Current directions in core–shell nanoparticle design†
Wolfgang
Schärtl
*
Institut für Physikalische Chemie, Johannes-Gutenberg Universitaet, Welderweg 11, 55099 Mainz, Germany. E-mail: schaertl@uni-mainz.de
First published on 29th March 2010
Abstract
Ten years ago I wrote a review about the important field of core–shell nanoparticles, focussing mainly on our own work about tracer systems, and briefly addressing polymer-coated nanoparticles as fillers for homogeneous polymer-colloid composites. Since then, the potential use of core–shell nanoparticles as multifunctional sensors or potential smart drug-delivery vehicles in biology and medicine has gained more and more importance, affording special types of multi-functionalized and bio-compatible nanoparticles. In this new review article, I try to address the most important developments during the last ten years. This overview is mainly based on frequently cited and more specialized recent review articles from leaders in their respective field. We will consider a variety of nanoscopic core–shell architectures from highly fluorescent nanoparticles (NPs), protected magnetic NPs, multifunctional NPs, thermoresponsive NPs and biocompatible systems to, finally, smart drug-delivery systems.
 Wolfgang Schärtl | Wolfgang Schärtl received his PhD in Chemistry in 1992 for his work on video microscopy of fluorescent nanoparticles, and Brownian dynamics computer simulations. As a post-doc, he joined the Hashimoto ERATO project, where he investigated the structure and dynamics of copolymer micelles in polymer melts. In 1995 he went to M. Schmidt at the University of Mainz as a research fellow, where he finished his “habilitation” in 2001 with a thesis on functional core–shell nanoparticles. Since 2005 he has held a permanent position as lecturer and researcher. His current research interests include nanoscopic optical tracers to investigate the dynamics in nanoparticle-polymer composites, and photoreactive nanoparticles as building blocks for supramolecular structures. |
Introduction
A spherical nanoparticle with a core–shell architecture is a feasible way to combine multiple functionalities on a nanoscopic length scale.1 Some older examples from our own previous work include the chemical incorporation of fluorescent or photoreactive organic dye molecules within a nanoporous core surrounded by a non-functional crosslinked shell,2 or surface coating of nanoparticles with synthetic polymer chains.3 These earlier nanoparticle architectures were mainly motivated by basic research interests, like optical tracer diffusion measurements in concentrated colloidal dispersions where the dye label should not influence the interparticle interactions,4 or optimized preparation of homogeneous nanoparticle-polymer composites. Since then, the focus in nanoparticle design has shifted to more complex nanoscopic core–shell architectures potentially useful for biomedical applications, for example as smart sensor materials or in the field of drug targeting. This interest in multifunctional biocompatible nanoscopic core–shell systems has triggered a strong synthetic progress in the combination of supramolecular surface chemistry and nanoparticle synthesis, with such a far-fetched ultimate goal like smart bombs, that is, nanoscopic vehicles which are capable of safely being incorporated within the human body, carrying a poisonous drug to tumor cells only, and releasing the drug load exactly at the location needed, thereby minimizing the collateral damage still so common in cancer therapy.This review is organized as following: first, the synthesis and application of multifunctional silica nanoparticles based on highly fluorescent tracer cores surrounded with a silica shell will be described, an overview mainly based on recent articles by Prof. Wiesner, Cornell, US,5 and Prof. Liz-Marzán, Vigo, Spain.6 In the second section, magnetic core nanoparticles surrounded with a protective or even biocompatible shell are discussed in some detail, following a recent contribution by Prof. Schüth, Mülheim, Germany.7 In the third section of this review, I will present the strategy of coating functional nanoparticles with poly(ethylene glycol) (PEG) as a feasible way to render nanoparticles biocompatible, focussing on contributions by Prof. Kataoka, Nagoya, Japan.8 A special system of nanoparticles solubilized in mutual media by a polymer coating are poly(N-isopropylacrylamide) (PNIPAM) coated gold clusters, first prepared in a well-defined way as core–shell nanoparticles by Prof. Tenhu, Helsinki, Finland, and co-workers.9,10 Importantly, this PNIPAM coating is thermoresponsive, thereby not only making the NPs compatible with water but also allowing the manipulation of the shell size with temperature. To conclude the issue of hydrophilic and biocompatible nanoparticles, a recent review article on hydrophilic nanoparticles prepared by phase transfer, by Prof. Parak, Munic, Germany,11 will be summarized in section 4. Finally, in the last section about so-called mechanized multifunctional core–shell nanoparticles, I will discuss some highlights from a recent very nice review by Prof. Stoddard, North Western University, US,12 on drug delivery nanoparticles, or, as he calls them, “smart bombs”.
Core–shell nanoparticles
1. Silica-embedded fluorescent nanoparticles – from highly fluorescent tracers to multifunctional sensor systems
Silica plays an important role in the preparation of core–shell nanoparticle systems due to its excellent physical and chemical properties: it is optically transparent, easily functionalized, and the simple and robust synthesis of silica particles from the monomer tetraethoxysilane (TEOS) has been established for many decades, most frequently following the Stöber procedure.13 Also, a variety of functional building blocks, trimethoxy- and triethoxysilanes, are commercially available. A very interesting and important application of silica nanoparticles is to use them as a host for functional organic molecules: for example, if fluorescent dye molecules are embedded in a silica (or polyorganosiloxane) matrix, the protective shell in combination with the immobilization of the fluorophores leads to a strong enhancement in fluorescence quantum yield.2 In addition, the local concentration of dye molecules embedded in a dispersible silica nanoparticle serving as a carrier may strongly exceed the solubility limit of the pure dye molecules. Taking both effects into account, highly fluorescent core–shell nanoparticles useful as optical tracers have been prepared by us and other groups, one famous recent example being the so-called C-dots developed by Professor Wiesner and co-workers at Cornell.5,14 These nanoscopic “superfluorescent” dyes have excellent optical properties like high quantum yield and strongly enhanced photoresistance towards bleaching, the latter mainly due to the fact that the silica shell protects the dye molecules from atmospheric oxygen, recommending C-dots for sensor systems in bio-medical applications. Interestingly, the C-dots nearly reach the luminescence performance of semiconductor quantum dots, which are much more difficult to prepare, and also may be toxic if not adequately covered with a protective shell. An additional advantage of the C-dot architecture compared to semiconductor nanocrystals is the absence of stochastic blinking, since one silica nanoparticle contains several organic dye molecules which emit independently. Also, the silica shell not only enhances the fluorescence quantum yield and photostability, but also spatially separates the organic dye molecules, thereby reducing the probability of interdye energy transfer and fluorescence quenching.Last but not least, the silica surface allows for chemically simple additional functionalization. For example, if one combines the fluorescent functionalization of the silica nanoparticles with an outer magnetic layer, the silica-dye architecture may provide the synthetic basis for more sophisticated multifunctional sensor systems. Fig. 1 sketches this approach from silica-embedded fluorescent dye molecules towards multifunctional, in this case magnetic, sensor nanoparticles.
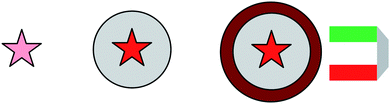 | ||
| Fig. 1 From single fluorescent organic dye molecules to super fluorescent silica-dye nanoparticles, to multifunctional core–shell sensor systems (here: magneto-optical sensors). | ||
Here, the dye label allows for optical sensing, while the magnetic shell allows for manipulation of the particles by magnetic fields. One potential application of such a nanoscopic multifunctional core–shell architecture would be its use in hypothermal therapy: the dye labels allow to track the particle location via their luminescence, and the magnetic shell serves to transfer heat to the position of the nanoparticles via magnetic excitation.
To confirm that the organic dye molecules embedded in the C-dots are not in contact with the environment of the nanoparticles but safely encapsulated inside the silica matrix, Wiesner and co-workers studied the dye fluorescence emission profiles in various solvents, and found negligible solvatochromic shifts. To prepare a variety of nanoscopic sensors, the particle emission wavelength could easily be tuned by incorporating different organic dye molecules within the respective silica nanoparticles. Also, organic dyes which perform best in a non-aqueous environment are rendered water-soluble by encapsulation in a silica matrix, leading to optimized hydrophilic fluorescence labels, which usually are much less expensive as well as more easily prepared than single dye molecules optimized for application in aqueous environment by complex synthetic modifications of hydrophobic precursor dyes. Importantly, the Stöber method13 allows a simple control of fluorescent core and non-fluorescent shell size by variation of the reactant concentration, both in the range from nanometres to microns. For example, Wiesner et al. prepared tetramethylrhodamine isothiocyanate (TRITC)-based nanoparticles with 50 nm to 1500 nm diameters, and pure silica shell thickness >10 nm in each case. Since this shell is thicker than the radius of radiation-less energy transfer by the Förster mechanism,15,16 there exists no energy transfer between different dye-labeled particle cores and therefore no loss in luminescence.
To create multifunctional particles simultaneously useful for sensing and drug loading, the mesoporosity of the silica matrix can be controlled in the pore size range 2–50 nm by adding amphiphilic structure-directing agents to the reaction mixture during particle growth.17,18 These mesoporous silica materials with extremely high internal particle surface are classified in the so-called M41S family, MCM-41 (hexagonal) and MCM-48 (cubic bicontinuous) being the most important structures. The size control of nanoscopic pores within the silica particles allows for encapsulation of substrate molecules of suitable size, a prerequisite for drug-delivery systems. Here, one should note that silica is not biodegradable, which, compared to conventional biodegradable drug carrier systems, affords the development of more sophisticated strategies for controlled drug release, for instance by the design of responsive nanoscopic gates at the outlet of the pores. This topic will be discussed in more detail in section 5 of this review. In this first section, I will briefly describe a more simple nanoscopic drug carrier system where the drug is simply released by diffusion out of the nanoscopic silica pores: using hexadecyltrimethylammonium bromide as the structure-directing agent, Wiesner and cow-orkers prepared fluorescent MCM-48 mesoporous silica nanoparticles with particle size 400 nm and a narrow pore size distribution of 2.74 ± 0.11 nm. Before the pores were filled with suitable substrate molecules, in this case camphotecin, a powerful chemotherapeutic drug which however lacks water solubility, the templating surfactant was removed by several washing steps. The purified MCM-48 particles then were loaded by dissolving the particles in a camphotecin dimethylsulfoxide solution, and free camphotecin was removed by washing of the loaded nanoparticles in phosphate-buffered saline solution. Drug release studies, where the camphotecin was simply diffusing out of the pores due to osmotic pressure, showed that the mesoporous nanoscopic silica system showed a release rate nearly identical to that of biodegradable poly(lactic-co-glycolic acid) (PLGA) polymer-based particles, which is surprising since the PLGA matrix is completely destroyed during the release process while the mesoporous silica particles remain intact.
Wiesner et al. also developed fluorescent C-dots 50 nm in size encapsulated in a gold shell, a system interesting due to the tunable plasmon resonance of the metal shell achieved by simply changing the ratio of core-to-shell thickness. To grow this thin gold shell, nanosized (1.5–3 nm) gold colloids were first adsorbed onto the silica particle surface via silica-bound amine groups, the amines serving as a ligand which attaches to the gold nanoparticle surface. These metal seeds then provide the nuclei for a homogeneous gold shell growth by solution-phase reduction of HAuCl4 in presence of hydroxylamine hydrochloride. Besides simple photoexcitation, the light absorption of the gold shell may also be used for local heating, and therefore be employed for photothermal therapy in cancer treatment. Here, the multifunctional particles serve both as sensors due to the fluorescence of the inner C-dots, and as stimuli-responsive actuators due to the outer gold shell. It has been shown that indeed cells can be killed by this approach.
The review article by Wiesner and co-workers concludes with a set of nice examples of the application of multifunctional C-dots in biology: 1. To combine fluorescence imaging and biological cell targeting, 30 nm TRITC-C-dots were exposed to Immunoglobulin E (IgE) antibodies which bind via adsorption to the silica surface. If these particles are brought into contact with Rat Basophilic Leukemia (RBL) cells, they bind selectively to the cell surface via a specific antibody-receptor interaction, as confirmed by fluorescence microscopy. 2. Alternatively, the C-dot sensors were used for local chemical sensing by employing analyte-specific effects on the wavelength, lifetime or quantum yield of fluorophore emission. Here, a so-called ratiometric approach has proved to be most effective. One combines two different dye species within a given nanoparticle, one of which serves as an internal standard, while the second is responsive to the analyte concentration. The ratio of the emission intensities of the two different dyes then allows one to determine the local concentration of the analyte. Wiesner et al. chose to incorporate the standard dye safely inside the core of the C-dot, where it is well protected from any influence of the environment. The sensor-dye then is incorporated covalently in the nanoporous particle shell, where it comes easily into contact with the analyte molecules. For proof of principle, TRITC was used as the internal standard and fluorescein isothiocyanate as a pH-sensitive sensor dye of a ratiometric C-dot-based core–shell nanoparticle. These particles were then used to monitor the local pH within RBL cells. Since these cells do not easily uptake 70 nm particles, phorbol 12,13-dibutyrate, a common reagent in DNA transfection, was added to facilitate endocytosis of the fluorescent sensor particles into intracellular vesicles. The TRITC in this case not only serves as internal standard for the ratiometric pH sensor, but also for imaging the cellular uptake and tracking the sensor particles within the cell. These nice particles allowed Wiesner et al. to map the pH, ranging from 5.0 to 7.5, within a single RBL cell.
To conclude the first section focusing on fluorescent core–shell nanoparticles, let me briefly review the recent work of the group of Prof. Liz-Marzán on composite silica nanospheres with magnetic and fluorescent functionalities.6 As mentioned, magnetic nanoparticles consisting of maghemite (γ-Fe2O3) and magnetite (Fe3O4) are becoming increasingly interesting for potential medical application in hyperthermic therapy, since they respond to a magnetic field by a local increase in temperature. To stabilize these tiny particles, and also to render them water-soluble, the simplest and most feasible strategy is to encapsulate them in a nanoscopic silica shell. The advantages of such silica shells, such as optical transparency, simple chemical functionalization, or simple tuning of particle size, have already been discussed. To combine optical sensing and magnetic response, Liz-Marzán et al. first prepared iron oxide nanoparticles encapsulated in a silica core of size 30 nm by an in situ Stöber synthesis. These precursor core–shell nanoparticles were then covered with a defined silica shell of thickness 70 nm, yielding an average total particle diameter of 170 nm. However, since the iron oxide particles do not form a stable dispersion and tend to aggregate, by this simple procedure several magnetic nanoparticles were encapsulated at once. To combine the magnetic functionality with optical sensor properties, fluorescent semiconductor CdTe quantum dots stabilized by thioglycolic acid were adsorbed via electrostatic attraction onto the silica shell, which had previously been modified by layer-by-layer assembly,19–22 that is, deposition of a precursor multilayer polyelectrolyte film to generate a uniform surface charge and smooth particle surface. These composite magnetic-luminescent particles were finally coated with an additional outer silica layer of thickness 20 nm to further improve the colloidal and chemical stability. As demonstrated, the final core–shell nanoparticles could be directed to specific locations via a magnetic field, while their movement could be tracked due to the fluorescence of the semiconductor quantum dots.
Finally, we should note another nice example of bifunctional magnetic-optical core–shell nanoparticles not involving silica as an inert protective matrix but only the functional materials themselves, developed by Klimov and co-workers.23 They deposited CdSe quantum dots of size 2–3 nm onto preformed Co nanocrystals of 11 nm size and stabilized by organic surfactant molecules, leading to a uniform core–shell nanosphere. These composite core–shell particles were soluble in hexane due to the trioctylphosphine ligands covering the CdSe nanocrystal surface.
Note that, whereas the preparation of tiny nanocrystals which are soluble in non-polar solvents is well-established by now,24 it is still non-trivial to obtain hydrophilic metal, semiconductor or metal oxide nanoparticles with physical properties, like fluorescence quantum yield etc. comparable to their hydrophobic counterparts. However, since such hydrophilic particles are promising candidates for biomedical applications, there has been an increasing interest in the synthesis of suitable hydrophilic nanocrystal core–shell systems during the last 10 years. Concerning the preparation and application of biocompatible fluorescent semiconductor nanocrystals, a few years ago Medintz et al. have published a frequently cited overview.25 In this review, I will address this issue more generally in the next three sections which describe recent developments in the synthesis of multifunctional and biocompatible nanoscopic core–shell particles, using a variety of different approaches and chemical structures.
2. From simply protected to biocompatible magnetic NPs
Whereas in the previous section our starting point was nanoparticles labelled with fluorescent molecules for sensing applications, here we focus on magnetic core–shell nanoparticles. The major role of the non-magnetic shell is to protect the functional core from damaging environments like oxygen, as well as to render the nanoscopic colloids water-soluble or even make them compatible with biological media, as sketched in Fig. 2. This section is mainly based on a recent review by Professor Schüth,7 who is a leading expert in carbon nanoparticles, and subsequent hybrid structures.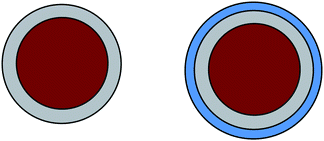 | ||
| Fig. 2 From simply protected to biocompatible magnetic nanoparticles. | ||
Magnetic nanoparticles are technologically very important: potential applications range from magnetic fluids to catalysis, biomedication and resonance imaging, to data storage. These particles show the best properties in a size regime 10–20 nm, where each particle behaves as a single magnetic domain with superparamagnetic behavior. An important limitation for any technological application is the colloidal and chemical stability of the particles under various conditions, such as an oxygen atmosphere, temperature, hydrophilic solution etc. To protect the particle surface against corrosion or degradation, various strategies all leading to a nanoscopic core–shell structure are feasible: grafting or coating with polymers or surfactants, or coating with an inorganic layer such as silica, discussed in detail in the first section, or carbon coating, which will be the new main focus in this section. Besides a simply protective coating, I will also address special functional coatings which influence the magnetic properties of the nanoscopic particle core.
Details of the superparamagnetic behavior of magnetic nanoparticles have been described elsewhere26,27 and will therefore not be repeated here, where we are more interested in the general design and structural properties of a core–shell architecture. Besides chemical degradation or oxydation, physical surface effects can decrease the magnetization of small particles. Also, if they get too close, magnetic particles can show cooperative switching, which is undesirable in storage applications. Here, an inert silica shell not only protects the particle surface, but also serves as a well defined spacer to keep individual magnetic nanoparticles apart. The role of other coating materials may be less obvious: gold-coated cobalt nanoparticles, for example, show a lower magnetic anisotropy than uncoated particles, whereas gold-coated iron particles show the opposite behavior.28 It seems to be state-of-the-art that magnetic core and metal coating may show interactions too complex to predict the resulting magnetic behavior. Even organic ligands may have unusual effects on the magnetic properties: donor ligands like amines do not influence the surface magnetization but cause the formation of rod-like magnetic nanoparticles due to their specific binding to certain crystal facets of the growing nanoparticle,28 whereas trioctylphosphine causes a reduction of the particle magnetization.29 In conclusion, even coating molecules expected to be inert may influence shape and/or magnetic properties of the core particles.
One special case is the coating of a magnetic core with another magnetic material, in which case a very strong effect on the magnetic core properties is to be expected. An example is the so-called exchange bias, i.e. a shift of the hysteresis loop along the field axis in systems with ferromagnetic–antiferromagnetic interfaces, an effect discovered first for Co cores surrounded with a CoO layer.30 To produce high-performance permanent magnetic nanoparticles, one can combine a soft magnetic phase (easily magnetized, such as Fe3Pt) and a hard magnetic phase (difficult to magnetize, such as Fe3O4) which then show magnetic exchange coupling.31,32 Finally, a core–shell structure where both materials are strongly magnetic allows a precise adjustment of the magnetic properties like anisotropy and magnetization via the dimensions of the core and shell, respectively.
Next, let us consider the synthetic approaches towards magnetic nanocrystals in more detail. Magnetic cores can be synthesized in a variety of ways. Co-precipitation of iron oxides from aqueous Fe2+/Fe3+ solutions by the addition of a base under inert atmosphere at room temperature is a facile and well-established procedure. The resulting magnetite particles, however, are easily oxidized and unstable in acidic solution. Other issues are size control and polydispersity of the magnetic cores. Here, organic surfactants such as oleic acid or trisodium citrate, the latter even serving as a reducing agent, are used to control the size and obtain stable magnetic nanoparticle dispersions.
Analogous to high-quality semiconductor nanocrystals,33–35 stabilized hydrophobic magnetic nanoparticles have also been prepared by thermal decomposition of organometallic compounds in high-boiling organic solvents in the presence of surfactants (fatty acids, oleic acid or hexadecylamine). Peng et al., for example, reported a general decomposition approach for the synthesis of size- and shape-controlled magnetic oxide nanocrystals by pyrolysis of metal fatty acid salts in non-aqueous solution.36 Water-soluble magnetite nanoparticles were prepared from FeCl3–6H2O as iron source and 2-pyrrolidone as coordinating solvent under reflux at 245 °C, in which case the reflux time serves to control the particle size.37 Not only nanoscopic oxides, but also metallic nanoparticles were prepared by thermal decomposition: iron particles have been synthesized by decomposition of [Fe(CO)5] in decalin in the presence of poly(isobutene), serving as a stabilizing polymer layer of thickness 7 nm whose respective amount controls the iron particle size from 2–10 nm, at 170 °C.38 In the case of decomposition of cobalt organyls, both the size and shape of the nanoscopic metal particles can be controlled via the choice of the correct amount and species of surfactant.39
Alternatively, microemulsions are used as a templating nanoreactors to prepare well-defined magnetic nanoparticles and even core–shell particles. For instance, gold-coated Co/Pt nanoparticles have been prepared in reverse micelles of cetyltrimethylammonium bromide with octane as the oil face and 1-butanol as the co-surfactant.40 One disadvantage of the microemulsion approach compared to the other strategies (co-precipitation and thermal decomposition) is the low yield compared to the large amount of solvents needed, making this approach non-practicable for the preparation of a larger amount of magnetic core–shell particles as needed for technical applications.
The fourth method to prepare magnetic particles is hydrothermal synthesis, a generalized method recently reported in Nature by Wang et al.41 The reaction mixture consists of a solid phase (metal linoleate), an ethanol–linoleic acid liquid phase, and a water–ethanol solution at different reaction temperatures typically 160–180 °C. The method is based on a phase transfer and separation mechanism occurring at the liquid–solid and the solid–solution interface, respectively, and it affords high pressure. Of all four methods so far described, it allows the best size and shape control for preparation of magnetic nanoparticles which are stabilized by surface-capping agents. However, since it was developed just recently, it is less well understood and established than the co-precipitation or thermal decomposition approach, which are nowadays most-frequently used to prepare defined magnetic core–shell nanoparticles on a larger scale.
Irrespective of how the stabilized magnetic nanoparticles have been prepared, usually they have to be protected from oxidation in air, or erosion by acids or bases. There exists two strategies in principle: coating with organic shells including surfactants and polymers, or coating with inorganic shells including silica, precious metals or oxides. The chemically most simple protection would be surface passivation by mild oxidation. For example, Boyen et al. showed that a controlled oxide layer was formed around Co nanoparticles exposed to an oxygen plasma.42 Similarly, Bönnemann et al. used synthetic air to smoothly oxidize freshly prepared Co nanoparticles to form a stable outer CoO layer, protecting the magnetic nanoparticles from further oxidation and thereby conserving their magnetic properties.43
To avoid aggregation of the nanoparticles and enhance the colloidal stability of ferrofluids, careful control of the surface charge and/or the use of specific surfactants is necessary. One strategy is the introduction of surface groups which become charged at a certain pH: for example, nowadays commercial ferrofluids are available which are stable either at pH <5 (acidic ferrofluid) or pH >8 (alkaline ferrofluid).
A polymer coating is also one established synthetic strategy to protect functional nanoparticles and enhance their colloidal stability by introducing a defined surface charge. Using simply the adsorption of polymers containing functional groups, such as carboxylic acids, phosphates and sulfates, to the surface of magnetite nanoparticles, such a polymer coating can be achieved. Suitable polymers are poly(pyrrole), poly(aniline), and polyesters, which all may interact with the surface of the magnetic nanoparticle due to their free electron pairs. Instead of adsorption, or grafting-onto, the polymer coating alternatively can be grown by a grafting-from polymerization from the nanoparticle surface. As one example, Vestal et al. prepared 9 nm core MnFe2O4 nanoparticles with an outer poly(styrene) shell of thickness 6 nm using 3-chloropropionic acid as a water-soluble ligand and initiator for atom transfer radical polymerization.44 Here, surface-grafting of the initiator is the crucial step to obtain a well-defined core–shell architecture. In general, an important drawback of polymer coatings is that, if they are too thin, they provide an insufficient barrier towards oxygen or small ions, and therefore cannot protect the magnetic nanoparticles from degradation. Also, a polymer coating usually is not very stable at elevated temperatures.
An alternative protection of magnetic nanoparticles from oxidation is a precious-metal coating. Pt-coated Co nanoparticles of total size <10 nm have been prepared by simply refluxing 6 nm Co colloids and Pt(II) di-hexafluoroacetylacetonate in a nonane solution in the presence of C12H25CN as a stabilizer.45 The reaction mechanism is redox transmetalation between Co0 and Pt2+, and these air stable particles can be redispersed in non-polar organic solvents due to their outer surfactant layer. Although gold is an ideal coating because of its low chemical reactivity, the direct coating of magnetic nanoparticles with Au is very difficult due to the dissimilar surfaces. One of the few successful examples are 11 nm iron nanoparticles coated with a 2.5 nm thin gold shell by a partial replacement reaction in a polar aprotic solvent.46 Gold-coated iron nanoparticles were also prepared by reverse microemulsion, where the micelles serve as nanoreactors.47 An alternative method consists of a combination of wet chemistry and laser irradiation: iron nanoparticles and gold powder were irradiated in a liquid medium, leading to 18 nm body centered cubic (bcc) iron single cores covered with a partially fused shell of 3 nm face centered cubic (fcc) gold nanoparticles.48 One additional big advantage of a gold coating besides its low chemical reactivity is that it easily can be further functionalized, using thiol groups as linkers. Thereby, multifunctional core–shell nanoparticles can be prepared in a well-defined way.
As an alternative frequently used coating, a silica shell not only protects the magnetic core, but since it can easily be chemically modified also may serve as a spacer to separate the core and additional functionalities, as for example organic dye molecules, to avoid unwanted interactions like luminescence quenching. As an example, magnetic nanoparticles were first coated with a thin silica shell, onto which the dye molecules were grafted.49 It is especially simple to coat magnetic iron oxide nanoparticles with a silica layer since the oxide surface easily binds to silica through OH surface groups. Lu et al. have shown that commercially available ferrofluids can be directly coated with silica by simple hydrolysis of tetraethoxysilane (TEOS).50 Since at pH >8 this silica surface is negatively charged, the resulting core–shell nanoparticles are dispersible in water. Importantly, the controlled coating of magnetic nanoparticles with a very thin silica shell on the nanometre scale still remains a challenge. A successful example is the preparation of homogeneous silica-coated Fe2O3 nanoparticles with a shell of controlled thickness between 1.8 and 30 nm, using the reverse microemulsion technique.51 Once again, the nanoreactor approach proves to be well-suited to the preparation of defined nanoscopic architectures, besides its major drawback concerning large-scale particle fabrication. In contrast to the simple silica coating of oxides, silica deposition onto metal nanoparticle surfaces is far more complicated. In addition, Fe and Co are readily oxidized in the presence of dissolved oxygen, which leads to a loss in magnetic properties. The strategy to overcome this problem is to create a glass-like (“vitreophilic”) nanoparticle surface by employing suitable primers. Alternatively, Co nanoparticles passivated by the mild oxidation method, and therefore possessing a thin outer layer of Co oxide which easily binds to the silicate building blocks, may be used as starting materials to prepare silica-coated magnetic Co nanoparticles.
An alternative similar in chemistry to the pure silica coating discussed so far is coating with a nanoscopic shell of trimethoxysiloxane to form an organosilicon layer.52 Compared to silica, this material, due to its higher porosity, has a lower density comparable to that of most solvents. Therefore, the resulting core–shell particles are usually stable against sedimentation even if their overall size exceeds several hundred nanometres, in which case most silica-based particles precipitate from dispersion within hours to days. In addition, the organosilicon nanoparticles possess a locally hydrophobic pore structure which may be advantageous for the encapsulation of hydrophobic guest molecules, and an organosilicon network can be functionalized at least as easily as the pure silica system. One example of embedding magnetic nanoparticles within an inert organosilicon matrix has recently been published by Utech et al.:53 oleic acid stabilized 6 nm magnetic maghemite particles were first dispersed in octadecyltrimethoxysilane, leading presumably to the formation of a double layer at the particle surface with trimethoxysilane groups at the outside. These reactive groups are then used as precursors to grow a shell consisting of methyltrimethoxysilane, dimethyldimethoxysilane and p-chloromethylphenyltrimethoxysilane around the magnetic particles. Here, the mixture of tri- and difunctional monomers is used to adjust the degree of crosslinking or pore structure of the nanoscopic coating. The chloromethylphenyltrimethoxysilane introduces functional chloromethyl groups into the particle shell which can be used, for example, to chemically fix organic dye molecules within the shell as successfully demonstrated before,2 leading to multifunctional protected magnetic nanoparticles. According to TEM images, the product consisted of both pure organosilicon particles and larger core–shell particles, the latter containing typically 1–3 magnetic nanoparticles in the center. This result indicates partial aggregation of the magnetic nanoparticles while they were dispersed in octadecyltrimethoxysilane, a problem frequently encountered in the preparation of silica- or silicon-based nanoscopic core–shell architectures.
Besides its mentioned benefits, the silica (or silicon) coating also has a few important drawbacks as a protective coating for magnetic nanoparticles: it is an insufficient barrier against oxygen due to its nanoporous structure, and it is not very stable under highly basic conditions. As a better alternative compared to the polymer and silica coatings discussed so far, coating with carbon-based materials has the major advantage of its higher chemical and thermal stability as well as biocompatibility. Well-developed graphitic carbon layers can be formed around metal nanoparticles by arc-discharge, laser ablation, and electron irradiation, using a variety of organic molecules as the carbon source. These carbon layers provide effective barriers against oxidation and acidic corrosion. Also, they are readily formed around metal nanoparticles, which conventionally are more difficult to coat than corresponding oxides, but have better magnetic properties. In a very simple way, Johnson et al. prepared polydisperse carbon-coated magnetic Fe and FeC nanoparticles in the size range 20 nm to 200 nm by direct pyrolysis of iron stearate under an Ar atmosphere.2 These particles, which were covered by 20 to 80 graphene layers, are stable under air up to 400 °C. Smaller carbon-coated nanoparticles were prepared by first covering Co nanoparticles with poly(furfurylalcohol), which was then carbonized by pyrolysis.54 Unfortunately, carbon-coated nanoparticles are often not obtained as well-defined monodisperse core–shell nanoparticles but as agglomerated clusters, since their formation mechanism and synthetic methods are not yet well-understood. Therefore, the preparation of dispersible well-defined carbon-coated core–shell nanoparticles still provides a major challenge in this field.
Finally, let us consider further functionalization of magnetic nanoparticles with catalytically active species, drugs, specific binding sites or other groups. The magnetic properties here allow a controlled manipulation of these multifunctional particles by external magnetic fields, as already sketched in Fig. 1. Our first examples were iron oxide nanoparticles coated with a silica shell subsequently functionalized with gold nanoparticles.55 The magnetic core silica shell nanoparticle was formed by the Stöber process, and then coated with a positively-negatively-positively charged polyelectrolyte trilayer by simply adsorbing the polymer chains due to electrostatic attraction. Finally, negatively charged citrate-stabilized gold nanoparticles were adsorbed onto the outer positive polyelectrolyte layer, and then a continuous gold shell was formed on the basis of these precursor seeds. The resulting multishell nanoparticles can be manipulated by magnetic fields, and have a strong resonance absorption in the visible and near-infrared range.
The preparation of magnetic nanoparticles directly coated with a functional polymer, on the other hand, is more difficult since it has not yet been possible to carry out a polymerization reaction on the surface of magnetic nanoparticles. One solution to this problem is a colloidal template polymerization: silica-coated magnetic nanoparticles prepared by the Stöber method were surface-functionalized with 3-(trimethoxysilyl)propyl methacrylate to introduce C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups, and N-isopropylacrylamide (NIPAM) and a crosslinker, N,N′-methylene bisacrylamide, were polymerized in the presence of these functional nanoparticles.56 This synthetic approach lead to thermoresponsive magnetic microspheres. Finally, hydrophobic nanoparticles can be converted into hydrophilic ones by ligand exchange, a procedure which will be reviewed in more detail in section 4.
C groups, and N-isopropylacrylamide (NIPAM) and a crosslinker, N,N′-methylene bisacrylamide, were polymerized in the presence of these functional nanoparticles.56 This synthetic approach lead to thermoresponsive magnetic microspheres. Finally, hydrophobic nanoparticles can be converted into hydrophilic ones by ligand exchange, a procedure which will be reviewed in more detail in section 4.
The review article by Schüth and co-workers concludes with a brief review of the application of magnetic nanoparticles in catalysis and biotechnology. As an example of a new catalyst, core–shell-type cobalt–platinum core–shell nanoparticles, with dodecyl isocyanide as stabilizers at the Pt surface, have been prepared by redox transmetalation.45,57 Here, the Pt atoms are used economically since they are all located close to or at the particle surface, while the magnetic Co core allows simple separation and recycling of the catalyst after the reaction. One example for biological applications are magnetic iron oxide nanoparticles grafted with dopamine used for protein separation. A more sophisticated separation system was prepared by Zhao et al., a genomagnetic nanocapturer for collection, separation and detection of trace amounts of DNA/RNA molecules with a single-base difference.58 These particles consist of a magnetic nanoparticle core, a silica coating as protecting and biocompatible layer, and avidin-biotin molecules as linkers for bioconjugating a molecular beacon as DNA probe. Another interesting field is magnetic drug delivery: magnetic particles with attached drug molecules are dragged to the target site by a magnetic field, held there during drug release, and removed at the end of the treatment. Finally, magnetic nanoparticles with a stable and biocompatible outer shell may become very important in hypothermia treatment, since heat is generated by the magnetic hysteresis loop if the particles are exposed to a varying magnetic field. Besides more and more promising examples found in recent research articles, however, the defined synthesis of such multifunctional particles still remains a major challenge, not mentioning biomedical applications.
3. Biocompatibilization/making hydrophilic nanoparticles by polymer coating
In this section, I will review the preparation of polymer-coated core–shell nanoparticles in more detail. We will first focus on poly(ethylene glycol) (PEG) based block copolymers, since PEG is an interesting coating material for nanoparticles for biological and medical applications due to its biocompatibility. A simple strategy to prepare colloidal nanospheres with a PEG coating is to utilize the self-organization of amphiphilic copolymer chains into micelles: in aqueous solution, block copolymers with a hydrophobic block and a hydrophilic PEG block form rather stable (compared to simple surfactants) micelles with very low critical micelle concentration (cmc). These micelles can be considered as amphiphilic core–shell nanoparticles of size typically 30–50 nm.59 The hydrophilic micellar core can be loaded with drug molecules, which usually stay there for longer times due the slow rate of dissociation of the micelles, and the outer shell can be functionalized to allow drug targeting, leading to a smart drug-delivery system with slow release.60 Also, metal and semiconductor nanoparticles have been successfully incorporated into the cores of such amphiphilic copolymer micelles, leading to a multifunctional core–shell architecture as sketched in Fig. 3.61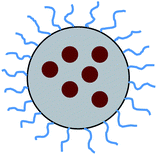 | ||
| Fig. 3 Amphiphilic copolymer micelle whose core is loaded with functional nanoparticles, e.g. Au, CdSe etc. | ||
A nice micellar copolymer system was developed by Kataoka and co-workers, who found that a mixture of PEG-block-poly(L-lysine) (PEG-b-PLL) and PEG-block-poly(α,β,-aspartic acid) (PEG-b-P(Asp)) spontaneously associates to form micelles with a core composed of a polyion complex of the oppositely charged PLL and P(Asp) segments. In contrast to micelles formed in aqueous solution from conventional amphiphilic copolymers, in which case the micellar core is hydrophobic and non-polar, these polar cores can easily be loaded with charged macromolecules, including even proteins and nucleic acids.62–65
The stabilization of gold nanoparticles for in vivo biomedical applications, for example with PEG-based copolymers, is essential, since the pure gold particles aggregate in high ionic strength milieu as well as adsorb biomolecules non-specifically, effects which lead to reduced sensitivity and selectivity of the colloidal nanosensor in a biological fluid. Here, the block copolymer micelles can directly be used as templates or nanoreactors to form noble metal nanoparticles in situ within the micellar core. In this case, the core has to be hydrophilic.
One general concept for the preparation of transition metal nanoparticles embedded within micellar cores, following the Pearson hard–soft concept,66 is to start from weakly coordinated metal ions, e.g. Pd(OAc)2 or Pd(ClO4)2, which are complexes of a soft acid (the transition metal ions) and a hard base (acetates, perchlorates, etc.). The formation of a more stable complex of the soft transition metal ion with a softer base, e.g. poly(vinylpyridine), to assemble the micellar core, is the driving force for solubilization of the metal ions inside the micellar core. A chemical reduction process then leads to the formation of nanoscopic metal particles. Use of water as a reaction medium is possible if double-hydrophilic block copolymers, where only one block can coordinate with the metal ions, are used to form the micelles. One example is PEG-b-poly(ethyleneimine) PEI, where the PEI block can complex noble metal ions from salts as AuCl3, PdCl2 or H2PtCl6.61 For example, addition of a gold salt to an aqueous PEG-b-PEI solution and subsequent reduction therefore lead to the formation of polydisperse micelles filled with gold nanoparticles. Here, it should be noted that precise size control and formation of stable micelles to avoid aggregation is non-trivial for this aqueous system of double-hydrophilic copolymers, compared to conventional amphiphilic copolymer micelles.
To prepare PEG-coated metal nanoparticles, besides micellization and in situ formation of the metal particle inside a micellar core, PEG polymer chains with suitable functional groups can also be grafted directly onto the particle surface. This grafting-onto of PEG may either take place via ligand exchange in a second step after the metal nanoparticle synthesis, or as a single-step process in situ during particle growth. For example, to prepare stable multifunctional core–shell gold nanoparticles, Kataoka and co-workers used heterobifunctional PEG derivatives, containing both mercapto and acetal terminal groups. The acetal moiety can readily be transformed into a reactive aldehyde group, while the SH group strongly binds to the gold particle surface. Bifunctional PEG chains of molar mass 3090 were successfully grafted onto gold nanoparticles of size 8.9 nm during particle formation, i.e. reduction of HAuCl4 with NaBH4. Next, lactose as a model ligand was connected to the distal end of the PEG chains.67,68 These core–shell gold nanoparticles then form reversible aggregates in a controlled manner in presence of the biomolecule lectin under physiological conditions, and this aggregate formation is monitored by a corresponding change in color from red (for single gold nanoparticles) to purple, a nice example for a biomedical assay based on optical detection.
A second example for such a gold nanoparticle-based biomedical assay is obtained if a cationic PEG-based block copolymer, PEG-b-poly(dimethylaminoethyl methacrylate) (PAMA), is used as a stabilizer during gold nanoparticle preparation.8 Here, again the metal–ligand coordination between the Au ions and PAMA acts as a driving force to form a micellar core, and the Au salt was reduced in situ to form monodisperse gold nanoparticles in the absence of additional reductive agents. The PEG chain ends then were functionalized with biotin molecules, and the resulting core–shell nanoparticles showed controlled aggregation in the presence of avidin. Due to their surface plasmon resonance, gold nanoparticles play an important role in signal enhancement of biosensing systems,69,70 thereby strongly increasing the sensitivity to detect the immobilized avidin compared to a conventional biotin-avidin-binding assay in absence of gold nanoparticles.
A facile way of preparing luminescent quantum dots for biological applications was developed analogous to the strategy just illustrated for the biofunctional core–shell gold nanoparticles: CdS nanoparticles stabilized by PEG-b-PAMA were formed at room temperature via selective precipitation by simply mixing the copolymer, a Cd(II) salt and Na2S in aqueous solution. The resulting nanoparticles were very stable even in concentrated salt solutions, and in addition could easily be modified for biomedical applications.71
As an alternative to the preparation of fluorescent nanoparticles coated with a selective copolymer, amino-derivatized polysaccharides (aminodextran or Amdex) were used as stabilizers, and the resulting Amdex-CdS nanoparticles were successfully coated with an antibody.72 These nanocomposites, however, were not stable but underwent aggregation in aqueous solution, leading to a dramatic loss of their photoluminescence. This underlines the importance of using selective copolymers like PEG-b-PAMA in the preparation of polymer-stabilized nanoparticles.
A special type of polymer-stabilized nanoparticles are noble metal nanoparticles coated with a thermosensitive water-soluble polymer, poly(N-isopropylacrylamide) (PNIPAM). Tenhu et al. used the grafting-onto method to coat gold nanoparticles with PNIPAM chains of molar mass 5000 g Mol-1 end-functionalized with SH groups, which were prepared in a controlled way by a living polymerization technique (RAFT) and subsequent reduction of dithioester end groups.73 Different reaction conditions were employed for the grafting, and it was found that again the single-step procedure, i.e. simply reducing HAuCl4 and forming nanoparticles in presence of the PNIPAM-SH, was the most facile and successful way, leading to 2 nm gold cores covered with a dense layer of PNIPAM chains of thickness about 4.5 nm. This value is a bit higher than expected for a random coil confirmation of the PNIPAM chains, indicating that the grafted PNIPAM adopts an extended coil configuration. The small size of the Au nanoparticles formed under these conditions, corresponding to a large surface curvature and high surface area, underlines the efficiency of the PNIPAM-SH polymer chains as a stabilizer compared to conventional alkane thiols. Also, comparison with PEG-SH showed that the PNIPAM-SH is an even better ligand and stabilizer. It should be noted, however, that these PNIPAM-coated Au nanoparticles were not stable but still tended to aggregate even in highly dilute aqueous dispersions.
A similar core–shell particle architecture with a nanoscopic gold core and thermosensitive PNIPAM polymer shell was recently presented by Li and co-workers, who instead of grafting-onto used the grafting-from approach in terms of surface-initiated atom transfer radical polymerization (ATRP) as a controlled polymerization technique to grow a defined polymer layer from a gold nanoparticle surface.74–76 Here, the citrate on the gold particle surface first has to be replaced with a disulfide ATRP initiator via ligand exchange, before the polymer layer is grown by ATRP. Using this grafting-from approach, Li et al. prepared a very interesting multifunctional core–shell system: spherical gold nanoparticles with a thermoresponsive poly(N-isopropylacrylamide) and poly(methoxy oligo(ethylene glycol) methacrylate) copolymer shell, which in aqueous dispersion show two transition temperatures near 33 and 55 °C, respectively.75 It should be noted that also pH-responsive core–shell gold nanoparticles were prepared in the Li group by surface-initiated ATRP grafting-from of poly(4-vinylpyridine) (PVP) chains.77,78
We conclude this section on core–shell nanoparticles coated with hydrophilic polymers with another multifunctional PNIPAM-based core–shell system, where the thermoresponsive behavior is used to control the activity of the nanocomposite as a chemical catalyst.79 Ballauff et al. have prepared a polymeric core–shell nanoparticle consisting of a PS latex core of size about 150 nm, surrounded by a crosslinked PNIPAM shell of thickness >70 nm at room temperature. Importantly, this shell will collapse if the temperature is increased to 34 °C due to the reversible phase transition of the PNIPAM. Therefore, the PNIPAM network could act as a nanoreactor which can be opened or closed in a controlled way by changing the reaction temperature. To prove this concept, Ballauff et al. incorporated 8.5 nm Ag nanoparticles in their PNIPAM shell network, formed by reduction of Ag+ ions, which were complexed by the PNIPAM groups of the core–shell polymer nanoparticles in the first step, with NaBH4 added in a second step. The reduction of 4-nitrophenol by NaBH4, where Ag serves as a catalyst, was used as a model reaction to monitor the catalytic activity of the Ag-PNIPAM-PS core–shell nanocomposites. Importantly, the reaction rate constant k depends both on reaction temperature and accessibility of the catalyst to the reactants, the later depending on the swollen state of the PNIPAM network. This leads to a minimum in k at the PNIPAM phase transition temperature 34 °C, where k decreases to less than 20% of its value at T = 25 °C, simply due to the diffusional barrier formed by the collapsed PNIPAM network shell, isolating the Ag catalyst from the reaction.
4. Biocompatible nanoparticles prepared from hydrophobic precursors by the phase-transfer strategy
In this section, we will consider in more detail how to transfer high-quality nanoparticles, like fluorescent quantum dots or magnetic NPs, from non-polar organic media, where they are easily prepared, into aqueous solution, and then how to functionalize them with biological molecules. This transfer and subsequent functionalization are necessary to use such nanoparticles in more sophisticated biomedical applications, like sensing, biological assays or controlled drug delivery.Highly fluorescent semiconductor nanocrystals are often prepared in organic solvents or even surfactants by thermolysis, since in contrast to water these organic media can be heated well above 100 °C. High reaction temperature and absence of water expand the range of materials which can be synthesized, and also lead to annealing out of crystal lattice defects, thereby for example increasing the fluorescence quantum yield of semiconductor nanocrystals. Also, the outer surfactant layer stabilizes the hydrophobic nanoparticles, which therefore can readily be dispersed in non-polar solvents. Here, the absence of aggregation, an undesired effect often found in aqueous nanocrystal systems, further improves the spectroscopic properties of the fluorescent particles.
Typically, CdSe semiconductor NPs are prepared at T = 250–300 °C by thermolysis of the respective inorganic precursors, and the resultant hydrophobic nanoparticles are stabilized by an outer layer of trioctylphosphine oxide (TOPO), sometimes mixed with other amphiphilic molecules including alkyl amines, phosphonic acids or carboxylic acids.33 The synthetic protocols to prepare these particles have been optimized during the last 10–15 years, leading to very monodisperse and well-defined nanocrystals. In addition, it also is possible to grow anisotropic nanocrystals of various shapes in a controlled way, using as stabilizers organic surfactant molecules which bind selectively to certain facets of the growing nanocrystals. Multishell nanocrystals are one further step in complexity of nanoparticle architecture: for example, an epitaxial shell of ZnS has been grown around spherical and rod-shaped CdSe nanoparticles, leading to an enhancement in fluorescence quantum yield.24,80–82 Despite this rapid progress in nanocrystal synthesis, the preparation of defined and stable hybrid materials, like nanocrystals coated with polymers or biomolecules, still provides a major challenge.
As mentioned, to use high-quality hydrophobic nanocrystals in biomedical applications, they first have to be transferred into water while maintaining their colloidal stability. The easiest method to obtain stable hydrophilic NPs is an exchange of the hydrophobic ligands with hydrophilic ones,83–86 the latter either containing charges or being hydrophilic polymer brushes, like PEG or dextran (see Fig. 4). Whereas the charges stabilize the colloidal particles against aggregation by Coulomb repulsion, a polymer coating of the NPs introduces a shorter-range steric repulsion. Since the binding of a ligand depends strongly on the chemical composition of the nanocrystal surface, and for successful exchange the hydrophilic ligand has to bind more strongly than the hydrophobic one, there exists no general protocol for hydrophobic–hydrophilic phase transfer that may work for any type of nanocrystals. For example, hydrophilic thiols like mercaptopropionic acid are typically used to transfer hydrophilic CdSe/ZnS nanocrystals from chloroform into water. However, since the SH group shows only a moderate binding affinity towards the ZnS surface, these hydrophilic nanoparticles are not stable, but form aggregates and even start to precipitate within a day.87 More stable hydrophilic CdSe/ZnS nanocrystals are obtained if a hydrophilic ligand with more than one binding site is used for the exchange.88–90
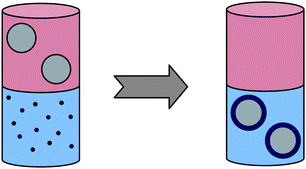 | ||
| Fig. 4 Preparation of biocompatible core–shell nanoparticles from their high-quality hydrophobic precursors by phase transfer. | ||
If the hydrophilic ligands could be crosslinked immediately after the exchange reaction, one would obtain far more stable hydrophilic nanocrystals. This leads to an advanced phase-transfer protocol, for example, first replacing the hydrophobic ligands with suitable silanes, and then crosslinking the hydrophilic layer to form a TEOS shell by polycondensation11,91–97 analogous to the well-known Stöber process. Still, since the first step is a ligand exchange, it is difficult to identify suitable silanes for any nanocrystal species. In conclusion, ligand exchange seems to be not the best strategy for the preparation of hydrophilic nanocrystals mainly due to its chemistry-specific nature.
A more general concept applicable to any type of hydrophobic nanocrystal and not involving ligand exchange is the addition of an extra layer onto the hydrophobic coating of the nanocrystals. Here, this additional outer layer is stabilized by hydrophobic interactions similar to lipid membranes and comparable to vesicular systems. Importantly, this double-layer concept leads to an increase in particle size compared to nanocrystals transferred into water by direct ligand exchange, but also yields more stable colloidal particles. In addition, a large variety of amphiphilic polymers can be used to form the outer double layer, allowing direct incorporation of different functionalities inside the nanoparticle shell.
As one example for this more general extra-layer concept, we have been able to transfer hydrophobic semiconductor core–shell nanocrystals from chloroform into water at high pH values by complexation of the hydrophobic ligand chains with water-soluble peramino-β-cyclodextrin.98 This complexation scheme should work for any type of hydrophobic nanoparticles which are stabilized with hydrocarbon chains. Still, the average particle size, as determined by dynamic light scattering, was much larger in the aqueous phase than in the organic phase, i.e. 45 nm compared to 7 nm, indicating partial aggregation of the NPs during the phase-transfer. However, an even stronger aggregation tendency was found if we transferred our nanocrystals by direct ligand exchange with liponic acid.
For sophisticated biomedical applications, not simply hydrophilic but biofunctionalized nanocrystals are needed. Such nanocrystal-biomolecule conjugates have already been prepared for various purposes, ranging from simple cell labeling to biochemical sensing. Suitable biological molecules can be attached to nanocrystals by two different strategies. The first would be a direct ligand exchange, using for example biomolecules with SH groups which can react with the surface of gold or CdSe nanocrystals by partially replacing the original phosphine and mercaptopropionic acid ligands.99–103 Alternatively, the biomolecules can be chemically bound to the stabilizing shell around the inorganic nanocrystal core.104 The biomolecules can also be bound to the nanoparticle by several interactions, ranging from simple adsorption,105,106 to electrostatic attraction between biomolecules and nanocrystal surfaces which are oppositely charged.89,107 The most defined and stable architecture, however, is obtained if the biomolecules are chemically bound to the stabilizing shell.92,104,108 As one more detailed illustrative example, consider the following biofunctionalized core–shell nanoparticle architecture recently presented by Li et al.:109 citrate-stabilized AuNPs were first functionalized with a carboxyl-terminated alkanethiol via ligand exchange. Next, the terminal carboxyl groups were subsequently bonded with side-chain amino groups of an enzymatic protein through an EDC/NHS coupling reaction. In enzyme activity assays, these bioconjugates display an enhanced thermostability and similar pH-dependence behavior compared to that of free enzyme. Note, however, that bioconjugate core–shell nanoparticles can be prepared with such simplicity, starting with a ligand exchange in the aqueous phase, only from well-defined hydrophilic precursors, like the well-known citrate-stabilized hydrophilic gold nanoparticles.
Next, let us briefly review the work of Parak et al. on the conjugation of single-stranded oligonucleotides onto hydrophobic Au nanoparticles as a more general illustrative example to prepare bioconjugate core–shell nanoparticles from hydrophobic precursors. The simplest strategy would be direct ligand exchange by adding SH-modified oligonucleotides to a solution of phosphine-stabilized Au nanoparticles, leading to partial replacement of the original phosphine ligands by the SH-nucleotides which bind very strongly to the Au surface. This concept has routinely been employed to functionalize hydrophilic citrate-stabilized Au nanoparticles with oligonucleotides.110 However, for some applications or the preparation of well-defined structures, like Au nanoparticle dimers or trimers with controlled interparticle spacing, it is desired to control the number of nucleotides binding to a single Au particle. If SH-oligonucleotides and Au nanoparticles are simply mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, however, a stoichometric distribution is obtained. To solve this problem, gel electrophoresis can be used to sort the oligonucleotide-Au hybrids by particle charge and size. The ligand exchange does not cause a significant change in particle charge, since both the original phosphine ligands and the exchanging oligonucleotides are negatively charged, but certainly the nanoparticle size strongly increases with the number of attached oligonucleotides. Once the Au-oligonucleotides were sorted by the number of oligonucleotide chains per nanoparticle, DNA-mediated dimers and trimers of Au nanoparticles could be prepared.111
:1 ratio, however, a stoichometric distribution is obtained. To solve this problem, gel electrophoresis can be used to sort the oligonucleotide-Au hybrids by particle charge and size. The ligand exchange does not cause a significant change in particle charge, since both the original phosphine ligands and the exchanging oligonucleotides are negatively charged, but certainly the nanoparticle size strongly increases with the number of attached oligonucleotides. Once the Au-oligonucleotides were sorted by the number of oligonucleotide chains per nanoparticle, DNA-mediated dimers and trimers of Au nanoparticles could be prepared.111
There also exists an upper limit for the number of oligonucleotides which can be attached to the surface of a single nanoparticle, defined by the stereochemistry of the system assuming a fully stretched chain configuration of the attached oligonucleotide chains. For example, a maximum of 115 or 157 single-stranded oligonucleotides of 12 bases could be attached to gold nanoparticles of diameter 13 and 15.7 nm, respectively.110,112 This coating layer of fully stretched DNA molecules is useful as a spacer extending a defined length from the gold nanoparticle surface: for instance, if a fluorescent molecule is attached to the outer chain end of the DNA molecules, such a defined spacer may be helpful in detailed studies of fluorescence quenching in the vicinity of gold surfaces in dependence of surface–dye distance.
Another important issue for biomedical applications of nanoparticles is the controlled particle uptake by living cells, using nanoparticles conjugated with biomolecules which bind to specific cell sites. For example, CdSe nanocrystals were modified with phalloidin which specifically binds to the actin network of fibroblasts, and this concept has successfully been used for multicolor labelling of living cells.108,113 Since the CdSe nanocrystals are very stable towards photobleaching, they can be used for single-molecule tracing to precisely follow the pathway of cellular uptake of nanoparticles: this uptake starts with endocytosis, and then the NPs are transported to vesicular compartments close to the nucleus of the cell where they remain. Both receptor-mediated and non-specific cellular nanoparticle uptake have been reported. Biocompatible fluorescent core–shell nanoparticles due to their high stability cannot only be used to selectively label cells, they also are passed on to both daughter cells after cell division, and therefore facilitate the long-term observation of the fate of individual living cells.
Finally, another nice application of such surface-modified fluorescent core–shell nanoparticles is the so-called phagokinetic track method: cells incorporate biocompatible fluorescent nanoparticles adsorbed to a surface very efficiently. If the cell is migrating across a surface containing a fluorescent NP layer, its migration path appears dark under the fluorescence microscope since it ingests the NPs on its way and thereby removes them from the surface.114
5. Multifunctional core–shell nanoparticles for drug delivery (“smart bombs”)
In this final section, I will review the most sophisticated type of multifunctional core–shell nanoparticles found in the current literature, smart drug-delivery systems. The concept for this type of biomedical application is illustrated in Fig. 5: first, a mesoporous nanoparticle loaded with a medical drug is selectively attaching to a specific cell type due to its selective surface coating. Having reached its target, the nanocarrier simply disintegrates, or opens supramolecular gates chemically attached close to the outlet of the particle’s mesopores, to release its cargo. This cargo release could be stimulated either externally by heat or light, or even better by the local chemical environment of the target, like pH or concentration of other specific ions. In addition to this smart drug delivery, the particle might serve as a sensor if it is labelled with fluorescent molecules to allow particle tracking, thereby verifying the controlled drug delivery. Such smart multifunctional particles are highly desired to overcome the disadvantages of current cancer therapy, where toxic substances not only kill the cancer cells but also severely damage other cells of the human body.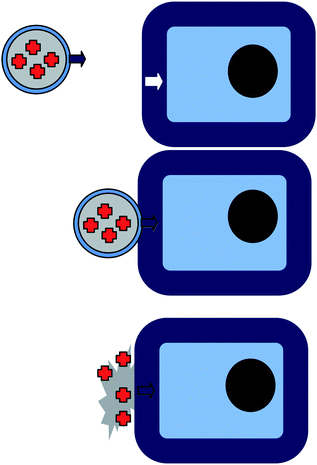 | ||
| Fig. 5 Multifunctional core–shell nanoparticles for drug delivery (“smart bombs”). | ||
Most drug-delivery systems currently approved by the American Food and Drug Administration (FDA) are either based on a bilayer liposome architecture as a nanoscopic container, or comprise therapeutic compounds linked to PEG. Multifunctional silica-based core–shell nanoparticles provide an alternative more sophisticated approach. The idea is to coat the surface of a drug-loaded mesoporous nanoparticle with a molecular machinery which allows controlled release of the embedded drug molecules. Such machinery could either be reversibly operating molecular nanovalves, or irreversible supramolecular structures, where the supramolecular structures are much simpler to prepare.
Combining the sol–gel process with templating surfactant molecules, silicate structures with controlled porosity, like the well-known MCM-41, have been prepared.17 The process originally led to large particles of hexagonally packed mesoporous silicates, but it has recently been improved to yield various topologies, e.g. mesoporous silica thin films or even nanoparticles.115–119 The mesoporous spherical silica nanoparticles are typically smaller than 200 nm in diameter, contain pores with average diameter 2 nm, and have already successfully been used as imaging agents, and as carriers for drugs and proteins.120–127 As mentioned before, silica can easily be functionalized by co-condensation with functional silanes, but also the particle surface can be modified by post-synthetic grafting involving the attachment of functional groups to the surface of the mesopores through silylation either before or after surfactant removal. By mixing the silica source with an aqueous solution of surfactant-coated nanocrystals, metal nanocrystals can easily be incorporated inside the mesoporous silica matrix, a procedure which was applied to iron oxide, gold and silver nanocrystals.128,129 Also, successful loading of silica particles with organic dye molecules like Rhodamin B, fluorescein and coumarins, as well as with various drugs and biomolecules has been demonstrated. The release behavior of conventional silica nanoparticles loaded with model drugs has been studied in some detail, and was also discussed in section 1 of this review: although the mesoporous silica nanoparticles exhibit sustained-release properties simply for steric reasons, they are not suitable to release the incorporated molecules in a controlled manner, that is, in response to an external stimulus. Instead, mesoporous silica particles with increased pore size and pore walls functionalized with “responsive gates” seem to be more promising.
The release of encapsulated drug molecules can be triggered by light as an external stimulus, as has been demonstrated for azobenzene-modified nanoparticles.130 Here, the cis–trans isomerization of azobenzene molecules conjugated along the pore walls serves in two different ways to trigger controlled release of the encapsulated cargo molecules: first as a photoswitchable gate, and second as a nanoimpeller. If the system is irradiated with light of wavelength 450 nm, both the cis–trans and the trans–cis isomerisation occur simultaneously, leading to a wagging motion of the azobenzene groups which causes an enhanced release rate of the encapsulated drug molecules. Lu et al. have used such nanoimpeller silica nanoparticles to deliver and release camphotecin into cancer cells.
As one further example of a reversible photoresponsive gate, the 2 + 2 photocycloaddition of coumarins attached to the pore outside of MCM-41 nanoparticles has been used to build a reversible gate: upon irradiation with UV light >310 nm the cycloaddition seals the pores, while irradiation at 250 nm cleaves the cyclobutane ring and thereby opens the pores.131,132 Here, it should be noted that we recently also have used a reversible 2 + 2 photocycloaddition to interconnect nanoscopic spherical polyorganosiloxane particles labelled at the surface with photoreactive molecules, namely coumarin133 or nitrocinnamate.134,135 This reversible interparticle connection was employed either to form microscopic capsules which then could be cleaved by UV irradiation,134 or to manipulate the viscosity of a colloidal dispersion by controlled cluster formation and destruction.135
An irreversible example of a photoresponsive release system are mesoporous silica nanoparticles modified with tiny inorganic nanocrystals, which are linked with a photocleavable group to block the pore opening. One analogous example of an irreversible chemical-responsive gate is a system of chemically labile disulfide bonds between the pore openings and inorganic nanocrystals.127,136 These weak bonds are cleaved upon addition of a reducing agent, and the nanocrystals blocking the pore openings are released from the silica particle surface. This type of release mechanism is called irreversible snap-top release, potentially activated by different chemical or physical stimuli. Very interesting examples of snap-top release are insulin-capped mesoporous silica nanoparticles:137 Zhao et al. developed a controlled release nanoparticle system which is capable of delivering insulin and cyclic adenosine monophosphate (cAMP) sequentially. Insulin is used to close the pores of MCM-41 loaded with cAMP. Brought in contact with glucose or other saccharides, first the insulin caps are removed, and then the cAMP is released from the open pores. This dual-drug delivery system is very attractive for biomedical applications, since the released cAMP additionally can induce insulin production, in addition to the amount of insulin directly released from the caps. Various enzyme-responsive snap-top systems have also been developed, as described in detail elsewhere. A UV-light responsive snap-top nanovalve system was developed by Kim et al. using cyclodextrin caps and o-nitrobenzyl ester moieties as photoreactive linkers, which are cleaved if irradiated at 365 nm.138 An alternative approach utilizes the difference in binding efficiencies of β-cyclodextrin to trans-azobenzene (high) and cis-azobenzene (extremely low), leading to a controlled de-threading of the cyclodextrin caps (and consequential release of the encapsulated cargo) upon trans–cis isomerization of the azobenzene stalks under irradiation at 351 nm.139
Next, let us consider a different type of responsive gates: so-called nanovalves. Mechanically interlocked molecules such as catenanes and rotaxanes provide the basis for the construction of such nanovalves for so-called mechanized silica nanoparticles. The rotaxanes consist of a dumbbell component encircled around its rod section by a ring, and bulky stoppers at the end of the rod prevent the ring from slipping off. The idea is to prepare a bistable rotaxane, where both the rod and the ring have two different complementary recognition sites with one preferred at equilibrium. If an external stimulus is applied, the preferred site changes, and the ring moves accordingly, leading to a reversible nanovalve. For proof of principle, Stoddart and co-workers first prepared mesoporous silica nanoparticles tethered with redox- and photoswitching2pseudorotaxanes, composed of 1,5-bis[2-(2-hydroxyethoxy)ethoxy]naphtalene (BHEEN) rods encircled by cyclobis(paraquat-p-phenylene) (CBPQT4+) rings, which act as irreversible stimuli-responsive gatekeepers to encapsulate or release cargo molecules.140 The gates remain closed when the CBPQT4+ rings encircle the BHEEN units on the rod due to [π⋯π] and [C–H⋯O] noncovalent bonding interactions. Upon addition of a chemical reducing agent, a CBPQT˙2+ bisradical is formed which no longer interacts with the BHEEN, leading to de-threading of the rings and therefore irreversible opening of the gates. This reduction and corresponding de-threading could also be achieved photochemically by irradiation at 365 nm in presence of the photosensitizer 9-anthracenecarboxylic acid (ACA) and triethanolamine as a sacrificial agent.
To prepare a reversibly operating nanovalve, bistable [2]rotaxanes, where the CBPQT4+ rings can be induced to shuttle between two different recognition sites, namely tetrathiofulvalene (TTF) and 1,5-dioxynaphtalene (DNP), have been employed.141 These rotaxanes were chemically attached onto the silica nanoparticle surface such that the free DNP units are closer to the pore opening while the bulky rings are associated with the more distant TTF units, that is, in the open state of the nanovalve. Next, the mesopores are loaded with a luminescent model drug, and the nanovalves are closed by oxidation of the TTF to TTF2+ with Fe(ClO4)3, since Coulomb repulsion now causes the CBPQT4+ rings to move to the second recognition site closer at the silica surface, thereby blocking the gate and entrapping the fluorescent cargo. The gate is opened again by reduction of the TTF2+ with ascorbic acid, as monitored by an increase in fluorescence intensity of the solution. Importantly, there is no description of how fluorescence from entrapped and released molecules is distinguished, so we assume that the nanoparticles somehow are separated from the solution, for example by dialysis or precipitation, before the fluorescence spectra are taken. The reversible nanovalve system can be optimized, in terms of release rate for cargo molecules of different sizes, by adjustment of the length of the silane linkers used to chemically attach the rotaxane rods to the silicate substrate. As an alternative to the redox-stimulated reversible nanovalve system, Stoddart et al. also developed a pH-sensitive rotaxane system,142 as well as one based on proton abstraction and competitive binding of complexing agents like dimethylammonium or metal ions.143 In 2009 a dual-controlled mechanized silica nanoparticle system containing both azobenzene-based nanoimpellers (inside the pores) and pH-responsive nanovalves (at the pore outlet at the particle surface) was developed.144 These particles contained two different types of nanovalves, one opening at acidic, the other opening at basic conditions, and only release their cargo if both pH (either <7 or >7) and irradiation are present as stimuli, that is, if the nanoimpeller is showing its wagging and the gates are open.
Having presented responsive gates based on molecular organic chemistry in some detail, let us finally consider some mesoporous silica particles with supramolecular gates based on polymers or dendrimers. Redox-activated polymer-capped MCM-41 nanoparticles have been prepared for example by functionalizing the surface of the mesoporous silica nanoparticles first with poly(N-acryloxysuccinimide), which then was crosslinked with cystamine to seal the pores.145 These crosslinks can be cleaved by disulfide reducing agents, leading to a controlled cargo release. Polyamines as outer coating have been used to prepare pH- and anion-driven supramolecular mechanized silica nanoparticles:146–148 hydrogen-bonding between these amines at neutral pH leads to a local clustering of the polymers and open pores, while at pH <7 charge repulsion leads to a more space-filling homogeneous polymer coating and therefore to closed pores. The anion-controlled release at pH <7 is based on the formation of stronger or weaker complexes of the polyammonium groups with the respective anions. Other supramolecular controlled-release core–shell nanoparticles include silica-supported lipid bilayers which successively may exchange lipids with oppositely charged free liposomes, leading to a protocell architecture with reduced bilayer defects and controlled surface charge to optimize the release inside cells compared to conventional liposomes.149 To deliver larger cargo molecules, polyamidoamine (PAMAM) dendrimers can be covalently attached to the surface of MCM-41 nanoparticles to complex plasmid DNA, yielding a gene transfection agent.150
To serve as drug-delivery systems, the biocompatibility of the mesoporous silica nanoparticles has to be optimized. Here, macrocycles like cyclodextrins (CD) and cucurbit[n]urils not only enhance the cell membrane permeability, but also can encapsulate drug molecules and thereby protect them from chemical or enzymatic degradation. Park et al. developed one of the first pH-responsive pseudo-rotaxane-based mechanized nanoparticle systems consisting of the three biocompatible components silica nanoparticles, poly(ethyleneimine) (PEI) and CD.151 Calcein as a model cargo is entrapped in the pores of the silica core, which are closed with surface-grafted pH-responsive PEI/CD polypseudorotaxanes. The cargo remains inside the pores as long as the PEI chains remain deprotonated (pH >11), whereas at lower pH the CD rings de-thread, opening the gates to release the calcein.
To conclude this section, the different concepts for controlled release discussed here are summarized in Fig. 6.
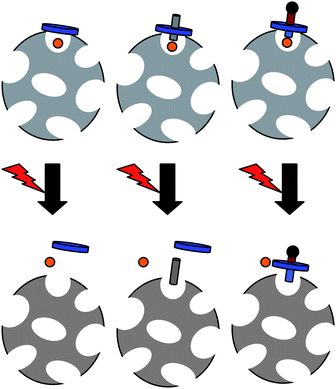 | ||
| Fig. 6 Concepts for controlled release from mesoporous core–shell silica nanoparticles upon external or local stimuli: snap-top, irreversible nanovalve and reversible nanovalve (from left to right). For simplification, just one pore and one gate are shown per particle. | ||
Conclusions
I have presented many different examples of core–shell nanoparticle architectures developed during the last decade. In particular, the desire to design highly sophisticated multifunctional nanoparticle architectures for biomedical applications has stimulated the recent progress in synthesis of core–shell nanoparticles with increasing structural complexity. The incorporated functionalities range from simple imaging and sensing, using fluorescent molecules, semiconductor nanocrystals or metal nanoparticles, to manipulation of local temperature in cancer hypothermal therapy (magnetic nanoparticles, light absorbing gold nanoparticles), and finally controlled drug delivery and release, combining clever supramolecular or organic chemistry with colloidal nanoscience. In most cases, mesoporous silica nanoparticles are used as carriers due to their simple chemical modification, optical transparency, biocompatibility, etc. Compared to the status when I wrote my last review on core–shell nanoparticles 10 years ago, the progress in multifunctional core–shell nanoparticle design is really overwhelming both in amount and quality of contributions being published in this field, of which only some highlights could be presented here. We certainly will see more and more biomedical applications of such highly sophisticated systems in the very near future, leading perhaps (and hopefully) to a revolution in the treatment of serious scourges of mankind like cancer or other diseases.References
- W. Schärtl, Adv. Mater., 2000, 12, 1899–1908 CrossRef CAS.
- C. Graf, W. Schartl, K. Fischer, N. Hugenberg and M. Schmidt, Langmuir, 1999, 15, 6170–6180 CrossRef CAS.
- G. Lindenblatt, W. Schartl, T. Pakula and M. Schmidt, Macromolecules, 2000, 33, 9340–9347 CrossRef CAS.
- C. Graf, W. Schartl, M. Maskos and M. Schmidt, J. Chem. Phys., 2000, 112, 3031–3039 CrossRef CAS.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- V. Salgueiriño-Maceira, M. A. Correa-Duarte, M. Spasova, L. M. Liz-Marzán and M. Farle, Adv. Funct. Mater., 2006, 16, 509–514 CrossRef CAS.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef CAS.
- M. Nuopponen and H. Tenhu, Langmuir, 2007, 23, 5352–5357 CrossRef CAS.
- J. Shan and H. Tenhu, Chem. Commun., 2007, 4580–4598 RSC.
- D. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, J. Phys. Chem. B, 2001, 105, 8861–8871 CrossRef CAS.
- K. K. Cotí, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC.
- W. Stober, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesner, Nano Lett., 2005, 5, 113–117 CrossRef CAS.
- T. Förster, Z. Naturforsch., A: Phys. Sci., 1949, 4, 321–327.
- T. Förster, Z. Elektrochem., 1960, 64, 157–165 CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- S. T. Dubas and J. B. Schlenoff, Macromolecules, 1999, 32, 8153–8160 CrossRef CAS.
- G. B. Sukhorukov, E. Donath, H. Lichtenfeld, E. Knippel, M. Knippel, A. Budde and H. Mohwald, Colloids Surf., A, 1998, 137, 253–266 CrossRef.
- G. B. Sukhorukov, E. Donath, S. Davis, H. Lichtenfeld, F. Caruso, V. I. Popov and H. Mohwald, Polym. Adv. Technol., 1998, 9, 759–767 CrossRef CAS.
- G. Decher and J. D. Hong, Ber. Bunsen-Ges., 1991, 95, 1430–1434 CAS.
- H. Kim, M. Achermann, L. P. Balet, J. A. Hollingsworth and V. I. Klimov, J. Am. Chem. Soc., 2005, 127, 544–546 CrossRef CAS.
- B. O. Dabbousi, J. RodriguezViejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- R. F. Ziolo, E. P. Giannelis, B. A. Weinstein, M. P. Ohoro, B. N. Ganguly, V. Mehrotra, M. W. Russell and D. R. Huffman, Science, 1992, 257, 219–223 CrossRef CAS.
- J. L. Dormann, Revue de Physique Appliquée, 1981, 16, 275–301 CrossRef CAS.
- P. M. Paulus, H. Bonnemann, A. M. van der Kraan, F. Luis, J. Sinzig and L. J. de Jongh, Eur. Phys. J. D, 1999, 9, 501–504 CrossRef CAS.
- N. Cordente, M. Respaud, F. Senocq, M. J. Casanove, C. Amiens and B. Chaudret, Nano Lett., 2001, 1, 565–568 CrossRef CAS.
- S. E. Inderhees, J. A. Borchers, K. S. Green, M. S. Kim, K. Sun, G. L. Strycker and M. C. Aronson, Phys. Rev. Lett., 2008, 101, 117202 CrossRef CAS.
- H. Zeng, J. Li, J. P. Liu, Z. L. Wang and S. H. Sun, Nature, 2002, 420, 395–398 CrossRef CAS.
- J. Nogues, J. Sort, V. Langlais, V. Skumryev, S. Surinach, J. S. Munoz and M. D. Baro, Phys. Lett., 2005, 422, 65–117 Search PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- X. G. Peng, J. Wickham and A. P. Alivisatos, J. Am. Chem. Soc., 1998, 120, 5343–5344 CrossRef CAS.
- S. O'Brien, L. Brus and C. B. Murray, J. Am. Chem. Soc., 2001, 123, 12085–12086 CrossRef CAS.
- N. R. Jana, Y. F. Chen and X. G. Peng, Chem. Mater., 2004, 16, 3931–3935 CrossRef CAS.
- Z. Li, Q. Sun and M. Y. Gao, Angew. Chem., Int. Ed., 2005, 44, 123–126 CrossRef CAS.
- K. Butter, A. P. Philipse and G. J. Vroege, J. Magn. Magn. Mater., 2002, 252, 1–3 CrossRef CAS.
- O. Song and Z. J. Zhang, J. Am. Chem. Soc., 2004, 126, 6164–6168 CrossRef CAS.
- E. E. Carpenter, C. T. Seip and C. J. O'Connor, J. Appl. Phys., 1999, 85, 5184–5186 CrossRef CAS.
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 437, 121–124 CrossRef CAS.
- H. G. Boyen, G. Kastle, K. Zurn, T. Herzog, F. Weigl, P. Ziemann, O. Mayer, C. Jerome, M. Moller, J. P. Spatz, M. G. Garnier and P. Oelhafen, Adv. Funct. Mater., 2003, 13, 359–364 CrossRef CAS.
- H. Bonnemann, W. Brijoux, R. Brinkmann, N. Matoussevitch, N. Waldofner, N. Palina and H. Modrow, Inorg. Chim. Acta, 2003, 350, 617–624 CrossRef CAS.
- C. R. Vestal and Z. J. Zhang, J. Am. Chem. Soc., 2002, 124, 14312–14313 CrossRef CAS.
- J. I. Park and J. Cheon, J. Am. Chem. Soc., 2001, 123, 5743–5746 CrossRef CAS.
- Z. H. Ban, Y. A. Barnakov, V. O. Golub and C. J. O'Connor, J. Mater. Chem., 2005, 15, 4660–4662 RSC.
- Q. X. Liu, Z. H. Xu, J. A. Finch and R. Egerton, Chem. Mater., 1998, 10, 3936–3940 CrossRef CAS.
- J. Zhang, M. Post, T. Veres, Z. J. Jakubek, J. W. Guan, D. S. Wang, F. Normandin, Y. Deslandes and B. Simard, J. Phys. Chem. B, 2006, 110, 7122–7128 CrossRef CAS.
- D. L. Ma, J. W. Guan, F. Normandin, S. Denommee, G. Enright, T. Veres and B. Simard, Chem. Mater., 2006, 18, 1920–1927 CrossRef CAS.
- Y. Lu, Y. D. Yin, B. T. Mayers and Y. N. Xia, Nano Lett., 2002, 2, 183–186 CrossRef CAS.
- D. K. Yi, S. S. Lee, G. C. Papaefthymiou and J. Y. Ying, Chem. Mater., 2006, 18, 614–619 CrossRef CAS.
- F. Baumann, M. Schmidt, B. Deubzer, M. Geck and J. Dauth, Macromolecules, 1994, 27, 6102–6105 CrossRef CAS.
- S. Utech, C. Scherer and M. Maskos, J. Magn. Magn. Mater., 2009, 321, 1386–1388 CrossRef CAS.
- A. H. Lu, W. C. Li, N. Matoussevitch, B. Spliethoff, H. Bonnemann and F. Schuth, Chem. Commun., 2005, 98–100 RSC.
- V. Salgueiriño-Maceira, M. A. Correa-Duarte, M. Farle, A. López-Quintela, K. Sieradzki and R. Diaz, Chem. Mater., 2006, 18, 2701–2706 CrossRef CAS.
- Y. H. Deng, W. L. Yang, C. C. Wang and S. K. Fu, Adv. Mater., 2003, 15, 1729–1732 CrossRef CAS.
- C. H. Jun, Y. J. Park, Y. R. Yeon, J. R. Choi, W. R. Lee, S. J. Ko and J. Cheon, Chem. Commun., 2006, 1619–1621 RSC.
- X. J. Zhao, R. Tapec-Dytioco, K. M. Wang and W. H. Tan, Anal. Chem., 2003, 75, 3476–3483 CrossRef CAS.
- Z. Tuzar, P. Kratochvil, K. Prochazka and P. Munk, Collect. Czech. Chem. Commun., 1993, 58, 2362–2369 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- L. Bronstein, M. Sedlak, J. Hartmann, M. Breulmann, H. Colfen and M. Antonietti, Abstr. Paper Am. Chem. Soc., 1997, 213, 33.
- K. Kataoka, H. Togawa, A. Harada, K. Yasugi, T. Matsumoto and S. Katayose, Macromolecules, 1996, 29, 8556–8557 CrossRef CAS.
- S. Katayose and K. Kataoka, Bioconjugate Chem., 1997, 8, 702–707 CrossRef CAS.
- S. Katayose and K. Kataoka, J. Pharm. Sci., 1998, 87, 160–163 CrossRef CAS.
- M. A. Wolfert, E. H. Schacht, V. Toncheva, K. Ulbrich, O. Nazarova and L. W. Seymour, Hum. Gene Ther., 1996, 7, 2123–2133 CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- S. Takae, Y. Akiyama, H. Otsuka, T. Nakamura, Y. Nagasaki and K. Kataoka, Biomacromolecules, 2005, 6, 818–824 CrossRef CAS.
- H. Otsuka, Y. Akiyama, Y. Nagasaki and K. Kataoka, J. Am. Chem. Soc., 2001, 123, 8226–8230 CrossRef CAS.
- L. A. Lyon, M. D. Musick and M. J. Natan, Anal. Chem., 1998, 70, 5177–5183 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- Y. Nagasaki, T. Ishii, Y. Sunaga, Y. Watanabe, H. Otsuka and K. Kataoka, Langmuir, 2004, 20, 6396–6400 CrossRef.
- I. Sondi, O. Siiman, S. Koester and E. Matijevic, Langmuir, 2000, 16, 3107–3118 CrossRef CAS.
- J. Shan, M. Nuopponen, H. Jiang, E. Kauppinen and H. Tenhu, Macromolecules, 2003, 36, 4526–4533 CrossRef CAS.
- D. X. Li, Q. He, Y. Cui, K. W. Wang, X. M. Zhang and J. B. Li, Chem.–Eur. J., 2007, 13, 2224–2229 CrossRef CAS.
- D. X. Li, Y. Cui, K. W. Wang, Q. He, X. H. Yan and J. B. Li, Adv. Funct. Mater., 2007, 17, 3134–3140 CrossRef CAS.
- D. X. Li, Q. A. He and J. B. Li, Adv. Colloid Interface Sci., 2009, 149, 28–38 CrossRef CAS.
- D. X. Li, Q. He, Y. Yang, H. Mohwald and J. B. Li, Macromolecules, 2008, 41, 7254–7256 CrossRef CAS.
- D. X. Li, Q. He, Y. Cui and J. B. Li, Chem. Mater., 2007, 19, 412–417 CrossRef CAS.
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., Int. Ed., 2006, 45, 813–816 CrossRef CAS.
- L. Manna, E. C. Scher, L. S. Li and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 7136–7145 CrossRef CAS.
- T. Mokari and U. Banin, Chem. Mater., 2003, 15, 3955–3960 CrossRef.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS.
- Y. F. Chen, T. H. Ji and Z. Rosenzweig, Nano Lett., 2003, 3, 581–584 CrossRef CAS.
- K. S. Mayya and F. Caruso, Langmuir, 2003, 19, 6987–6993 CrossRef CAS.
- A. C. Templeton, M. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
- S. F. Wuister, I. Swart, F. van Driel, S. G. Hickey and C. D. Donega, Nano Lett., 2003, 3, 503–507 CrossRef CAS.
- J. Aldana, Y. A. Wang and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 8844–8850 CrossRef CAS.
- S. Kim and M. G. Bawendi, J. Am. Chem. Soc., 2003, 125, 14652–14653 CrossRef CAS.
- H. Mattoussi, J. M. Mauro, E. R. Goldman, G. P. Anderson, V. C. Sundar, F. V. Mikulec and M. G. Bawendi, J. Am. Chem. Soc., 2000, 122, 12142–12150 CrossRef CAS.
- F. Pinaud, D. King, H. P. Moore and S. Weiss, J. Am. Chem. Soc., 2004, 126, 6115–6123 CrossRef CAS.
- Y. Kobayashi, M. Horie, M. Konno, B. Rodriguez-Gonzalez and L. M. Liz-Marzán, J. Phys. Chem. B, 2003, 107, 7420–7425 CrossRef CAS.
- W. J. Parak, D. Gerion, D. Zanchet, A. S. Woerz, T. Pellegrino, C. Micheel, S. C. Williams, M. Seitz, R. E. Bruehl, Z. Bryant, C. Bustamante, C. R. Bertozzi and A. P. Alivisatos, Chem. Mater., 2002, 14, 2113–2119 CrossRef CAS.
- M. Alejandro-Arellano, T. Ung, A. Blanco, P. Mulvaney and L. M. Liz-Marzán, Pure Appl. Chem., 2000, 72, 257–267 CrossRef CAS.
- M. A. Correa-Duarte, M. Giersig and L. M. Liz-Marzán, Chem. Phys. Lett., 1998, 286, 497–501 CrossRef CAS.
- L. M. LizMarzan and A. P. Philipse, J. Colloid Interface Sci., 1995, 176, 459–466 CrossRef CAS.
- A. Schroedter and H. Weller, Angew. Chem., Int. Ed., 2002, 41, 3218–3221 CrossRef CAS.
- A. Schroedter, H. Weller, R. Eritja, W. E. Ford and J. M. Wessels, Nano Lett., 2002, 2, 1363–1367 CrossRef CAS.
- W. Schärtl, R. G. Xie, T. Ren, M. Sejfic, G. Wenz, R. Heisel, C. Scherer, M. Maskos, K. Fischer and T. Basché, J. Lumin., 2009, 129, 1428–1434 CrossRef.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- W. J. Parak, T. Pellegrino, C. M. Micheel, D. Gerion, S. C. Williams and A. P. Alivisatos, Nano Lett., 2003, 3, 33–36 CrossRef CAS.
- G. P. Mitchell, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1999, 121, 8122–8123 CrossRef CAS.
- M. E. Akerman, W. C. W. Chan, P. Laakkonen, S. N. Bhatia and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12617–12621 CrossRef CAS.
- S. J. Rosenthal, A. Tomlinson, E. M. Adkins, S. Schroeter, S. Adams, L. Swafford, J. McBride, Y. Q. Wang, L. J. DeFelice and R. D. Blakely, J. Am. Chem. Soc., 2002, 124, 4586–4594 CrossRef CAS.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- K. Hanaki, A. Momo, T. Oku, A. Komoto, S. Maenosono, Y. Yamaguchi and K. Yamamoto, Biochem. Biophys. Res. Commun., 2003, 302, 496–501 CrossRef CAS.
- R. Mahtab, H. H. Harden and C. J. Murphy, J. Am. Chem. Soc., 2000, 122, 14–17 CrossRef CAS.
- H. Mattoussi, J. M. Mauro, E. R. Goldman, T. M. Green, G. P. Anderson, V. C. Sundar and M. G. Bawendi, Phys. Status Solidi B, 2001, 224, 277–283 CrossRef CAS.
- X. Y. Wu, H. J. Liu, J. Q. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. F. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS.
- D. X. Li, Q. He, Y. Cui, L. Duan and J. B. Li, Biochem. Biophys. Res. Commun., 2007, 355, 488–493 CrossRef CAS.
- L. M. Demers, C. A. Mirkin, R. C. Mucic, R. A. Reynolds, R. L. Letsinger, R. Elghanian and G. Viswanadham, Anal. Chem., 2000, 72, 5535–5541 CrossRef CAS.
- D. Zanchet, C. M. Micheel, W. J. Parak, D. Gerion and A. P. Alivisatos, Nano Lett., 2001, 1, 32–35 CrossRef CAS.
- P. Sandström, M. Boncheva and B. Åkerman, Langmuir, 2003, 19, 7537–7543 CrossRef.
- M. Bruchez Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- W. J. Parak, R. Boudreau, M. Le Gros, D. Gerion, D. Zanchet, C. M. Micheel, S. C. Williams, A. P. Alivisatos and C. Larabell, Adv. Mater., 2002, 14, 882–885 CrossRef CAS.
- Q. Cai, Z. S. Luo, W. Q. Pang, Y. W. Fan, X. H. Chen and F. Z. Cui, Chem. Mater., 2001, 13, 258–263 CrossRef CAS.
- J. Kobler, K. Moller and T. Bein, ACS Nano, 2008, 2, 791–799 CrossRef CAS.
- Y. F. Lu, H. Y. Fan, A. Stump, T. L. Ward, T. Rieker and C. J. Brinker, Nature, 1999, 398, 223–226 CrossRef CAS.
- Y. F. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. L. Gong, Y. X. Guo, H. Soyez, B. Dunn, M. H. Huang and J. I. Zink, Nature, 1997, 389, 364–368 CrossRef CAS.
- S. P. Naik, W. Fan, T. Yokoi and T. Okubo, Langmuir, 2006, 22, 6391–6397 CrossRef CAS.
- T. Heikkila, J. Salonen, J. Tuura, M. S. Hamdy, G. Mul, N. Kumar, T. Salmi, D. Y. Murzin, L. Laitinen, A. M. Kaukonen, J. Hirvonen and V. P. Lehto, Int. J. Pharm., 2007, 331, 133–138 CrossRef CAS.
- M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef CAS.
- M. Vallet-Regí, Chem.–Eur. J., 2006, 12, 5934–5943 CrossRef CAS.
- Q. Yang, S. H. Wang, P. W. Fan, L. F. Wang, Y. Di, K. F. Lin and F. S. Xiao, Chem. Mater., 2005, 17, 5999–6003 CrossRef CAS.
- Y. F. Zhu, J. L. Shi, W. H. Shen, H. R. Chen, X. P. Dong and M. L. Ruan, Nanotechnology, 2005, 16, 2633–2638 CrossRef CAS.
- J. Andersson, J. Rosenholm, S. Areva and M. Linden, Chem. Mater., 2004, 16, 4160–4167 CrossRef.
- C. Charnay, S. Begu, C. Tourne-Peteilh, L. Nicole, D. A. Lerner and J. M. Devoisselle, Eur. J. Pharm. Biopharm., 2004, 57, 533–540 CrossRef CAS.
- C. Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C. H. Shin, J. G. Park and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688–689 CrossRef CAS.
- I. Gorelikov and N. Matsuura, Nano Lett., 2008, 8, 369–373 CrossRef CAS.
- J. Lu, E. Choi, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421–426 CrossRef CAS.
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350–353 CrossRef CAS.
- N. K. Mal, M. Fujiwara, Y. Tanaka, T. Taguchi and M. Matsukata, Chem. Mater., 2003, 15, 3385–3394 CrossRef CAS.
- C. Graf, W. Schartl and N. Hugenberg, Adv. Mater., 2000, 12, 1353–1356 CrossRef CAS.
- X. F. Yuan, K. Fischer and W. Schartl, Langmuir, 2005, 21, 9374–9380 CrossRef CAS.
- X. Yuan, M. Schnell, S. Muth and W. Schartl, Langmuir, 2008, 24, 5299–5305 CrossRef CAS.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS.
- Y. N. Zhao, B. G. Trewyn, I. I. Slowing and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398–8400 CrossRef CAS.
- C. Park, K. Lee and C. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275–1278 CrossRef CAS.
- D. P. Ferris, Y. L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686–1688 CrossRef CAS.
- R. Hernandez, H. R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS.
- T. D. Nguyen, H. R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS.
- T. D. Nguyen, K. C. F. Leung, M. Liong, C. D. Pentecost, J. F. Stoddart and J. I. Zink, Org. Lett., 2006, 8, 3363–3366 CrossRef CAS.
- K. C. F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Chem. Mater., 2006, 18, 5919–5928 CrossRef CAS.
- S. Angelos, Y. W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344–11346 CrossRef CAS.
- R. Liu, X. Zhao, T. Wu and P. Y. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS.
- R. Casasús, E. Aznar, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto and P. Amorós, Angew. Chem., Int. Ed., 2006, 45, 6661–6664 CrossRef CAS.
- R. Casasús, E. Climent, M. D. Marcos, R. Martínez-Máñez, F. Sancenon, J. Soto, P. Amorós, J. Cano and E. Ruiz, J. Am. Chem. Soc., 2008, 130, 1903–1917 CrossRef CAS.
- R. Casasús, M. D. Marcos, R. Martínez-Máñez, J. V. Ros-Lis, J. Soto, L. A. Villaescusa, P. Amorós, D. Beltrán, C. Guillem and J. Latorre, J. Am. Chem. Soc., 2004, 126, 8612–8613 CrossRef CAS.
- J. W. Liu, X. M. Jiang, C. Ashley and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 7567–7569 CrossRef CAS.
- D. R. Radu, C. Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216–13217 CrossRef CAS.
- C. Park, K. Oh, S. C. Lee and C. Kim, Angew. Chem., Int. Ed., 2007, 46, 1455–1457 CrossRef CAS.
Footnote |
| † Dedicated to Professor Manfred Schmidt on the occasion of his 60th birthday |
| This journal is © The Royal Society of Chemistry 2010 |