Coating and dispersion of ceramic nanoparticles by UV-ozone etching assisted surface-initiated living radical polymerization†
Toshihiko
Arita
*
Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aobaku, Sendai 980-8577, Japan. E-mail: tarita@tagen.tohoku.ac.jp; Fax: (+81) 22-217-5631; Tel: (+81) 22-217-5630
First published on 18th August 2010
Abstract
Commercially available unmodified ceramic nanoparticles (NPs) in dry powder state were surface-modified and dispersed in almost single-crystal size. The surface-initiated living radical polymerization after just UV-ozone soft etching enables one to graft polymers onto the surface of ceramic NPs and disperse them in solvents. Furthermore, a number of NPs were dispersed with single-crystal sizes. The technique developed here could be applied to almost all ceramic NPs including metal nitrides.
Composite materials, especially, inorganic–polymer composites are quite attractive because they exhibit promising new properties which are believed to be trade-off functions.1 In order to solve the trade-off, polymer grafting to the surface of inorganic materials seems to be essential. Because most of the polymer materials are in the metastable state, composite materials where polymers are simply blended with inorganic materials are not stable, i.e., their components will segregate. When polymer chains are grafted to inorganic NPs, segregation is inhibited because the polymer species are perfectly mixed (without phase separation). However, the handling of inorganic NPs is not so simple. Because of their high surface energy density, NPs easily agglomerate and/or aggregate causing a loss of nanosized functionalities.2
Many studies have been reported on grafting polymer chains to NPs, however, all of the examples need special reagents to connect the inorganic surfaces and polymers, e.g., silane coupling agents,3–5 thiols,6,7 organic ligands,8–10 fatty acids,11 phosphonic acid12etc. For example, using a silane coupling agent, one of the most promising surface modifiers of NPs, one can modify the surface of NPs and disperse many kinds of NPs. However, silane coupling agents are expensive and limited by their storage and handling requirements. Here, a new and quite simple method to produce directly polymer-grafted ceramic NPs is proposed. This technique drastically increases the number of combinations of ceramics and polymers that can be grafted and reduces the number of the steps needed to prepare polymer-grafted ceramic NPs.
Commercially available powder ceramic NPs were prepared and spread on a glass shale. The shale was introduced into a UV-ozone etcher (TC-003/C, Meiwafosis Co., Ltd.) and exposed to UV-ozone radiation for 45 min. After the exposure, the surface layer of powder was immediately transferred into a test tube with a screw cap and transferred into the oxygen- and moisture- free glove box. Then, ta previously prepared solution for living radical polymerization (LRP) was added to the test tube and it inserted into an aluminium block heater already heated to the desired temperature (see the ESI†). After a scheduled time had passed, the test tube was removed from the glove box and the polymerization was quenched by adding an oxygen-rich good solvent for the synthesized polymer. The NPs were washed with the same solvent by dispersion and centrifugation at least five times to remove any unbound polymer from the solution. The characterization of the NPs was carried out by transmission electron micrography (TEM), dynamic light scattering (DLS), attenuated total reflection Fourier-transfer infrared spectroscopy (ATR-FTIR), thermogravimetry (TG), X-ray photoelectron spectroscopy (XPS) and gel permeation chromatography (GPC).
Using this simple fabrication, colloidally stable NPs were obtained. For example, the dispersion of poly(methyl methacrylate) (PMMA)-grafted α-Al2O3 NPs in chloroform gives a stable colloidal solution as depicted in Fig. 1. The α-Al2O3 NPs could be separated by just evaporating the solvent and they formed α-Al2O3 cake, not powder. The α-Al2O3 cake could be redispersed in chloroform and dried again and again. These results demonstrate that the polymer was successfully grafted to the surface of α-Al2O3. Without the grafted polymer chains, the NPs could not stably disperse in chloroform and form a tough cake.
 | ||
| Fig. 1 Images of unmodified α-Al2O3 powder, dispersion of PMMA-grafted α-Al2O3 in chloroform (5 wt%) and a thin cake of PS-grafted α-Al2O3. The coin represents the scale (d = 20 mm). | ||
The average particle size of PMMA-grafted α-Al2O3 NPs measured by DLS is about 80 nm as shown in Fig. 2(a). The DLS size indicates that some dispersed NPs underwent aggregation or agglomeration. However, the initial averaged particle size (APS) (from the NP supplier, see the ESI†) of α-Al2O3 NPs was 40–80 nm, this implies that the NPs were dispersed from a dry powder state to only a few NP aggregates. The DLS results of poly(styrene) (PS)-grafted TiN and AlN NPs also suggest the size of the dispersants in the solutions were slightly larger than the APS of the NPs. In addition, the sizes of the dispersants were less than 100 nm and their dispersions were quite stable for more than 1 day. The dispersion of PS-grafted TiN NPs in chloroform was quite stable. A 5 wt% solution of the PS-grafted TiN NPs suspension did not show any precipitation for 3 months. This was the most stable and highest NP containing dispersion obtained in this study. The stability of the dispersion depended on the core ceramic material, the NP size, the polymer material and the polymer molecular weight. NPs with a smaller APS and those which were non-magnetic tended to show better dispersion. In addition, with increasing NP content, the dispersion became less stable (ESI†).
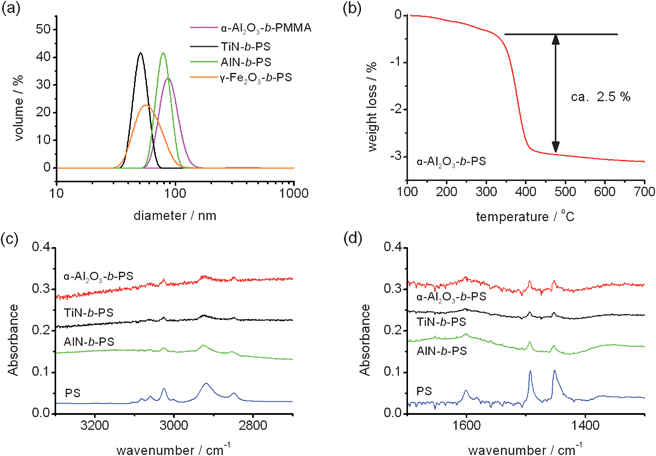 | ||
| Fig. 2 DLS sizes of the PMMA-grafted α-Al2O3 NPs, PS-grafted TiN, AlN and γ-Fe2O3 NPs in chloroform (a). The concentrations of the NPs were 0.1 wt% except for the γ-Fe2O3 NPs (0.05 wt%). The TG curve of the PS-grafted α-Al2O3 NPs is also depicted (b). ATR-FTIR spectra of the PS-grafted α-Al2O3,TiN and AlN NPs. The wavenumber regions where the C–H vibration appears (c) and C–C vibration appears (d) were individually depicted because the peaks corresponding to PS-grafted NPs were very small. | ||
To examine the surface nature and interaction between the ceramic NPs and polymer chains, the ATR-FTIR spectra of polymer-grafted NPs were analyzed. Fig. 2(c) and (d) show the FTIR spectra of PS-grafted TiN, AlN and α-Al2O3 NPs. Bands observed in the 3000–2800 cm−1 and 3100–3000 cm−1 region were attributed to the C–H stretching mode of alkyl and aromatic groups, respectively. The bands at 1600 and 1500–1430 cm−1 correspond to the C–C vibration frequency of the aromatic group. The ATR-FTIR analysis also supports that PS was successfully grafted to the surface of the ceramic NPs.
The amount of grafted polymer was measured by TG analysis. As an example shown in Fig. 2(b), the TG curve of the PS-grafted α-Al2O3 NPs showed a weight reduction around 400 °C and the total weight loss was 2.5%. PS thermally decomposes at 400 °C at atmosphere pressure and α-Al2O3 is stable at 800 °C so the weight loss can be regarded as the amount of PS included in the PS-grafted α-Al2O3 NPs. From the weight loss, the number of PS chains grafted on α-Al2O3 NPs was calculated as 154 chains per NP. This can be converted to the graft density of PS chains to the α-Al2O3 surface, that is, 0.0045 chains nm−2. As shown in the ESI† (TG curve of the PS and TiN mixture which was prepared using the same procedure as PS-grafted TiN NPs without the UV-ozone surface treatment), if the polymer chains were not grafted to NPs, they would have been eliminated by the five cycles of dispersion and centrifugation (see the ESI†).
For further analysis of the surface chemistry of the ceramic NPs, XPS spectra were also analyzed. The XPS spectra shown in Fig. 3 clearly show the amount of carbon was reduced after the UV-ozone treatment and significantly increased after the graft polymerization of MMA. In addition, the amounts of Al and O decreased after the graft polymerization. These imply the presence of PMMA on the α-Al2O3 NPs after the graft polymerization. It also shows there is no significant change in the Al 2p and O 1s XPS spectra before and after the UV-ozone treatment. These observations are consistent with the previous analyses of FTIR and TG, i.e., the graft density was very small.
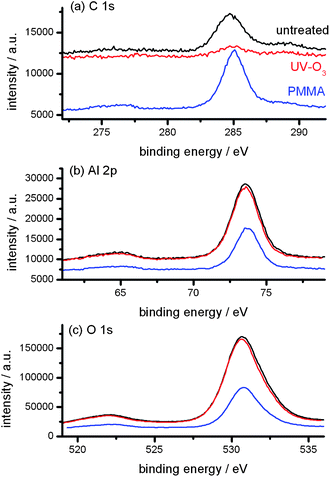 | ||
| Fig. 3 C 1s (a), Al 2p (b) and O 1s (c) XPS spectra of untreated (black), UV-ozone treated (red) and PMMA-grafted (blue) α-Al2O3 NPs. | ||
TEM was employed to examine the size and morphology of the NPs. Fig. 4 depicts the TEM images of the directly polymer-grafted ceramic NPs. As one can clearly see from the images, most of the NPs are dispersed into single nanometre sizes. These good dispersions could never be achieved if the polymer chains were not grafted onto the surface of the NPs. The initial state of the NPs was powder, this implies that the polymer chain has to penetrate into the gaps between aggregated NPs to disperse them. This is unlikely to happen if styrene does not polymerize from the NPs surface because the affinity between styrene and ceramics is poor. It is more reasonable to think the polymerizing PS chains from the NPs might disperse NPs. This is quite interesting. The particle size information obtained from the TEM image does not directly reflect the entire distribution, however, it implies some of the NPs were dispersed with single-crystal sizes from an aggregated powder of NPs.
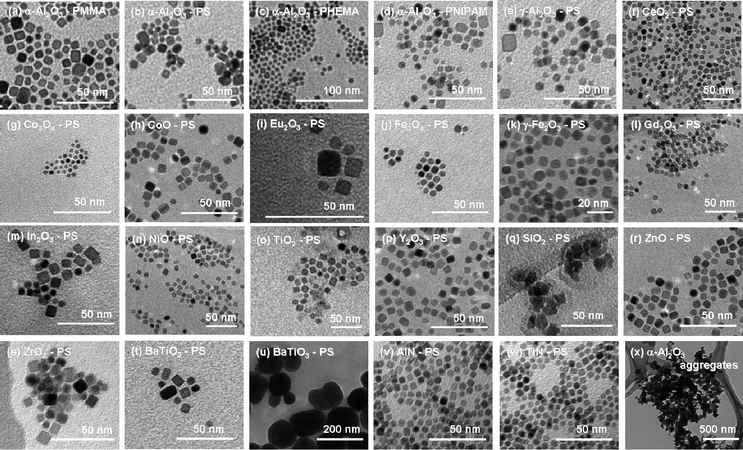 | ||
| Fig. 4 TEM images of the directly polymer-grafted NPs. The image of the unmodified α-Al2O3 NPs (x) is also depicted for comparison. | ||
The predicted chemical bonding between NPs and polymers will be discussed here. Although no direct evidence for covalent modification by polymers is presented in this paper, circumstantial evidence supporting this was presented above, e.g., the difference of TG weight loss of polymer-grafted and simply blended NP and polymer mixtures. This implies the polymer chains were grafted through a chemical bond. It is well known that oxygen atoms in an excited state (O(1D)) are generated during the UV-ozone etching process.13 O(1D) is quite reactive and can oxidize many materials. Meanwhile, it is also well known that ceramic surfaces are terminated by O and O–H groups.14,15 When O(1D) generated by UV-ozone etching attacks an O–H terminated site on the ceramic surface, the site will be oxidized and may be converted to peroxide (O–O–H). The peroxide may initiate radical polymerization by homolytic fragmentation of the O–O bond of the peroxide. This is the reaction that we expected to occur and the idea behind this study. The rate of polymerization for the solution of monomer with UV-ozone-treated NPs was always much faster than those without UV-ozone treated NPs. This supports effective propagating radical production from the surface of UV-ozone-treated NPs.
The yield of well-dispersed NPs can be varied by the species of the core material, the condition of the UV-ozone exposure and the polymerization, however, well-dispersed NPs were obtained for all the core materials tested in this study. The polymer-grafted NPs synthesized did not include any binder between the NPs and polymers so they should free from the problems caused by ligand-type surface modifiers, e.g., reduction of the core fraction in the composite, coloring etc.10,12 This method could be a promising new technique to utilize NPs in a powder (dry) state for inorganic–polymer hybrid materials, especially, for coating, paints, calcination, sintering, semiconductors and so on.
In conclusion, the surface modification of NPs in a powder (dry) state by surface-initiated living radical polymerization was performed using an extraordinarily simple method. The method developed in this study enables one to prepare directly polymer-grafted NPs with more or less no limitation of the combination between ceramic NPs and grafted vinyl polymers.
Acknowledgements
This work was supported by a Scientific Research Grant from the Ministry of Education, Science, Sports, and Culture of Japan. The author thanks Grant-in-Aid for Young Scientists (B) No. 19750094 for financial support.References
- Hybrid Materials, ed. G. Kickelbick, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- Nanoparticles, ed. G. Schmid, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- J. Pyun, S. Jia, T. Kowalewski, G. D. Patterson and K. Matyjaszewski, Macromolecules, 2003, 36, 5094–5104 CrossRef CAS.
- R. Matsuno, K. Yamamoto, H. Otsuka and A. Takahara, Macromolecules, 2004, 37, 2203–2209 CrossRef CAS.
- E. Marutani, S. Yamamoto, T. Ninjbadgar, Y. Tsujii, T. Fukuda and M. Takano, Polymer, 2004, 45, 2231–2235 CrossRef CAS.
- H. Oh and P. F. Green, Nat. Mater., 2009, 8, 139–143 CrossRef CAS.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS.
- C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111–123 RSC.
- R. Matsuno, K. Yamamoto, H. Otsuka and A. Takahara, Chem. Mater., 2003, 15, 3–5 CrossRef CAS.
- M. Iijima, M. Kobayakawa, M. Yamazaki, Y. Ohta and H. Kamiya, J. Am. Chem. Soc., 2009, 131, 16342–16343 CrossRef CAS.
- B. Hojjati and P. A. Charpentier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3926–3937 CrossRef CAS.
- T. Arita, K. Moriya, K. Minami, T. Naka and T. Adschiri, Chem. Lett., 2010, 39, 961–963 CrossRef.
- J. M. Dohan and W. J. Masschelein, Ozone: Sci. Eng., 1987, 9, 315–334 CAS.
- Surface Coatings for Advanced Materials, ed. R. P. Agarwala, Trans. Tech., Zurich, 1997 Search PubMed.
- Prospect of Surface Treatment and Modification Technology in Inorganic Materials: Metal, Ceramics and Glass, ed. E. Kamijo, Y. Suzuki and A. Fujisawa, CMC Publishing Co., Ltd, Tokyo, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XPS spectra and the list of the directly polymer grafted nanoparticles. See DOI: 10.1039/c0nr00317d |
| This journal is © The Royal Society of Chemistry 2010 |