Uniform and controllable preparation of Au–Ag core–shell nanorods using anisotropic silver shell formation on gold nanorods†
Yoshifumi
Okuno
,
Koji
Nishioka
,
Ayaka
Kiya
,
Naotoshi
Nakashima
,
Ayumu
Ishibashi
and
Yasuro
Niidome
*
Department of Applied Chemistry, Faculty of Engineering, Motooka 744, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: ynidotcm@mail.cstm.kyushu-u.ac.jp; Fax: +81 92 802 2843; Tel: +81 91 802 2841
First published on 23rd June 2010
Abstract
Anisotropic and controllable silver shell formation on gold nanorods was realized in a micellar solution of hexadecytrimethylammonium chloride. Uniformity of the anisotropic Au–Ag core–shell particles contributes separation of four extinction bands. The ability to manipulate the shapes and sizes of these nanoparticles offers a wide-range control of the surface extinction from the visible to the near infrared regions (450–800 nm).
Introduction
Metal shell formation on nanoparticles can optimize the electronic, catalytic, and optical properties of nanoparticles.1 Gold and silver are frequently used for shell or core materials,1–11 because shell formation of these elements is a useful way to control the spectroscopic properties of nanoparticles. The distinct surface plasmon (SP) bands of gold and silver are dependent on their sizes and shapes and contribute to enhancing the optical properties of nanoparticles.12,13The most typical metal nanoparticles that disperse in water are probably gold nanoparticles.14,15 Gold nanoparticles are frequently used as probe materials for analytical or bio-chemical applications, due to their chemical stabilities, the SP bands locating in visible and near infrared (IR) regions, and facile surface modification using thiol compounds.16,17 There are many publications reporting preparation of gold nanoparticles. The preparation of spherical,18 thin triangular,19 cubic,20 wire-like,21 or rod-shaped22 gold nanoparticles have been reported. As uniform gold nanoparticles can be synthesized in various shapes and sizes, gold is a preferable material to prepare core nanoparticles.
Silver nanoparticles show large SP bands in ultra-violet (UV) and visible regions.23–25 The distinct SP bands of silver nanoparticles contribute to effective enhancements for Raman scattering,26 infrared absorption,27 and fluorescence28 of organic molecules. Consequently, silver can be used as a functional material because of its remarkable optical properties. Various methods to obtain spherical, cubic, or octahedral silver nanoparticles have been reported;23,29,30 however, preparation of anisotropic silver nanoparticles in a uniform shape has proven challenging. Spectroscopic properties of the previously prepared anisotropic silver nanoparticles indicated that the uniformities observed for anisotropic nanoparticles were not as good as those of anisotropic gold nanoparticles.31–40 To improve the uniformity of anisotropic silver nanoparticles, a new approach to control the growth of silver nanocrystals is required.
In this work, gold nanorods were wrapped with silver shells to obtain uniform anisotropic nanoparticles that showed the spectroscopic characteristics of silver. The gold nanorods are uniform rod-shaped gold nanoparticles that show distinct optical characteristics originating from their anisotropic shapes.41 Formation of uniform silver shells on gold nanorods has been expected to be a useful method to prepare uniform anisotropic silver nanoparticles. On the basis of this concept, several preparation methods of Au–Ag core–shell nanorods have previously were reported.42–45 However, in many cases, the resultant shells were not uniform on gold nanorods. Due to the insufficient homogeneity of the silver shells, their SP bands showed large half-bandwidths, and minor bands were found as shoulders to the major peaks. Liu et al. reported that uniform silver shells could be formed on gold nanorods in the presence of water-soluble polymers.45 Their core–shell nanorods showed two extinction bands that were assigned to longitudinal and transverse SP oscillations. Xia et al. reported uniform octahedral silver nanoparticles in which gold nanorods were incorporated.46 These were Au@Ag core–shell nanoparticles that showed one broad peak at ∼460 nm and a shoulder peak at ∼350 nm. Previously, we have also reported silver shell formation in hexadecyltrimethylammonium chloride (CTAC) and hexadecyltrimethylammonium bromide (CTAB) mixed micellar solutions.47 We showed that the silver shell formation in a CTAC solution was much faster than that in a CTAB solution. Fast nucleation and rapid growth of silver metals on gold nanorods in a CTAC solution contributed to the fast formation of the shell, but the fast reactions did not improve the uniformity of the silver shells.47 Rapid nucleation is advantageous for the formation of a uniform shell; however, if the subsequent shell growth is competitively fast against the nucleation, the formation of an inhomogeneous silver shell occurs. As a possible strategy to obtain uniform Au–Ag core–shell nanorods, the growth rate of the shell should be suppressed. Two types of experimental procedures that contained intrinsic processes to suppress the chemical reactions of silver ions were tried in this work.
Results and discussion
Silver chloride as a silver-ion source
The first procedure involved using silver chloride (AgCl) particles suspended in water as a silver-ion source to obtain silver shells. The AgCl particles, the diameters of which were 600–900 nm, were expected to feed AgCl2− ions gradually in the reaction solutions containing concentrated CTAC molecules (80 mM). An AgCl-suspended solution (10 mM, 0.25 mL) and an ascorbic acid solution (0.1 M, 0.5 mL) were added to a CTAC solution (80 mM, 10 mL) containing a certain amount of gold nanorods at room temperature. The reaction solution showed drastic spectral changes when the pH of the reaction solutions was adjusted to 5.4 by the addition of a sodium hydrochloride (NaOH) solution. The spectral changes are shown in Fig. 1(a). At 180 min after the addition of the AgCl-suspended solution the spectral changes had stopped, and subsequently the solution showed four bands at 346, 400, 447 and 549 nm. During the shell formation, the longitudinal SP bands of gold nanorods at around 900 nm moved to the shorter wavelength regions. The two bands at around 400 and 447 nm also changed their peak positions and intensities. The two bands moved to the longer wavelength regions, and showed similar changes in the intensities with those of the longitudinal SP bands in the longest wavelength regions. The band at around 346 nm, on the other hand, did not show remarkable peak shifts. The spectral changes of the four bands have simultaneously stopped at 180 min after the addition of the suspended AgCl solution.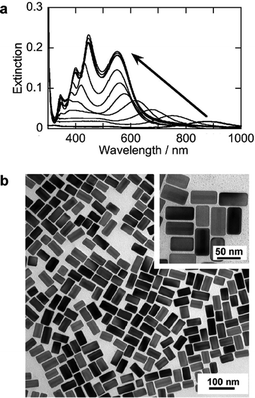 | ||
| Fig. 1 Extinction spectra and TEM images of Au–Ag core–shell nanorods prepared by using AgCl particles at room temperature. (a) spectral changes of a reaction solution after the addition of AgCl particles and NaOH solutions (0, 10, 30, 60, 90, 120, 150, 180, 210, 240, 270 min). (b) a TEM image of gold–silver core–shell nanorods (reaction time: 180 min). The extinction spectra plotted against wavenumbers were shown in Fig. S1 of the ESI.† | ||
In transmission electron microscopic (TEM) images (Fig. 1(b)), the gold nanorods were observed in semi-transparent layers that were consistent with those of previously observed gold–silver core–shell nanoparticles.42–44 A high-resolution TEM image and a energy dispersive X-ray spectrum are shown in Fig. S2 of the ESI.† They indicate a lattice image of metal–silver shells and the presence of silver and gold in a nanorod, respectively. These data reveal the formation of the Au–Ag core–shell nanorods with uniform shapes. (Additional TEM images are presented in Fig. S3 of the ESI.†) It should be noted that the growth of the silver shells was anisotropic. As shown in the inset of Fig. 1(b). The silver shells in the transverse direction were thicker than those in the longitudinal direction. For palladium, it is often the case that the metal palladium grows faster along the [110] planes of gold than along the [100] planes.48 In a CTAC solution, the surfaces of gold nanorods probably affected the crystallography of the silver shells. In a CTAB solution, silver shell formation frequently gave dumb-bell and symmetric (boat-like) silver shells.42–44 It was shown that CTAB molecules formed bilayers on gold nanorod surfaces.49 The same procedure as that of Fig. 1 using a CTAB solution also gave dumb-bell or boat-like silver shells (see Fig. S4 of the ESI†). Thus, the anisotropic silver shell formation was also affected by the molecular assemblies on gold nanorods. It was found that CTAC layers on gold nanorods were advantageous for the formation of uniform core–shell nanorods.
The TEM images showed no remarkable by-product in Fig. 1 and Fig. S3.† Thus, the spectroscopic properties in Fig. 1(a) should be assigned to the optical responses of the core–shell nanorods with anisotropic silver shells. The four bands in the extinction spectra (Fig. 1(a)) were also observed in previous research efforts;39,47 however, they were not clearly isolated from each other; minor peaks were observed as shoulders of the major peaks. (An absorption spectrum and a TEM image of Au–Ag core–shell nanorods prepared by our previous method47 are shown in Fig. S5 of the ESI.†) At the present stage of this work we have not yet determined the origins of the four peaks; however, the isolated four SP peaks in Fig. 1(a) indicate that the new procedure using AgCl particles gives very uniform core–shell nanorods.
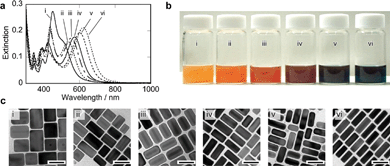
Fig. 2 shows extinction spectra at 180 min after the addition of AgCl particles at different pH conditions. At low pH conditions (pH = 4.1 (i) and 4.5 (ii)), longitudinal SP bands of gold nanorods shifted to shorter wavelength regions. Time dependent spectral changes and TEM images are shown in Fig. S6 of the ESI.† These spectral changes and TEM observations indicated that the silver shell formation proceeded even at the lower pH conditions, but the rate of the shell formation was very slow. At pH = 5.0 (iii) and 5.4 (iv), the shell formation has stopped by 180 min following the addition of the AgCl particles, and at that time point, the four SP bands that are characteristic of the Au–Ag core–shell nanorods that were observed. This pH range was shown to be appropriate for preparing silver shells. In contrast, at the higher pH conditions (pH = 5.7 (v) and 6.2 (vi)), the spectral changes stopped within 10 min and resulted in indistinct profiles in the absorption spectra (Fig. S6 of the ESI†). This indicates that the reduction of Ag ions at the higher pH is very rapid and this reduction rate hindered the formation of uniform Au–Ag core–shell nanorods.
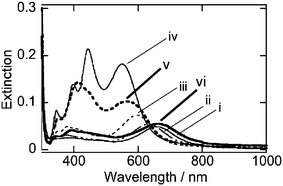 | ||
| Fig. 2 Extinction spectra at 180 min after the addition of AgCl particles. pH = 4.1 (i), 4.5 (ii), 5.0 (iii), 5.4 (iv), 5.7 (v), 6.2 (vi). | ||
Low pH reactions with silver nitrate
In the second procedure, silver nitrate was used as a silver ion source for the formation of silver shells. This procedure has previously been used in our paper,47 but the pH of the reaction solutions is regulated to control the reduction of the silver ions. Fig. 3(a) shows extinction spectra at 180 min after the addition of the silver nitrate solutions at different pH conditions. Under acidic conditions (pH = 3.0 (i), 3.8 (ii), and 4.4 (iii)), the changes in the spectra indicate that the shell formations are very slow. At higher pH conditions (pH = 4.6 (iv), 4.9 (v) and 5.4 (vi)), silver ions appear to be quickly reduced, but large SP bands are observed at ∼410 nm. This band is assigned to colloidal silver nanoparticles. Thus, at the higher pH conditions, the formation of the colloidal silver nanoparticles competes against the formation of the silver shells on gold nanorod surfaces.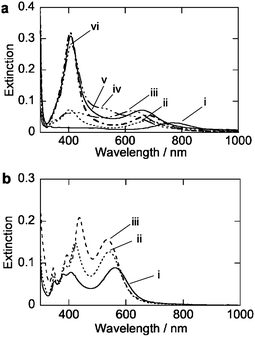 | ||
| Fig. 3 Extinction spectra of Au–Ag core–shell nanorods prepared by using AgNO3 solutions. (a) Extinction spectra at 180 min after the addition of AgNO3 solutions at room temperature. pH = 3.0 (i), 3.8 (ii), 4.4 (iii), 4.6 (iv), 4.9 (v), 5.4 (vi). (b) Extinction spectra at 180 min after the addition of AgNO3 solutions at 60, 70, 80 °C. | ||
In acidic conditions, reduction of silver ions by the ascorbic acid is very slow because ascorbate is active for the reduction.8 The dissociation constant (pKa) of ascorbic acid is pKa = 4.2, and at this pH, the reaction speeds and the resultant morphologies of the silver shells are drastically changed.
Heating of the reaction solutions accelerates the silver shell formation at lower pH conditions (pH = 3) using AgNO3. Fig. 3(b) shows extinction spectra at after 90 min at 60 (vii), 70 (viii), and 80 (ix) °C. At 60 °C (vii), the spectrum shows the four distinct SP bands that are assigned to the formation of the silver shells. TEM observations indicated that silver shells were formed on gold nanorods (Fig. S7 of the ESI†). At higher temperatures (70 (vii) and 80 (ix) °C), the reaction rates of the formation of the silver shells were faster than those at 60 °C. Moreover, the profiles of the SP bands and shapes of silver shells formed at these two high temperatures were not affected. These results indicated that the reaction temperatures could accelerate the shell growth processes without degrading the uniformity of the core–shell nanorods. The control of the shell growth processes was found to be an important factor in obtaining a silver shell.
Characteristics of the shell formation
The thickness of the silver shells can be controlled by the relative amounts of silver ions and gold nanorods. Fig. 4 shows the extinction spectra, photographs of the reaction solutions, and TEM images of the core–shell particles that were prepared by using AgCl particles at pH = 5.4 at room temperature. The results of shell formation using AgNO3 solutions (pH = 3.0, 70 °C) at the different Ag/Au ratios (7.1–57) are shown in Fig. 5. The spectra in Fig. 4 and 5 indicated that good reproducibility of the two developed synthesis procedures was possible (intensities of the longitudinal SP bands were not normalized) and the solutions showed dramatic changes in color from orange to green (Fig. 4(b) and 5(b)). The TEM images clearly indicated the formation of silver shells on the gold nanorods. It was shown that the both procedures examined in this work were useful methods to realize controllable and reproducible silver-shell formation on gold nanorods.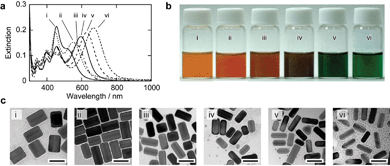 | ||
| Fig. 4 Extinction spectra, macroscopic photographs, and TEM images of Au–Ag core–shell nanorods prepared by using different amount of AgCl particles. (a) Extinction spectra at 180 min after the addition of AgCl particles at room temperature. Molar ratios of Ag/Au were 57 (i), 28.5 (ii), 19 (iii), 14.2 (iv), 9.5 (v), and 7.1 (vi). Concentration of silver ions was constant (0.25 mM). (b) Macroscopic photographs of reaction solutions. (c) TEM images of the core–shell nanorods. The scale bars indicate 50 nm. | ||
 | ||
| Fig. 5 Extinction spectra, macroscopic photographs, and TEM images of Au–Ag core–shell nanorods prepared by using different amount of AgNO3 solutions. (a) Extinction spectra at 180 min after the addition of AgNO3 solutions at 60 °C. Molar ratios of Ag/Au were 57 (i), 28.5 (ii), 19 (iii), 14.2 (iv), 9.5 (v), and 7.1 (vi). Concentration of silver ions was constant (0.25 mM). (b) Macroscopic photographs of reaction solutions. (c) TEM images of the core–shell nanorods. The scale bars indicate 50 nm. | ||
Even on long and short gold nanorods (aspect ratio = 7 and 4) and spherical gold nanoparticles, uniform core–shell nanorods could be prepared using the described methods (Fig. S8 in the ESI†). Consequently, the slow reactions of the AgCl particles or the AgNO3 solutions at low pH are useful to control the growth of anisotropic silver shells on various gold nanoparticles. Using these methods, we can control the anisotropic synthesis of silver shells on gold nanorods with uniform shapes.
Fig. 6 indicates the characteristics of our methods. The vertical and horizontal axes of Fig. 6 are the transverse diameters and longitudinal lengths of the nanoparticles. Aspect ratios (transverse width/longitudinal length) are shown as five straight lines. If there were spherical (isotropic) nanoparticles of different particle sizes, they would be plotted on the line of the aspect ratio = 1, that is, the most-inclined line. In the case of the synthesis of the silver nanorods that were reported in previous papers,33,34 the shapes of these nanorods are distributed in the green area. The longitudinal lengths of the silver nanorods are shown to be controlled over a wide range, but the transverse diameters are restricted from 10 to 35 nm. Xia and co-workers reported preparations of silver nanowires35–38 and silver nanobars.50 The blue and yellow areas in Fig. 6 indicate the distribution of the silver nanowires and nanobars produced by these groups. In the previous cases, it was shown that the control of transverse diameters was not as good as that of the longitudinal length. Consequently, the green, blue, and yellow areas are long rectangles in the horizontal direction.
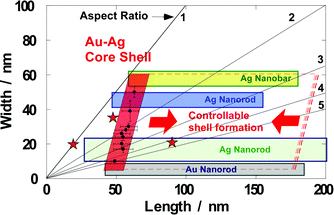 | ||
| Fig. 6 Schematic illustration of controllability of anisotropic metal nanoparticles. The size of the gold–silver core–shell particles is controllable in the transverse direction (red quadrangle). It is very different from the size distributions of the previous methods, shown as green, blue, and yellow rectangles.33–38,47 Red stars are plotted from the TEM images in Fig. S8 in the ESI.† | ||
In Fig. 6, the sizes of our core–shell nanorods by the two methods are also plotted. The methods used herein control the thickness of the silver shells in the transverse direction of the core–shell nanorods. In the longitudinal direction, growth of the silver shells is suppressed. This anisotropic shell formation can be seen in the TEM images in Fig. 1(b), 4(c), 5(c), and S6–8.† The plots of our core–shell nanorods (within the red quadrangle) indicate very different controllability from the previous results (green, blue, and yellow rectangles). Thus, our methods propose a novel strategy to manipulate the shapes of anisotropic silver nanoparticles. In our methods, various gold nanoparticles can be used as seed particles (Fig. S8 in the ESI†). In particular, as seed nanoparticles, gold nanorods can be prepared over wide ranges without degrading their uniformities (gray rectangle).51 As such, various gold nanorods can be used as core particles for the formation of silver shells, and this will facilitate the design of desirable shapes for core–shell particles.
Experimental
Gold nanorods in a CTAB solution, which were prepared by a photochemical method,52 were obtained from a joint research project of Dai-Nihon-Toryo Co. Ltd. and Mitsubishi Materials Corp. The aspect ratio of the gold nanorods was about 5 (51 ± 7 nm and 9.7 ± 1.1 nm in longitudinal and transverse directions, respectively). Longer (73 ± 11 nm × 10 ± 1.7 nm, aspect ratio = 7) and shorter (42 ± 5 nm × 9.2 ± 1.2 nm, aspect ratio = 4) rods were also used. CTAB and CTAC were obtained from Tokyo Kasei and used without further purification. The gold nanorod solution was centrifuged at 8000 × g for 60 min, and the precipitates were redispersed in a CTAC solution (80 mM). This procedure was repeated twice, and then the concentration of the gold nanorods was set to be 33 mM (0.22 mM as Au atoms). The absorbance of the nanorod solution was 0.5 at 900 nm and corresponds to the top peak of the longitudinal SP band. An appropriate amount of the nanorod solution (0.1–2.4 mL) and an ascorbic acid solution (100 mL, 0.5 mL) was added in a CTAC solution (10 mL, 80 mM). This is the reaction solution used for the formation of the silver shells. The AgCl particles or a silver nitrate solution was added to the reaction solution. AgCl nanoparticles were prepared by the addition of silver nitrate (17 mg) in a CTAC solution (10 mL, 80 mM). Dynamic light scattering measurements indicated that the sizes of the AgCl particles were distributed between 500–950 nm. A sodium hydroxide solution (0.5 M, 0.06–4.0 mL) and silver ions (10 mM, 0.25 mL) were added to the reaction solution with vigorous stirring. The sodium hydroxide solution was added to control the pH of the reaction solutions.Spectral changes of each solution were monitored by sampling a small portion of the reaction solutions in a thin optical cell (optical path length: 1 mm). TEM observations were performed using a JEM 2010 (JEOL, operated at 120 kV). A high-resolution image was obtained by JEM-2010FEF (JEOL, operated at 200 kV).
Conclusion
The uniform Au–Ag core–shell nanorods were obtained by retarding the silver shell growth on gold nanorods. The colloidal solutions of the Au–Ag core–shell nanorods showed the four extinction bands that originated from the anisotropic silver shell formation. The spectroscopic characters and the TEM images of the core–shell nanorods indicated that our methods gave very reproducible and controllable core–shell nanorods. This work provides a novel strategy to facilitate the precise control of the anisotropic silver-shell formation. The monodisperse anisotropic silver nanoparticles, which have never previously been produced, will contribute to novel optical phenomena that can be induced by anisotropic silver nanostructures.Acknowledgements
This work was supported in part by a KAKENHI (Grant-in-Aid for Scientific Research) on the Priority Area “Strong Photon-Molecule Coupling Fields (No. 470)” and a Grant-in-Aid for the Global COE Program, “Science for Future Molecular Systems” from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- S. Kalele, S. W. Gosavi, J. Urban and S. K. Kulkarini, Curr. Sci., 2006, 91, 1038–1052 CAS.
- C. Wang, S. Peng, R. Chan and S. Sun, Small, 2009, 5, 567–570 CrossRef CAS.
- Y. Yang, J. Shi, G. Kawamura and M. Nogami, Scr. Mater., 2008, 58, 862–865 CrossRef CAS.
- Y. Huang, Y. Yang, Z. Chen, X. Li and M. Nogami, J. Mater. Chem., 2008, 43, 5390–5393 Search PubMed.
- A. M. Gobin, M. H. Lee, N. J. Halas, W. D. James, R. A. Drezek and J. L. West, Nano Lett., 2007, 7, 1929–1934 CrossRef CAS.
- M. Tsuji, N. Miyamae, S. Lim, K. Kimura, X. Zhang, S. Hikino and M. Nishio, Cryst. Growth Des., 2006, 6, 1801–1807 CrossRef CAS.
- S. S. Shankar, A. Rai, A. Ahmad and M. Sastry, J. Colloid Interface Sci., 2004, 275, 496–502 CrossRef CAS.
- C.-C. Huang, Z. Yang and H.-T. Chang, Langmuir, 2004, 20, 6089–6092 CrossRef CAS.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. e. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS.
- L. R. Hirsch, J. B. Jackson, A. Lee, N. J. Halas and J. L. West, Anal. Chem., 2003, 75, 2377–2381 CrossRef CAS.
- L. Lu, H. Wang, Y. Zhou, S. Xi, H. Zhang, J. Hu and B. Zhao, Chem. Commun., 2002, 144–145 RSC.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, Berlin, 1994 Search PubMed.
- J. Turkevitch, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- B. L. Cusing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS.
- P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 CrossRef.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145 CrossRef.
- J. E. Millstone, G. S. Métraux and C. A. Mirkin, Adv. Funct. Mater., 2006, 16, 1209–1214 CrossRef CAS.
- C.-J. Huang, Y.-H. Wang, P.-H. Chiu, M.-C. Shih and T.-H. Meen, Mater. Lett., 2006, 60, 1896–1900 CrossRef CAS.
- B. D. Busbee, S. O. Obare and C. J. Murphy, Adv. Mater., 2003, 15, 414–416 CrossRef CAS.
- Y.-Y. Yu, S.-S. Chang, C.-L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef CAS.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3396 CrossRef CAS.
- A. Hatta, T. Ohshima and W. Suëtaka, Appl. Phys. A: Mater. Sci. Process., 1982, 29, 71–75 CrossRef.
- K. Aslan, J. R. Lakowicz and C. D. Geddes, Anal. Bioanal. Chem., 2005, 382, 926–933 CrossRef CAS.
- A. Taleb, C. Petit and M. P. Pileni, Chem. Mater., 1997, 9, 950–959 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS.
- A. Tao, P. Sinsermsuksakul and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 4597–4601 CrossRef CAS.
- R. Jin, Y. C. Cao, E. Hao, G. S. Métraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487–490 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Commun., 2001, 617–618 RSC.
- K. K. Caswell, J. N. Wilson, U. H. F. Burnz and C. J. Murphy, J. Am. Chem. Soc., 2003, 125, 13914–13915 CrossRef CAS.
- Y. Sun, B. Gates, B. Mayers and Y. Xia, Nano Lett., 2002, 2, 165–168 CrossRef CAS.
- Y. Sun, B. Mayers, T. Herricks and Y. Xia, Nano Lett., 2003, 3, 955–960 CrossRef CAS.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736–4745 CrossRef CAS.
- Y. Sun and Y. Xia, Adv. Mater., 2002, 14, 833–837 CrossRef CAS.
- B. Pietrobon, M. McEachran and V. Kitaev, ACS Nano, 2009, 3, 21–26 CrossRef CAS.
- B. Wiley, Y. Sun and Y. Xia, Acc. Chem. Res., 2007, 40, 1067–1076 CrossRef CAS.
- J. Pérez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzán and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870–1901 CrossRef CAS.
- Y. Xiang, X. Wu, D. Liu, Z. Li, W. Chu, L. Feng, K. Zhang, W. Zhou and S. Xie, Langmuir, 2008, 24, 3465–3470 CrossRef CAS.
- C. S. Ah, S. D. Hong and D.-J. Jang, J. Phys. Chem. B, 2001, 105, 7871–7873 CrossRef CAS.
- Z. Yang and H.-T. Chang, Nanotechnology, 2006, 17, 2304–2310 CrossRef CAS.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2004, 108, 5882–5888 CrossRef CAS.
- E. C. Cho, R. H. C. Camargo and Y. Xia, Adv. Mater., 2010, 22, 744–748 CrossRef CAS.
- Y. Okuno, K. Nishioka, N. Nakashima and Y. Niidome, Chem. Lett., 2009, 38, 60–61 CrossRef CAS.
- Y. Xiang, X. Wu, D. Liu, X. Jiang, W. Chu, Z. Li, Y. Ma, W. Zhou and S. Xie, Nano Lett., 2006, 6, 2290–2294 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368–6374 CrossRef CAS.
- B. J. Wiley, Y. Chen, J. M. McLellan, Y. Xiong, Z.-Y. Li, D. Ginger and Y. Xia, Nano Lett., 2007, 7, 1032–1036 CrossRef CAS.
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff and J. Gao, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS.
- Y. Niidome, K. Nishioka, H. Kawasaki and S. Yamada, Chem. Commun., 2003, 2376–2377 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6. See DOI: 10.1039/c0nr00130a |
| This journal is © The Royal Society of Chemistry 2010 |