 Search PubMed.
- The Handbook: A Guide to Fluorescent Probes and Labeling Technologies 10th edn, Molecular Probes, Eugene, 2009 Search PubMed.
- G. Sun, A. Hagooly, J. Xu, A. M. Nyström, Z. Li, R. Rossin, D. A. Moore, K. L. Wooley and M. J. Welch, Biomacromolecules, 2008, 9, 1997–2006 CrossRef CAS.
- The λmax, abs for CCFNP2 was shifted to 500 nm, i.e., a 12 nm red-shift.
- For the PEO45-based PEO-b-PVBA system, we have already found that extending of the PVBA block segment length to 26 repeating units causes a dramatic morphological switch from nanoparticles to vesicles.57 Therefore, the hydrophilic PEO block was replaced with longer PEO113 and the hydrophobic PVBA block was correspondingly lengthened to 46 repeating units to retain the block length ratio constant at ca. 2.5 (hydrophilic vs. hydrophobic)..
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of block copolymer precursors, dynamic emission spectra of CCFNP1, DLS and TEM characterizations of CCFNP8, and UV–vis and fluorescence spectra of CCFNP8. See DOI: 10.1039/b9nr00304e |
| This journal is © The Royal Society of Chemistry 2010 |