Tuning intermolecular non-covalent interactions for nanowires of organic semiconductors
Lang
Jiang
ac,
Jianhua
Gao
a,
Yanyan
Fu
a,
Huanli
Dong
*a,
Huaping
Zhao
a,
Hongxiang
Li
b,
Qingxin
Tang
d,
Keqiu
Chen
d and
Wenping
Hu
*a
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100080, China. E-mail: dhl522@iccas.ac.cn; huwp@iccas.ac.cn
bShanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China
cGraduate University of Chinese Academy of Sciences, Beijing, 100039, China
dDepartment of Physics, Hunan University, Changsha, 410082, China
First published on 26th October 2010
Abstract
Anthracene and its derivatives are used to demonstrate a simple way to cast assemble nanowires of organic semiconductors with tuning of intermolecular non-covalent interactions by molecular design. The tuning of intermolecular interactions could be achieved by (i) decreasing intermolecular hydrophobic interactions by linking hydrophilic side chains to anthracene rings, (ii) increasing intermolecular interaction for self-assembly with the assistance of hydrogen bonds, and (iii) enhancing molecular π–π interaction by increasing the conjugated dimension of the compounds.
Introduction
Covalent interactions (bonds) hold atoms together with large bond energies on the order of ∼102 kcal mol−1. Compared with covalent interactions, non-covalent interactions are much weaker. However, the cumulative energies of non-covalent interactions could be huge. For example, huge numbers of small non-covalent forces drive the spontaneous folding or unfolding of proteins and nucleic acids. Moreover, the weak non-covalent interactions (hydrogen bonding, π-stacking, electrostatic interactions, van der Waals forces etc.) could be tuned by designing molecules so that they can self-assemble one by one into desired architectures. It gives the potential advantages of constructing building blocks using synthetic chemistry at multi levels,1,2 and opens the “bottom-up” route for the synthesis of organic nanostructures.3–7 Discrete nanoparticles with controlled chemical composition are readily synthesized.8–10 However, the strategy still poses substantial intellectual challenges. For example, although there are countless examples of self-assembly, the basic rules that govern these assemblies, the self-assembling processes with design, and the nature of non-covalent forces during the self-assembly processes, are still being studied. In addition, the ideas for information processing and electrical transduction require either fundamental redesign in going from the macroscopic systems presently used to self-assembled systems, or the development of new types of organic molecules that show appropriate properties.11–13Organic semiconductors have been attracting worldwide attention and experiencing a rapid development recently.14–18 The conjugated materials wherein the π delocalized electrons are responsible for the intra-molecular conduction. They may often be solution-processed, allowing the fabrication of devices on plastic substrates such as Kapton (polyimide) or PET (polyethylene terephthalate). And their low cost is ideal for large-area applications. In addition, the most attractive point of organic semiconductors is the incorporation of molecular functionality by design, which opens the door for synthesizing desired materials with desired functions under control. Here, by using anthracene and its several derivatives as candidates (Scheme 1) we demonstrate a simple way to tuning intermolecular non-covalent interactions for the assembly of nanowires of organic semiconductors. As we know, acenes, which consist of n-linked benzene rings (n = 3: anthracene, n = 4: tetracene, n = 5: pentacene), are promising candidates for electronic devices because their planar shapes facilitate crystal packing and the extended π system over molecules enables the intermolecular overlap of π systems. Hole mobility of single crystals of acenes were found to reach 3 cm2 V−1 s−1 for anthracene,19–21 1 cm2 V−1 s−1 for tetracene22–24 and several tens of cm2 V−1 s−1 for pentacene25–27 at room temperature. However, the instability and insolubility of pentacene limit its practical applications regardless its outstanding electrical properties (e.g., field-effect properties). Recent progresses on anthracenes have demonstrated not only high stability, but also good electrical properties even in thin film states.28–30 Hence, here we use anthracene and its derivatives as the candidate compounds to demonstrate how to tune intermolecular non-covalent interactions to self-assemble nanowires of organic semiconductors.
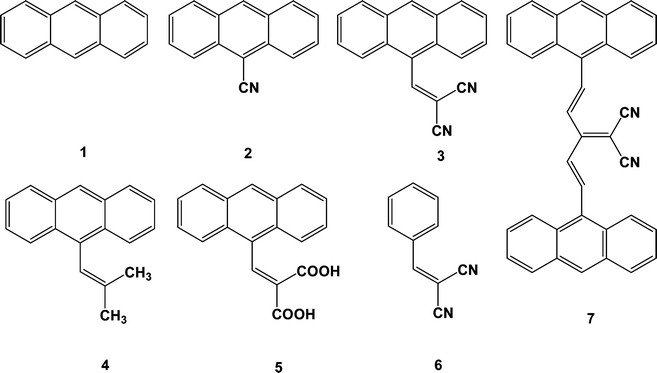 | ||
| Scheme 1 Molecular structures of anthracene and its derivatives. | ||
Experimental
Anthracene and its six derivatives (Scheme 1) could be divided into two groups, group one (compounds 1, 2, 3, 4, 5) were changed to examine the intermolecular interaction induced by molecular side chain for self-assembly, group two (compounds 3, 6, 7) was used to examine the role of conjugated dimension for self-assembly. Herein, compounds 1, 2 were purchased from Aldrich directly. Compounds 4,31,325,33634 were synthesized according to reference as described elsewhere. Compounds 3 and 7 were synthesized as described below.Compound 3
0.372 g (5.6 mmol) malononitrile and a drop of piperidine added into the solution of 0.393 g 9-anthraldehyde (1.9 mmol) in 50 ml CH3CN. The mixture was then heated to reflux for 0.5 h. After cooling, the solvent was evaporated. The residue was purified by column chromatography (CH2Cl2) to afford 0.3 g product (yield: 62%). MS (EI) m/z, 254. 1H-NMR (300MHz, CDCl3): δ 8.94 (s, 1H), 8.66 (s, 4H), 8.09 (d, 2H), 7.94 (d, 2H), 7.70–7.54 (m, 4H).Compound 7
A three-neck flask was charged with 1,5-bis(9-anthryl)-1,4-pentadien-3-one (0.91 g, 2.1 mmol), malononitrile (0.66 g, 10 mmol, ammonium acetate (3.85 g, 50 mmol), glacial acetic acid (3 mL) and 50 mL toluene. The reaction mixture was heated to reflux and stirred vigorously for 2 days. Then it was cooled to room temperature, CH2Cl2 (100 mL) was added. CH2Cl2 layer was washed with water to remove ammonium acetate and glacial acetic acid. The organic phase was dried over MgSO4, filtered, and evaporated to dryness. The crude product was washed by petroleum ether and further purified by silica gel column chromatography using petroleum ether/methylene chloride (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent. After crystallized from toluene, pure product was obtained as red solid. Yield, 0.68 g (67%). MS (EI) m/z 482.1H NMR (400 MHz, CDCl3) 8.59 (d, 2H), 8.54 (s, 2H), 8.36 (d, 4H), 8.07 (d, 4H), 7.57 (m, 8H), 7.46 (d, 2H). Anal. calcd for C36H22N2 (%): C: 89.60; H: 4.60; N: 5.80. Found: C: 89.28; H: 4.71; N: 5.69.
:1) as eluent. After crystallized from toluene, pure product was obtained as red solid. Yield, 0.68 g (67%). MS (EI) m/z 482.1H NMR (400 MHz, CDCl3) 8.59 (d, 2H), 8.54 (s, 2H), 8.36 (d, 4H), 8.07 (d, 4H), 7.57 (m, 8H), 7.46 (d, 2H). Anal. calcd for C36H22N2 (%): C: 89.60; H: 4.60; N: 5.80. Found: C: 89.28; H: 4.71; N: 5.69.
The substrates used in the experiments were successively cleaned with pure water, hot acetone, hot ammonia/hydrogen peroxide solution (ammonia/hydrogen peroxide/water = 1:1:5), pure water, pure ethanol, and finally with argon plasma treated for 15 s. After that, anthracene and its derivatives solutions were cast on the SiO2/Si or glass substrates directly, then, kept the substrates in the Petri dishes at room temperature over night; after the solvent evaporation, small crystals were obtained on substrates. The products were characterized by XRD (D/max2500) and SEM (Hitachi S-4300 SE).
Results and discussion
Anthracene (compound 1) always produces small crystals, hexagonal or elongated hexagonal small crystals, as shown in Fig. 1a. The controllable self-assembly of anthracene itself into one-dimensional (1D) micro- and nanowires is highly challenging. The main reasons lie in the controlling of the molecular assembly for the growth along the π-stacking direction against the lateral association of other directions. Usually, the growth along the π-stacking direction (driven by the intermolecular π–π interaction and hydrogen bonds etc.) dictates the self-assembly of 1D morphology, while the growth in other directions favors the bulk assemblies (driven by the strong, solvophobically favorable hydrophobic interactions between anthracene molecules). The competition of the two growths decides the organizing states of organic molecules. Seeing the molecular packing in anthracene crystal, the molecular inter distance along b axis is around 6.09 Å (Fig. 1b), indicating the weak π–π interaction between molecules, i.e., the equal growth probability of anthracene molecules along π-stacking direction or other directions, so that anthracene molecules self-assemble into hexagonal or elongated hexagonal small crystals.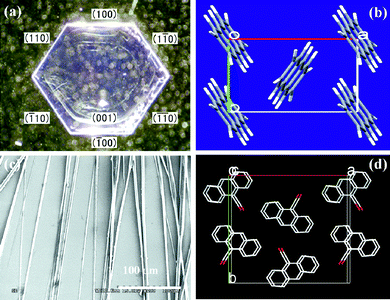 | ||
| Fig. 1 (a) Optical images of cast crystal of compound 1, (b) molecular stack of compound 1 crystal, (c) SEM images of cast crystal of compound 2 from CH2Cl2 (saturated solution), (d) molecular stacks of compound 2 crystal. | ||
It is expected that the growth competition of anthracene between the π-stacking direction and other directions could be tuned so that molecules assemble along the π-stacking direction instead of other directions. For this target, several approaches are considered: (1) decreasing intermolecular hydrophobic interactions such as by linking hydrophilic side chains to anthracene rings, (2) increasing intermolecular interaction for self-assembly with the assistance of other non-covalent interactions such as hydrogen bonds, or (3) increasing the conjugated dimension of anthracene macro rings for π-stacking.
Comparing with anthracene, 9-anthracenecarbonitrile (compound 2) has a cyano group linking to anthracene ring to act as the side chain. It was interesting that the self-assembled products of 2 are microwires instead of hexagonal or elongated hexagonal small crystals as that of anthracene (see Fig. 1c), indicating the linking of the cyano group have tuned the molecular interactions and resulted in the change of the molecular stacking of compound 2,35,36i.e., the tuning of the growth competition between the π-stacking direction and other directions. From the crystalline structures of 2 (Fig. 1d), it is obvious that there are strong π interactions along the c axis of 2 molecules, so that the molecular stacking of 2 tends to along this direction. It is probably an important reason why compound 2 assembles into microwires instead of bulk crystals as that of anthracene. Notably, CH2Cl2 is used as the solvent for the assembly of compound 1 and 2 as well as the following other compounds so that the effect of different solvents can be avoided during the self-assembly process.37
Compound 2 demonstrates the linking of a cyano group to anthracene ring is effective to tune the molecular interactions and change the molecular stacking. Encouraging by these, compound 3 was further synthesized with (1) the expansion of the conjugation dimension, (2) the enhancement of hydrophilicity, and (3) the increase of molecular interactions with the link of the conjugated dicyanovinyl group. Theoretical calculation suggests that the molecules of compound 3 can form larger planar conjugated dimers due to the hydrogen bonding interactions between dicyanovinyl groups and anthracene rings (Fig. 2). Here, the geometry optimizations for both single molecule and dimer are done by using Gaussian 03 program at hybrid density functional theory (DFT) B3LYP level with 6-31G basis sets. As shown in Fig. 2, two molecules of compound 3 can form a dimer with the shortest distance around 2.41 Å, and two molecules are at the same plane to generate a larger planar conjugated dimer with the maximal dihedral angle of the two molecules is less than ±5°. The length of the CH⋯NC contacts in the dimer is much shorter than that of the van deer waals distance. Hence, the existence of intermolecular hydrogen bonds, the enlarged conjugated dimension, and short molecular contacts are prospective for the efficient self-assembly of 3.
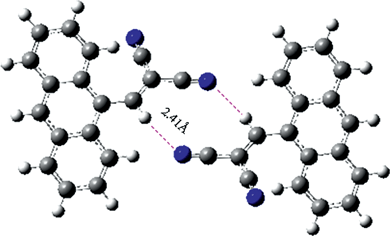 | ||
| Fig. 2 The formation of the dimer between compound 3 by theoretical calculation. | ||
A single crystal of compound 3 with dimensions of 0.5 × 0.2 × 0.2 mm was obtained with a slowly solvent volatilization. X-Ray diffraction (XRD) patterns of the crystal suggest it belongs to P21/c space group with a = 32.461(7) Å, b = 4.1267(8) Å, c = 20.067(4) Å, β = 107.58°, V = 2562.57 Å3, z = 8. It is obvious from Fig. 3 that the molecules of compound 3 indeed easily form larger planar conjugated dimers under intermolecular hydrogen bonding interactions between dicyanovinyl groups and anthracene rings with the shortest distance between two molecules is at 2.6 Å, which agrees well with theoretical calculation (2.41 Å). The large planar dimers result in a co-facial molecular π stacking along b axis, i.e., compound 3 molecules can stack along the b axis with strong intermolecular π–π and hydrogen bonding interactions, while molecules in other directions are linked by weak van der Waals interactions.
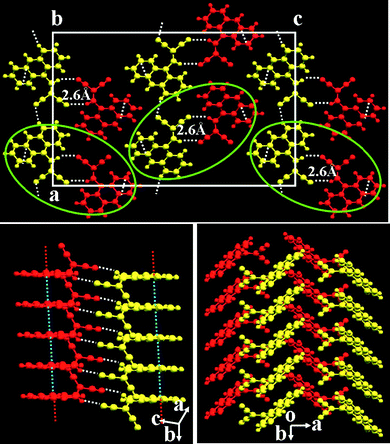 | ||
| Fig. 3 Crystal structures of compound 3. | ||
Indeed, ideal one-dimensional micro- and nanowires can be obtained by cast assembling the CH2Cl2 solution of compound 3 on substrates (Fig. 4). The self-assembly process is surprisingly efficient and rapid, and the cast products are always very clean, beautiful and uniform at constant conditions. The length of the wires could be approached several hundred micrometres to millimetres. And the assembly of the wires could be in large area and exhibits very weak substrate dependence, no matter on glass or silicon or any other arbitrary substrates. These results vividly demonstrate that the linking of the conjugated dicyanovinyl group to anthracene ring is effective to tune the molecular stacking because of the expansion of the conjugation dimension by planar dimers and the increase of molecular interactions by hydrogen bonds.
 | ||
| Fig. 4 Cast products of compound 3 in large area on any substrate. | ||
This conclusion is confirmed by compound 4, wherein methyl groups are used to replace the nitrile groups of compound 3 to eliminate the formation of the conjugated dimers and hydrogen bonds. The assembly process is the same as that of 3, but the typical products shown in Fig. 5a exhibit no micro- or nanowires, indicating the poor 1D self-assembled ability of 4. It is because the methyl groups are not a benefit for the formation of hydrogen bonds and dimers as those of nitrile groups, i.e., the importance of intermolecular hydrogen bonds for self-assembly. If the methyl groups are replaced by using carboxylic acid groups as that of compound 5, to expect the recovery of hydrogen bonds again. The results were shown in Fig. 5b. Although the nanowires are not as good as that of 3, nice nanowires are available in the cast products because carboxylic acid groups are useful for the formation of hydrogen bonds, it indicates the necessary of hydrogen bonds for the cast assembling micro- and nanowires of the anthracene derivatives.
 | ||
| Fig. 5 SEM images of cast products from CH2Cl2 (saturated solution) for compound 4 (a), 5 (b), 6 (c), and 7 (d–f). | ||
It is believed that the main challenge of assembling anthracene derivatives into 1D structures lies in the controlling of the molecular assembly for the growth along the π-stacking direction. Therefore, the conjugated dimension for π-stacking should be also key important for the efficient assembly. In order to further demonstrate this concept, the role of the conjugated dimension of anthracene derivatives during the cast assembly process is further studied by replacing the anthracene ring of compound 3 with phenyl (compound 6). The typical cast products of compound 6 are shown in Fig. 5c. Although microwires were still observed, the products were obvious not as good as those of compound 3 with large-aspect ratio. The results suggested that the decrease of the conjugated dimension would reduce the 1D assembled ability of the molecules. Reversibly, with the enhancement of the π–π interactions by enlarging the conjugated dimension of the molecular macro rings (compound 7), excellent and long wires were assembled efficiently and rapidly by cast process (Fig. 5d–f). Typical lengths of the micro- or nanosized wires of compound 7 ranged from hundreds of microns to several millimetres and diameter at tens of nanometres to several microns. The results of compound 6, 7 prove that the enhancement of molecular π–π interaction by increasing the π conjugated dimension is also efficient for the self-assembly of micro- and nanowires of anthracene derivatives by cast assembly process.
Conclusion
In summary, it is demonstrated by using anthracene and its six derivatives that the tuning of molecular structure is efficient for controlling self-assembly of micro- and nanostructures of the compounds. 1D micro- and nanostructures are efficiently assembled during the cast process with the co-assistance of molecular non-covalent interactions of hydrophilic interactions, π-stacking and hydrogen bonds. The co-assistance of the molecular interactions is the most important and is the main driving force for the rapid cast assembly, which could be tuned by (i) linking hydrophilic side chains on anthracene ring to decrease the intermolecular hydrophobic interactions, (ii) increasing molecular interactions with hydrogen bonds or with expanded molecular conjugation dimensions. The morphology of the these 1D nanostructures can be highly repeated when we use the same cast assemble condition. And these 1D micro- and nanostructures would be promising candidates as semiconductor materials for organic photo/electronic devices.Acknowledgements
The authors acknowledge the financial support from NSFC (20721061, 50725311, 60801037), Ministry of Science and Technology of China (2006CB806200, 2006CB932100, 2009CB930400) and Chinese Academy of SciencesNotes and references
- V. Palermo and P. Samori, Angew. Chem., Int. Ed., 2007, 46, 4428–4432 CrossRef CAS.
- R. van Hameren, P. Schon, A. M. Van Buul, J. Hoogboom, S. V. Lazarenko, J. W. Gerritsen, H. Engelkamp, P. C. M. Christianen, H. A. Heus, J. C. Maan, T. Rasing, S. Speller, A. E. Rowan, J. A. A. W. Elemans and R. J. M. Nolte, Science, 2006, 314, 1433–1436 CrossRef CAS.
- R. M. D. Hansen, J. Kjelstrup-Hansen and H. G. Rubahn, Nanoscale, 2010, 2, 134 RSC.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS.
- G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049–1052 CrossRef.
- B. Olenyuk, J. A. Whiteford, A. Fechtenkötter and P. J. Stang, Nature, 1999, 398, 796–799 CrossRef CAS.
- J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1444 CrossRef CAS.
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- N. B. Bowden, M. Weck, I. S. Choi and G. M. Whitesides, Acc. Chem. Res., 2001, 34, 231–238 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- Y. M. Sun, Y. Q. Liu and D. B. Zhu, J. Mater. Chem., 2005, 15, 53–65 RSC.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS.
- D. Braga and G. Horowitz, Adv. Mater., 2009, 21, 1473–1486 CrossRef CAS.
- M. Mas-Torrent and C. Rovira, Chem. Soc. Rev., 2008, 37, 827–838 RSC.
- L. Jiang, W. P. Hu, Z. M. Wei, W. Xu and H. Meng, Adv. Mater., 2009, 21, 3649–3653 CrossRef CAS.
- L. Jiang, H. L. Dong and W. P. Hu, J. Mater. Chem., 2010, 20, 4994–5007 RSC.
- L. Jiang, J. Gao, E. Wang, H. Li, Z. Wang, W. Hu and L. Jiang, Adv. Mater., 2008, 20, 2735–2740 CrossRef CAS.
- J. Y. Lee, S. Roth and Y. W. Park, Appl. Phys. Lett., 2006, 88, 252106 CrossRef.
- Y. Xia, V. Kalihari, C. D. Frisbie, N. K. Oh and J. A. Rogers, Appl. Phys. Lett., 2007, 90, 162106 CrossRef.
- C. Reese, W. J. Chung, M. M. Ling, M. Roberts and Z. N. Bao, Appl. Phys. Lett., 2006, 89, 202108 CrossRef.
- D. Jurchescu, J. Baas and T. T. M. Palstra, Appl. Phys. Lett., 2004, 84, 3061 CrossRef CAS.
- V. Podzorov, S. E. Sysoev, E. Loginova, V. M. Pudalov and M. E. Gershenson, Appl. Phys. Lett., 2003, 83, 3504 CrossRef CAS.
- V. C. Sundar, J. Zaumseil, V. Podzorov, E. Menard, R. L. Willett, T. Someya, M. E. Gershenson and J. A. Rogers, Science, 2004, 303, 1644–1646 CrossRef CAS.
- K. Ito, T. Suzuki, Y. Sakamoto, D. Kubota, Y. Inoue, F. Sato and S. Tokito, Angew. Chem., Int. Ed., 2003, 42, 1159–1162 CrossRef CAS.
- H. Meng, F. P. Sun, M. B. Goldfinger, G. D. Jaycox, Z. G. Li, W. J. Marshall and G. S. Blackman, J. Am. Chem. Soc., 2005, 127, 2406–2407 CrossRef CAS.
- H. Meng, F. P. Sun, M. B. Goldfinger, F. Gao, D. J. Londono, W. J. Marshal, G. S. Blackman, K. D. Dobbs and D. E. Keys, J. Am. Chem. Soc., 2006, 128, 9304–9305 CrossRef CAS.
- A. A. Frimer, J. Weiss, H. E. Gottlieb and J. L. Wolk, J. Org. Chem., 1994, 59, 780–792 CrossRef CAS.
- H. D. Becker and K. Andersson, J. Org. Chem., 1983, 48, 4542–4549 CrossRef CAS.
- F. F. Louis and L. H. Jonathan, J. Am. Chem. Soc., 1938, 60, 2555–2559 CrossRef.
- H. K. Oh, J. H. Yang, H. W. Lee and I. Lee, J. Org. Chem., 2000, 65, 2188–2191 CrossRef CAS.
- T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619–621 CAS.
- S. Ito, M. Wehmeier, J. D. Brand, C. Kuebel, R. Edsch, J. Rabe and K. Muellen, Chem.–Eur. J., 2000, 6, 4327–4342 CrossRef CAS.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |