Polypeptide-guided assembly of conducting polymer nanocomposites†
Mahiar
Hamedi
a,
Jens
Wigenius
a,
Feng-I.
Tai
b,
Per
Björk
a and
Daniel
Aili‡
*b
aDivision of Biomolecular and Organic Electronics Department of Physics, Chemistry and Biology, Linköping University, SE-581 83, Linköping, Sweden
bDivision of Molecular Physics, Department of Physics, Chemistry and Biology, Linköping University, SE-581 83, Linköping, Sweden. E-mail: danai@ifm.liu.se
First published on 6th August 2010
Abstract
A strategy for fabrication of electroactive nanocomposites with nanoscale organization, based on self-assembly, is reported. Gold nanoparticles are assembled by a polypeptide folding-dependent bridging. The polypeptides are further utilized to recruit and associate with a water soluble conducting polymer. The polymer is homogenously incorporated into the nanocomposite, forming conducting pathways which make the composite material highly conducting.
The development of nanoelectronics has resulted in enormous advancements in fabrication techniques that have enabled mass-production of CMOS circuits with feature sizes below 45 nm.1 There is a large interest in new methods to further push the size limits, lower the production costs and facilitate the design of more advanced three-dimensional structures beyond today's 2.5 dimensional architectures. Self-assembly is probably the most important scheme in this development and is currently applied to many different areas and classes of nanoelectronics.2 This technique enables fabrication of structures well below 10 nm in feature size and allows for incorporation of novel nanomaterials, such as metallic and semiconducting nanoparticles with many interesting optical and electrical properties.3 The controlled self-assembly of electroactive nanocomposites is of great interest for the development of novel functional materials for biosensors,4,5 electrochromic/plasmonic hybrid devices,6 and polymer/nanoparticle-based memories.7–9
In the field of molecular self-assembly, biomolecules are widely employed as nanoscale building blocks because of their excellent molecular recognition properties, programmability, and chemical and structural versatility.10 Biomolecules are, however, poor conductors and have consequently to be modified in order to be of interest for electronic applications. Water soluble conjugated/conducting polymers (CPs) that can associate to various biomolecules have been developed for this purpose. CPs that interacts with DNA have been employed to create supramolecular systems that mimic digital logic operations,11,12 and for assembly of aligned luminescent nanowires.13 In addition to DNA, proteins offer many interesting structural and chemical features and previously have been used as templates for assembly of CPs to obtain nanoelectronic functionalities.14 Most proteins are however very fragile and can easily and irreversibly lose their native conformation, which severely limits their applicability as scaffolds for CPs. One inherently very stable protein structural state is the amyloid fibril. Amyloid-like protein fibrils have shown strong affinity for certain classes of ionic CPs, which has enabled self-assembly of luminescent and conducting CP–amyloid nanowires.15–17 The growth of amyloid fibres is however not easily controlled and often results in fibres with broad size distributions. Moreover, they are also not easily combined with other materials for assembly of more complex hybrid/composite nanostructures. In this perspective, the use of designed polypeptides with controllable assembly and folding properties seems very attractive. Polypeptides are generally extremely robust and are in terms of chemical and structural properties very flexible, which facilitate their use as molecular building blocks in nanoscale architectures.
In this communication we demonstrate a novel method for self-assembly of a water soluble conducting polymer (PEDOT-S) into supramolecular nanocomposites using well defined nanoscale building blocks comprising designed synthetic polypeptides and polypeptide-decorated gold nanoparticles as schematically outlined in Fig. 1. The two synthetic polypeptides used here, JR2E and JR2K, are 42-residue polypeptides that are de novo designed to fold into a helix–loop–helix motif and heterodimerize into four-helix bundles at neutral pH.18–21 At neutral pH, the glutamic acid rich polypeptide JR2E has a net charge of −5 whereas JR2K is rich in lysine residues and has a net charge of +11. Charge repulsion prevents homodimer formation at pH 7 and the peptide exists as random coil monomers when kept separate.18,19 Incorporation of a cysteine residue in the loop region (position 22) yielded the polypeptides JR2EC and JR2KC. The thiol group enabled specific and site directed immobilization of JR2EC on planar gold surfaces19 and gold nanoparticles.22,23 The cysteine-containing peptides can also exist in an oxidized form where two monomers are linked through a disulfide bond. The oxidized peptides, JR2EC2 and JR2KC2, can self-assemble into several micrometre long nanofibres as a result of heteroassociation and folding into disulfide-linked four-helix bundles.24 The fibre length can be controlled by capping the fibre growth using peptides without the cysteine residue. We have recently demonstrated that these fibres can be decorated with luminescent conjugated polymers.25 Addition of JR2KC2 to gold nanoparticles decorated with JR2EC results in a reversible heteroassociation and folding-dependent bridging of the nanoparticles.26 The polypeptides were prepared on the solid phase and purified on HPLC as described in ESI†.
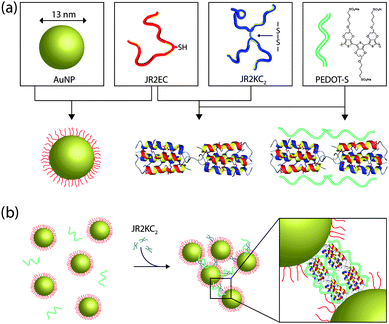 | ||
| Fig. 1 (a) Schematic illustration showing the basic nanoscale building blocks (from left to right): gold nanoparticles (AuNP), the designed polypeptides JR2EC and JR2KC2, and the conducting polyelectrolyte PEDOT-S. JR2EC is immobilized on the gold nanoparticles via a thiol residue in the loop region. Two JR2EC monomers can heteroassociate with JR2KC2 and fold into two disulfide-linked four-helix bundles, which can be utilized for assembly of JR2EC-decorated gold nanoparticles. The heterotrimeric complex is utilized as a scaffold for PEDOT-S for self-assembly of a conducting nanocomposite as illustrated in (b). | ||
The conducting polymer poly(3,4-ethylenedioxythiophene) (PEDOT) is among the most stable class of highly conducting CPs. In the present study, a new generation of this polymer, alkoxysulfonate poly(3,4-ethylenedioxythiophene) (PEDOT-S), is utilized (Fig. S1†).27 PEDOT-S is self-doped in pristine state with conductivities above 1 S cm−1. The anionic side chains make the polymer soluble in water and facilitate the interaction with various biomolecules. PEDOT-S has previously been demonstrated to assemble onto preformed amyloid fibrils forming conducting nanowire networks and transistors.28
Circular dichroism (CD) spectroscopy was employed in order to initially analyze the interaction between PEDOT-S and the polypeptides and the influence of PEDOT-S on the secondary structure of the polypeptides during self-assembly (Fig. 2a). When kept separately, JR2E and JR2K displayed typical random coil CD spectra. In the presence of PEDOT-S at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (50 µm), JR2K demonstrated a small increase in helicity whereas JR2E remained as a random coil. The interaction between PEDOT-S and the lysine rich JR2K is presumably mainly electrostatic resulting in enough shielding of charge repulsion between the polypeptide monomers to allow for a certain extent of folding. Folding of JR2K in the presence of a negatively charged CP has previously been reported.18 In the absence of PEDOT-S, heterodimerization results in a mean residue molar ellipticity at 222 nm ([Θ]222 nm) of about −21000 deg cm2 dmol−1. Addition of JR2K to a suspension of JR2E and PEDOT-S did not significantly affect the helicity, demonstrating that the peptides are able to fold properly in the presence of the polymer. If instead allowing PEDOT JR2K to first interact with PEDOT-S before introducing JR2E, a significantly lower helicity was obtained, [Θ]222 nm ≈ −14500 deg cm2 dmol−1. This indicates that PEDOT-S can associate to the JR2K monomers and affect their ability to heterodimerize and fold. The concentration of the peptides was assumed to be constant during measurements as no aggregation or precipitation was observed.
:1 molar ratio (50 µm), JR2K demonstrated a small increase in helicity whereas JR2E remained as a random coil. The interaction between PEDOT-S and the lysine rich JR2K is presumably mainly electrostatic resulting in enough shielding of charge repulsion between the polypeptide monomers to allow for a certain extent of folding. Folding of JR2K in the presence of a negatively charged CP has previously been reported.18 In the absence of PEDOT-S, heterodimerization results in a mean residue molar ellipticity at 222 nm ([Θ]222 nm) of about −21000 deg cm2 dmol−1. Addition of JR2K to a suspension of JR2E and PEDOT-S did not significantly affect the helicity, demonstrating that the peptides are able to fold properly in the presence of the polymer. If instead allowing PEDOT JR2K to first interact with PEDOT-S before introducing JR2E, a significantly lower helicity was obtained, [Θ]222 nm ≈ −14500 deg cm2 dmol−1. This indicates that PEDOT-S can associate to the JR2K monomers and affect their ability to heterodimerize and fold. The concentration of the peptides was assumed to be constant during measurements as no aggregation or precipitation was observed.
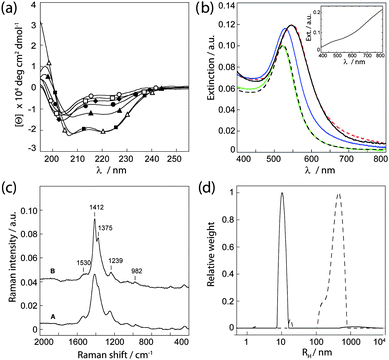 | ||
| Fig. 2 (a) CD spectra of JR2E and JR2K at pH 7 in the presence and absence of PEDOT-S. (○) JR2E, (□) JR2E + PEDOT-S, (◆) JR2K, (●) JR2K + PEDOT-S, (▲) JR2K + PEDOT-S + JR2E, (■) JR2E + PEDOT-S + JR2K and (△) JR2E + JR2K. (b) UV-vis spectra of (black broken line) AuNP–JR2EC, (green) AuNP–JR2EC + PEDOT-S, (blue) AuNP–JR2EC + PEDOT-S + JR2KC2, (black) AuNP–JR2EC + JR2KC2 + PEDOT-S, and (red broken line) AuNP–JR2EC + JR2KC2. Inset: PEDOT-S. (c) Raman spectra of A: PEDOT-S and B: AuNP–JR2EC + PEDOT-S + JR2KC2, cast and dried on a glass substrate. (d) Size distributions from DLS of (solid line) AuNP–JR2EC and PEDOT-S and (broken line) AuNP–JR2EC + PEDOT-S + JR2KC2. | ||
Gold nanoparticles with an average diameter of ∼13 nm were prepared by reduction of gold chloride with sodium citrate in aqueous solution.29 JR2EC was immobilized on the particles via the thiol residue in the loop region and unbound peptides were removed by repeated centrifugations. Spherical gold nanoparticles display a pronounced extinction band in the visible wavelength range due to the localized surface plasmon resonance (LSPR). A small shift in the LSPR peak position, from ∼520 nm to ∼524 nm, was seen after addition of the peptide, indicative of a successful immobilization. In the presence of PEDOT-S, these particles remained dispersed and no further changes in the LSPR peak were seen (Fig. 2b). This further confirms the observations from the CD spectrum of JR2E and PEDOT-S that no interactions between the two molecules occur and that the unspecific association of PEDOT-S to the gold nanoparticles is negligible. This is most likely due to the strong electrostatic charge repulsion between the two highly negatively charged species. The UV-vis spectrum of aqueous PEDOT-S dispersion typically shows a pale blue colour characterized by a broad bi-polaron absorption between 600 and 1000 nm, often referred to as a “free carrier tail” (Fig. 2b, inset).30 No changes in the spectra of PEDOT-S were seen in the presence of the peptides or peptide-decorated gold nanoparticles (data not shown), and the absorption of PEDOT-S was for clarity subtracted from the presented UV-vis spectra of the gold nanoparticles.
Aggregation of gold nanoparticles results in a substantial redshift of the LSPR peak position as well as a peak broadening, caused by the near-field coupling between adjacent particles. The magnitude of the redshift is highly dependent on the particle separation and the size of the aggregates. The smaller the distance and the larger the aggregates, the larger the resulting redshift.31,32 Specific and controlled aggregation of the JR2EC-decorated particles can be induced in a number of ways by exploiting the folding properties of the peptides. A rapid and extensive particle aggregation is observed in the presence of JR2KC2 due to a heteroassociation- and folding-dependent bridging of the particles.26 The resulting optical shift (Δλ > 30 nm) was not influenced by addition of PEDOT-S after addition of JR2KC2. Addition of PEDOT-S before addition of JR2KC2 also resulted in extensive particle aggregation but the resulting redshift was less pronounced (Δλ ≈ 10 nm), indicating either a larger particle separation upon aggregation or smaller aggregates (Fig. 2b). The particles eventually precipitated, resulting in a colourless solution and a purple precipitate, suggesting the presence of large aggregates. UV-vis spectra of the supernatant showed no presence of PEDOT-S indicating that all of the PEDOT-S had associated to the peptides. This was further confirmed by Raman spectroscopy (Fig. 2c). In the Raman spectrum of PEDOT-S, one strong peak at 1412 cm−1 and a few weaker bands were observed. The peaks are assigned as follows: 1530 cm−1 (asym C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C str, sym CαCβ(–H) str), 1412 cm−1 (sym CαCβ(–O) str), 1239 cm−1 (Cα–Cα′ (inter-ring) str) + Cβ–H bend, 986 cm−1 (oxyethylene ring def).33 An almost identical spectrum, but showing a slightly narrower band at 1412 cm−1, was obtained for the nanocomposite, demonstrating that the polymer was present in the composite and that its structure was not significantly affected by the association to the polypeptides.
C str, sym CαCβ(–H) str), 1412 cm−1 (sym CαCβ(–O) str), 1239 cm−1 (Cα–Cα′ (inter-ring) str) + Cβ–H bend, 986 cm−1 (oxyethylene ring def).33 An almost identical spectrum, but showing a slightly narrower band at 1412 cm−1, was obtained for the nanocomposite, demonstrating that the polymer was present in the composite and that its structure was not significantly affected by the association to the polypeptides.
Dynamic light scattering experiments further confirmed that gold nanoparticles functionalized with JR2EC did not assemble with PEDOT-S until after addition of JR2KC2 (Fig. 2d). In aqueous solution, PEDOT-S mainly exists as single chains or aggregates of only a few polymer chains.27 Dispersions of PEDOT-S and the JR2EC modified particles showed a rather narrow size distribution with a hydrodynamic radius of approximately 10 nm, mainly reflecting the size of the peptide functionalized particles as the particles are significantly better scatterers than the dispersed polymer. The addition of JR2KC2 shifted and broadened the size distribution to about 500 nm within 5 minutes, indicating rapid initiation of the self-assembly process and extensive particle aggregation (Fig. 2d). The normalized autocorrelation functions are presented in Fig. S2†. DLS also confirmed the interaction of JR2KC2 with PEDOT-S, as assemblies with a broad size distribution and an average hydrodynamic radius of 150 nm were observed (Fig. S2†).
Transmission electron microscopy (TEM) was used to analyze the structure of the nanocomposites. In the absence of PEDOT-S, large aggregates with a small and uniform interparticle separation (4.6 ± 0.2 nm) were seen (Fig. 3a). This is a consequence of the folding-dependent bridging when JR2KC2 associates with the immobilized JR2EC.27 Addition of JR2KC2 to a suspension of JR2EC functionalized nanoparticles containing PEDOT-S also resulted in large particle aggregates with a slightly larger and less uniform interparticle separation (Fig. 3b), as was indicated by the UV-vis spectra. The association of PEDOT-S to the peptides thus clearly affects the assembly of the particles resulting in less organized aggregates. This is presumably due to the association of PEDOT-S to JR2KC2 which, as was indicated in the CD spectra, may induce a certain extent of homoassociation of the polypeptides and consequently an increase in particle separation. The conducting nature of PEDOT-S allows for larger assemblies of the polymer to be visualized directly in TEM.28 No aggregates of PEDOT-S were however observed here (Fig. 3b), further indicating that the polymer is evenly distributed in the nanocomposite.
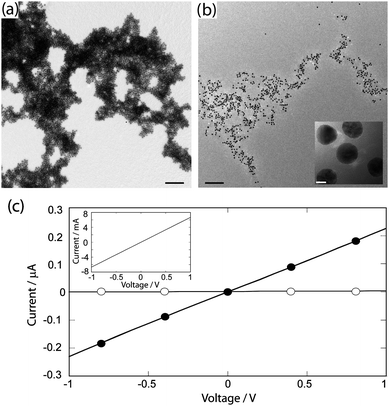 | ||
| Fig. 3 Electron micrographs of the self-assembled nanocomposites (a) without PEDOT-S and (b) with PEDOT-S. Scale bars: 200 nm, inset 5 nm. (c) Current–voltage characteristics of the nanocomposites: (○) without PEDOT-S and (●) with PEDOT-S as measured on inter-digitated gold electrodes. Inset: current–voltage characteristics of pure PEDOT-S. | ||
Conductivity measurements of the composite material were carried out by casting the self-assembled structures from water solution onto inter-digitized gold electrodes, followed by measurement of current–voltage characteristics (CV). The same technique was utilized when depositing the nanocomposites on the electrodes as on the TEM-grids, resulting in very thin films. The gold nanoparticle–polypeptide complex alone did not show any conductivity because of the separation (∼5 nm) between the gold nanoparticles induced by the non-conducting four-helix bundles (Fig. 3c). Despite displaying less dense assemblies, the PEDOT-S-containing nanocomposite was clearly conducting. Fully ohmic conductivity behaviour was seen for up to 1 V sweeps. The ohmic behaviour suggests that PEDOT-S remained conducting after the self-assembly process, and that the CP did not undergo noticeable oxidation/reduction during the voltage sweeps. As no unspecific binding of PEDOT-S to the particles was observed (Fig. 2), this strongly indicates that the resulting electrical conductivity throughout the bulk structure is a result of the association of PEDOT-S to the heterotrimeric polypeptide complex, which provides conducting nanobridges between the gold nanoparticles. The overall conduction should hence be a combined result of inter-chain, and intra-chain hopping in the CP chains, as well as conduction through the gold nanoparticles and electron transfer between the gold nanoparticle and CPs. The conductivity was lower in the nanocomposite than when casting pure PEDOT-S on the electrodes (Fig. 3c, inset), which most likely is due to the lower amount of PEDOT-S present in the composite materials as compared to in the pure polymer solution as well as to the resistance at the polymer–nanoparticles interface.
Conclusions
The controlled self-assembly of highly conducting conjugated polymers (CPs) into a supramolecular electroactive nanocomposites using synthetic designed polypeptides and polypeptide-decorated gold nanoparticles has been demonstrated. The polypeptide functionalized gold nanoparticles are assembled using a second set of polypeptides designed to heteroassociate and fold with the immobilized polypeptides. The CPs further associate to the polypeptides without significantly affecting their secondary structure, resulting in formation of extensive conducting networks. The proposed strategy for fabrication of electroactive nanocomposites enables a high level of control over the assembly process and the spatial arrangement of the nanoscale building blocks, as well as a large flexibility with respect to material composition. An efficient integration of conducting polymers into well defined nanocomposites will facilitate the development of novel components for bioorganic electronics, optoelectronics and biosensors and enable means for realizing an electronic interface at the nanoscale.Acknowledgements
The authors thank Roger Karlsson for kindly providing the PEDOT-S, and Prof. Lars Baltzer, Dr Johan Rydberg and Dr Karin Enander for introducing us to the field of synthetic polypeptides, and Anna Herland for discussions. Financial support from the Knut and Alice Wallenberg Foundation (KAW), Forum Scientium, the Swedish Research Council (VR) and the Strategic Research Foundation (SSF) through the OBOE centre and the programmes OPEN and NanoSense is also gratefully acknowledged. The authors are grateful for the support from Prof. Olle Inganäs and Prof. Bo Liedberg.Notes and references
- ITRS, International technology roadmap for semiconductors 2009 edition, 2010,available: http://www.itrs.net/Links/2009ITRS/Home2009.htm, last accessed 22 mars Search PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS.
- J. Li, Curr. Opin. Colloid Interface Sci., 2009, 14, 61 CrossRef CAS.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028 RSC.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS.
- M. A. G. Namboothiry, T. Zimmerman, F. M. Coldren, J. W. Liu, K. Kim and D. L. Carroll, Synth. Met., 2007, 157, 580 CrossRef CAS.
- R. J. Tseng, J. X. Huang, J. Ouyang, R. B. Kaner and Y. Yang, Nano Lett., 2005, 5, 1077 CrossRef CAS.
- G. Zotti, B. Vercelli and A. Berlin, Chem. Mater., 2008, 20, 6509 CrossRef CAS.
- A. Prakash, J. Ouyang, J. L. Lin and Y. Yang, J. Appl. Phys., 2006, 100, 054309 CrossRef.
- N. C. Seeman, Mol. Biotechnol., 2007, 37, 246 Search PubMed.
- B. M. Frezza, S. L. Cockroft and M. R. Ghadiri, J. Am. Chem. Soc., 2007, 129, 14875 CrossRef CAS.
- Y. L. Tang, F. He, S. Wang, Y. L. Li, D. B. Zhu and G. C. Bazan, Adv. Mater., 2006, 18, 2105 CrossRef CAS.
- P. Björk, A. Herland, I. G. Scheblykin and O. Inganäs, Nano Lett., 2005, 5, 1948 CrossRef.
- P. Björk, A. Herland, M. Hamedi and O. Inganäs, J. Mater. Chem., 2010, 20, 2269 RSC.
- K. P. R. Nilsson, A. Herland, P. Hammarström and O. Inganäs, Biochemistry, 2005, 44, 3718 CrossRef CAS.
- A. Herland, P. Björk, K. P. R. Nilsson, J. D. M. Olsson, P. Åsberg, P. Konradsson, P. Hammarström and O. Inganäs, Adv. Mater., 2005, 17, 1466 CrossRef CAS.
- A. Herland, P. Björk, P. R. Hania, I. G. Scheblykin and O. Inganäs, Small, 2007, 3, 318 CrossRef CAS.
- K. P. R. Nilsson, J. Rydberg, L. Baltzer and O. Inganäs, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10170 CrossRef CAS.
- K. Enander, D. Aili, L. Baltzer, I. Lundström and B. Liedberg, Langmuir, 2005, 21, 2480 CrossRef CAS.
- S. Olofsson and L. Baltzer, Folding Des., 1996, 1, 347 CrossRef CAS.
- S. Olofsson, G. Johansson and L. Baltzer, J. Chem. Soc., Perkin Trans. 2, 1995, 2047 RSC.
- D. Aili, K. Enander, J. Rydberg, I. Nesterenko, F. Björefors, L. Baltzer and B. Liedberg, J. Am. Chem. Soc., 2008, 130, 5780 CrossRef CAS.
- D. Aili, K. Enander, J. Rydberg, I. Lundstrom, L. Baltzer and B. Liedberg, J. Am. Chem. Soc., 2006, 128, 2194 CrossRef CAS.
- D. Aili, F. I. Tai, K. Enander, L. Baltzer and B. Liedberg, Angew. Chem., Int. Ed., 2008, 47, 5554 CrossRef CAS.
- J. Wigenius, P. Björk, M. Hamedi, D. Aili, Macromol. Biosci. DOI:10.1002/mabi.200900463.
- D. Aili, K. Enander, L. Baltzer and B. Liedberg, Nano Lett., 2008, 8, 2473 CrossRef CAS.
- R. H. Karlsson, A. Herland, M. Hamedi, J. A. Wigenius, A. Åslund, X. Liu, M. Fahlman, O. Inganäs and P. Konradsson, Chem. Mater., 2009, 21, 1815 CrossRef CAS.
- M. Hamedi, A. Herland, R. H. Karlsson and O. Inganäs, Nano Lett., 2008, 8, 1736 CrossRef CAS.
- G. Frens, Nature (London) Phys. Sci., 1973, 241, 20.
- T. Kim, J. Kim, Y. Kim, T. Lee, W. Kim and K. S. Suh, Curr. Appl. Phys., 2009, 9, 120 CrossRef.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, New York, 1995, vol. 25 Search PubMed.
- A. A. Lazarides and G. C. Schatz, J. Phys. Chem. B, 2000, 104, 460 CrossRef CAS.
- S. Garreau, G. Louarn, J. P. Buisson, G. Froyer and S. Lefrant, Macromolecules, 1999, 32, 6807 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Structure of PEDOT-S, DLS data and experimental details. See DOI: 10.1039/c0nr00299b |
| ‡ Current address: Department of Materials and Institute of Biomedical Engineering, Imperial College London, SW7 2AZ, London, UK. |
| This journal is © The Royal Society of Chemistry 2010 |