Addressing the magnetic properties of sub-monolayers of single-molecule magnets by X-ray magnetic circular dichroism
Fabrizio
Moro
*ab,
Valdis
Corradini
b,
Marco
Evangelisti
c,
Roberto
Biagi
b,
Valentina
De Renzi
b,
Umberto
del Pennino
b,
Julio C.
Cezar
d,
Ross
Inglis
e,
Constantinos J.
Milios
ef and
Euan K.
Brechin
e
aSchool of Chemistry, University of Nottingham, University Park, NG1 2RD, UK. E-mail: fabrizio.moro@nottingham.ac.uk
bCNR-Institute of Nanoscience S3 and Dipartimento di Fisica, Università di Modena e Reggio Emilia, via G. Campi 213/A, 41100, Modena, Italy
cInstituto de Ciencia de Materiales de Aragón, CSIC—Universidad de Zaragoza, Departamento de Física de la Materia Condensada, 50009, Zaragoza, Spain
dEuropean Synchrotron Radiation Facility, BP 220, F-38043, Grenoble Cedex, France
eSchool of Chemistry, University of Edinburgh, EH9 3JJ, Edinburgh, UK
fDepartment of Chemistry, University of Crete, 710 03, Voutes, Herakleion, Greece
First published on 12th October 2010
Abstract
We report on a comparative study of electronic and magnetic properties of Mn6 single-molecule magnets (SMMs) grafted on gold surface. Two derivatives with spin-ground states S = 4 and S = 12 have been functionalized with 3-tp-CO2− (3-thiophene carboxylate, tpc) ligands and characterized as thick films (TFs) as well as sub-monolayers (sMLs) by synchrotron based techniques. X-ray absorption spectroscopy at the Mn L2,3 edges shows the modification of the spectral lineshape in the sMLs with respect to the TFs suggesting that the local symmetry at the Mn sites changes once the molecules are deposited on gold surface. In spite of this, the expected MnIII oxidation state is preserved. X-ray magnetic circular dichroism (XMCD) spectra show that the total magnetic moment is only given by spin part because of the quenched orbital moment. Moreover, variable temperature and variable field XMCD spectra reveal an effective decrease of the Mn spin moment for both derivatives.
Introduction
Among the several objects which can be realised in nanomagnetism, single-molecule magnets (SMMs) have recently attracted much interest because of their potential application as molecular devices.1–8 They combine a large spin-ground state with a large and negative magnetic anisotropy giving rise to an anisotropy energy barrier leading to slow relaxation of the magnetization at low temperatures.9 In principle these properties might be used to store information in an individual molecular unit, although it has been hindered so far by two main issues. Firstly, the superparamagnetic blocking temperature is still too low because, even in the best examples (e.g. Mn12, ref. 10), it reaches only 2–3 K.9,11,12 This issue might be overcome by larger values of the spin-ground state and magnetic anisotropy, the latter being extremely difficult to achieve by controlled synthesis. Secondly, the retention of magnetic properties at the molecular level following the deposition of spatially isolated SMMs on substrates is far from being achieved.13 The interaction with a surface may modify the intrinsic properties of molecules, leading to the reduction of the magnetic core and the removal of the hysteresis loop—as has already been observed for Mn12ac on gold.14–16Recently the synthesis of SMMs with the general formula [MnIII6O2(R-sao)6(O2CR′)2L4–6], in short Mn6 (where R = H, Me or Et; R′ = H, Me, Ph; L = EtOH, MeOH, H2O; saoH2 = salicylaldoxime), has been reported. Milios et al.17–19 proved that the use of derivatised oxime ligands and bulky carboxylates deliberately increases the value of the spin-ground state of Mn6-based systems from spin S = 4 to S = 12 in a stepwise fashion, and in so doing enhances the energy barrier for magnetization reversal (Ueff) establishing a new record of 86.4 K. The representative core of the [MnIII6O2(sao)6(O2CR)2(ROH)4] derivative20,21 contains a non-planar [MnIII6(µ3-O2−)2(µ2-OR)2]12+ unit of two off-set, stacked [MnIII3(µ3-O2−)]7+ triangular subunits linked by two central oximato O-atoms (Fig. 1a). This class of compounds shows a ferromagnetic (FM) exchange interaction between the two antiferromagnetically (AF) coupled [MnIII3] triangles leading to an S = 4 spin-ground state.22 A number of important structural changes leading to different values of S and Ueff take place in the related complexes [MnIII6O2(R-sao)6(O2CR′)2(EtOH)4(H2O)2] with R = Et, Me, and Ph,23 where the increased steric bulk and non-planarity of the R-sao2− ligands cause a shortening (by ∼1 Å) of the phenolato oxygen-square pyramidal Mn distance and a severe twisting of the Mn–N–O–Mn moieties within each MnIII3 subunit (Fig. 1b). The structural changes force a switch in the dominant magnetic exchange interactions from AF to FM, thus stabilizing an S = 12 spin-ground state.19,23 However, all these derivatives are characterized by weak exchange interaction (J < 1 cm−1), leading to the population of low-lying excited states (S = 11, 10,…) and to increased numbers of possible pathways for the reversal of the magnetization via quantum tunnelling resulting in a reduction of the energy barrier. This problem has been successfully solved by further replacement of the carboxylates via simple metathesis type reactions that produce identical cores except for the Mn–N–O–Mn torsion angles (αv) which are modified according to the steric bulk of the (oxime) ligand. The complexes with the largest torsion angles show an increase in the magnitude of the pairwise exchange parameter (J) and an increase in the effective Ueff up to 86.4 K,19,23i.e. notably higher than that of Mn12ac (61 K).
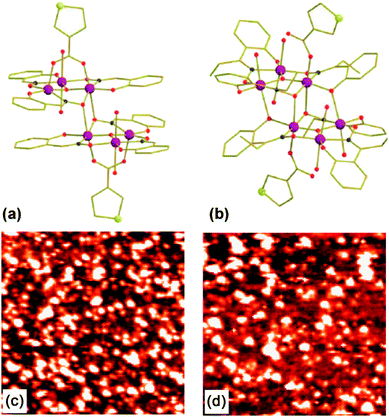 | ||
| Fig. 1 Mn6 structures (with 3-tp-CO2− functionalizations) of the derivatives 1 with S = 4 (a) and 2 with S = 12 (b). The large and medium circles depict the MnIII ions and the S atoms respectively. While dark and clear small circles indicate N and O atoms. The lines link the C atoms. H atoms are omitted for clarity. STM images (65 × 65 nm2) in constant-current mode of the Au(111) surface immersed in a solution of 1 (c) and 2 (d). | ||
In previous work,24 we succeeded in self-assembling two Mn6 derivatives [MnIII6O2(sao)6(O2C-tp-3)2(EtOH)4] (1) and [MnIII6O2(Et-sao)6(O2C-tpc-3)2(EtOH)4(H2O)2] (2) with spin-ground states S = 4 and S = 12 respectively on Au(111) surface. For both compounds, the functionalization is made by two 3-tp-CO2− units pointing in the opposite direction (to each other) and being perpendicular to the [MnIII3(µ3-O2−)]7+ triangular plane, with the sulfur atoms in outermost position on the thiophene ring (Fig. 1a and 1b). This favours the direct grafting on gold by covalent bond with the molecular axis which should be preferentially normal to the surface. The investigation, carried out by means of scanning tunnelling microscopy (STM), shown in Fig. 1c and 1d, and X-ray photoemission spectroscopy (XPS), suggests that Mn6 molecules preserve their integrity when deposited by liquid phase. The electronic structure of Mn6 on gold surface was also studied by means of resonant photoemission spectroscopy (ResPES) at the Mn L3 edge aimed to single out the Mn 3d derived states in the valence band.25 Moreover, magnetic measurements on powder pellets clearly showed that the 3-tp-CO2− functionalization does not significantly alter the magnetic properties of either compounds.24
The magnetic characterization of grafted SMMs is the key point which needs to be addressed. Unfortunately standard magnetometry techniques do not have enough sensitivity for detecting the magnetism of sub-monolayer quantities of grafted clusters. In terms of high resolution and element selectivity, X-ray absorption spectroscopy (XAS) and magnetic circular dichroism (XMCD) are excellent tools. The analysis of isotropic XAS spectra at the L2,3 edges of transition metals provides unambiguous information about the oxidation state and local symmetries of the selected chemical element. On the other hand, XMCD directly probes the local spin and orbital magnetic moments as well as the sign of the exchange interactions. In particular cases, the application of the XMCD sum rules allows the quantification of the orbital and magnetic moments in an element-specific way. Herein we address the structural, electronic and magnetic properties of the sub-monolayers (sMLs) and thick films (TFs) of 1 and 2, hereafter referred to as sML1, sML2, TF1 and TF2, by means of XAS and XMCD measurements in order to elucidate how the molecular properties are affected after the deposition on surface.
Experimental
The TFs of the Mn6tpc derivatives 1 and 2, and those of the pristine non-functionalized Mn6benz compounds, [Mn6O2(O2CPh)2(sao)6(EtOH)4] (TF3) and [Mn6O2(O2CPh)2(Et-sao)6(EtOH)6] (TF4), were obtained by a drop-casting procedure on a highly ordered pyrolytic graphite (HOPG) substrate. The sML samples were prepared by immersion of flame annealed gold on mica surface in a solution of the derivatives 1 and 2 in CH2Cl2 (1 mM) for 10 min. After the adsorption process the samples were rinsed with solvent in order to remove the excess molecular layers and quickly introduced into the sample chamber.26,27 The solvent used, deposition time and solution concentration were optimized in order to obtain a homogeneous sML distribution of isolated molecules. STM was used to check that the desired two-dimensional distribution of nanometric entities was actually achieved (Fig. 1c and 1d).24XAS measurements at the Mn L2,3 edges were carried out at the ID08 Dragon beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) in Total Electron Yield (TEY) detection mode and normalized to the incident photon flux. The photon source was an Apple II undulator delivering a high flux (1013 photons per s) of ∼100% of circularly polarized light.
The sML samples were fixed to a molybdenum sample holder by means of tantalum wires to ensure a proper thermal and electrical contact, and transferred into a liquid-helium cryomagnet kept under UHV (10−10 mbar). The direction of both impinging beam and magnetic field generated by superconducting coils is perpendicular to the sample surface (z axis). The XMCD spectrum is the difference between XAS spectra taken with the helicity of the incident photon (P) antiparallel (σ↑↓) and parallel (σ↑↑) to the field direction and normalized to the isotropic edge height. In order to avoid systematic errors, σ↑↓ (σ↑↑) absorption spectrum was obtained by averaging a set of spectra recorded with the field orientated parallel and anti-parallel to the photons direction.28 The beam flux was attenuated by more than one order of magnitude in order to avoid sample damaging induced by radiation exposure. In these conditions the spectra never showed any sign of degradation during the experiment.
Results and discussion
XAS spectra taken with right (σ↑↑) and left (σ↑↓) circularly polarized light and the relative XMCD signal (σ↑↓–σ↑↑) at the Mn L2,3 edges for TF1 and TF2 are shown in Fig. 2 and compared with the analogous Mn6benz derivatives TF3 and TF4 with spins 4 and 12, respectively (see Experimental section). The 2p core hole spin–orbit interaction splits the spectrum into two parts, corresponding to L3 (2p3/2) and L2 (2p1/2) edges centred around 641 and 652 eV respectively, with a separation of about 11 eV. The L2 edge features are more broadened than the L3 edge, owing to the increased probability of the Coster–Kronig Auger decay channel. Hereafter we focus only on L3 because of its fine structure. The L3 edge is further split into the t2g and eg sub-bands by the octahedral ligand field. The fine structure of the absorption edges is due to a combination of the electron–electron interaction within the Mn ions, the crystal field felt by the Mn ions and the hybridization of Mn3d, O2p and N2p orbitals. The strong similarity between the XAS and XMCD spectra of TF1, TF2, TF3 and TF4 samples clearly suggests that the tpc functionalization does not significantly affect the electronic and magnetic properties at the Mn sites in agreement with the magnetic measurements previously reported.24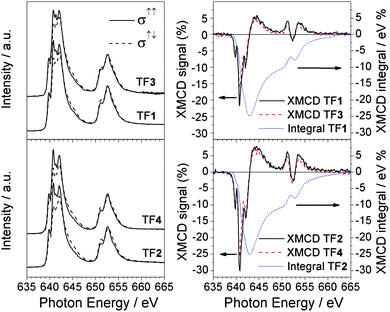 | ||
| Fig. 2 Comparison between Mn L2,3 XAS (left panel) and XMCD (right panel) spectra of TF1, TF2, TF3 and TF4 samples. The XMCD integrals (dotted curve) relative to the tpc functionalized Mn6 compounds (TF1 and TF2) are also reported. All the spectra were taken at 10 K with an applied magnetic field of 5 T. | ||
In Fig. 3 the isotropic Mn L2,3 absorption spectra of TF1, TF2, sML1 and sML2 are compared with a series of reference Mn oxide compounds. The absorption spectra of the TF1 and TF2 have almost identical lineshape suggesting that the Mn ions present the same local symmetries and comparable crystal field intensities in the two complexes. Indeed the overall site geometry extracted from crystallographic data of the Mn ions in 1 and 2 is the same.24 The local symmetry of each Mn ion (Fig. 1) is represented by a distorted octahedron with six apical atoms, five O-atoms and one N-atom. The main difference is that all the Mn ions are six-coordinate in 2, while four Mn ions are six-coordinate and the other two are five-coordinate in 1 (one apical oxygen is missing). Despite the obvious inequality of N and O, the very similar bond lengths and angles around the magnetic ion lead to an almost perfect equivalence of the four in-plane atoms. On the other hand, their average apical-in-plane distance ratio is 1.17 for 1 and 1.19 for 2 with a percentage difference of only 1.4%. In general, such small variations of the local symmetry around the selected chemical element cannot be observed in XAS spectra. For instance, in the case of other Mn-based complexes reported by Noh et al.,29 no significant variation of the spectral lineshape was found for changes below 6% in the apical-in-plane ratio.
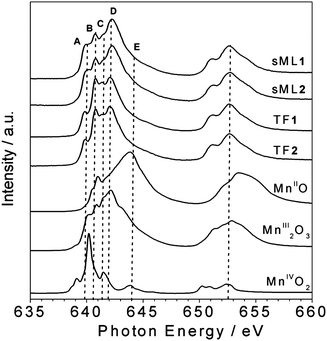 | ||
| Fig. 3 Comparison between the isotropic Mn L2,3 XAS spectra of sMLs and TFs of 1 and 2 and those of reference compounds. The main features A (640 eV), B (640.8 eV), C (641.5 eV), D (642.2 eV) and E (644.2 eV) are indicated. | ||
Although the XAS spectra of TF1 and TF2 are almost identical, interestingly those of sML1 and sML2 (Fig. 3) show sensible discrepancy at the L3 edge with respect to the corresponding TFs, the L2 edge being quite similar. Notably, these modifications are identical for both sML1 and sML2 suggesting that the same kind of perturbation affects the two differently functionalized compounds when the Mn6 molecules are deposited on gold surface. We will show in the following discussion, based on comparison of XAS spectra with those of reference manganese oxide compounds, that these modifications can be ascribed to a local structural modification of the grafted clusters, when they are deposited on the substrate.
The absorption spectra of MnO (MnII), Mn2O3 (MnIII) and MnO2 (MnIV), presented in Fig. 3 as references, were measured in similar experimental conditions as TF1 and TF2. Particular attention was paid to the energy alignment of all the spectra. The main spectral features are labelled as A (640 eV), B (640.8 eV), C (641.5 eV), D (642.2 eV) and E (644.2 eV) to facilitate the comparison. For both TF and sML the MnIII components (B, C and D) are dominant while the MnIV (E) is negligible. By comparing the lineshapes of MnO (MnII) and Mn2O3 (MnIII) spectra with those of TF1 and TF2, we can safely conclude that the fraction of reduced MnII ions must be extremely small.
The superimposition of normalized XAS and XMCD spectra taken for TF1 and sML1 reported in Fig. 4 allows a more accurate analysis. Three features, B, C and D, are dominant in TF1, while only D is dominant in sML1. In addition an apparent transfer of spectral weights from B and C to A in sML1 seems to occur with respect to TF1. At first glance this can be explained in terms of a structural modification of the Mn sites. The comparison between XMCD spectra also shows features at the same energy of A, B, C and D. The decrease of feature B gives evidence for a reduction of the magnetic moment associated with the MnIII ions, as will be discussed later. The same discussion follows for XAS and XMCD spectra of TF2 and sML2 being almost identical to those of TF1 and sML1 respectively.
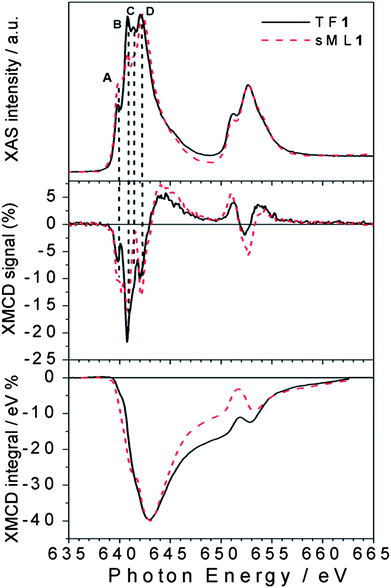 | ||
| Fig. 4 Direct comparison between normalized XAS and XMCD spectra of TF1 and sML1. The different A (640 eV), B (640.8 eV), C (641.5 eV) and D (642.2 eV) features observed for the TF and the sML suggest deviations of the local symmetries (see the text). In the bottom panel the corresponding XMCD integrals are also shown. | ||
In general, the interpretation of absorption and dichroic spectra is not trivial because the lineshape changes drastically with the strength of the crystal field and spin–orbit interaction. Notwithstanding, the origin of the spectral differences between TFs and sMLs can be explained by looking at well-known MnIII based reference systems. In Fig. 5 the isotropic Mn L2,3 absorption spectra of TF1 and TF2 are compared with those of Piedmontite30 and ZnMn2O4,29 whereas the spectra of sML1 and sML2 are compared with those of MnIII2O3 and ZnGa1Mn1O4.29 The similarity between Piedmontite, ZnMn2O4, and TF1 and TF2 absorption spectra let us suppose that the Mn ions posses large tetragonal distortions (i.e. Jahn–Teller distortion) as well known for polycrystalline spinel manganite oxides. This is also confirmed by the strong distortion of ZnMn2O4 (apical-in-plane ratio of 1.18) comparable to those of TF1 and TF2 (apical-in-plane ratio of 1.17–1.19 respectively). Conversely, the XAS spectra of ZnGa1Mn1O4 and in particular of Mn2O3 (MnIII) with almost perfect octahedral symmetry (apical-in-plane ratio of 1.06) are more similar to sML1 and sML2. This suggests that the absorption of the 3-tp-CO2− functionalized Mn6 clusters on gold surface induces a structural modification of the Mn sites reducing the elongation of the O–Mn–O axes, with respect to the bulk conditions, leading to an almost octahedral symmetry. Anticipating the variable temperature, variable field (VTVH) XMCD results, these changes imply modification of the distances and bonding angles between the Mn–Mn ions leading to modified exchange couplings. These modifications could be ascribed to the interaction with gold and/or to the lack of isotropic interactions with all the surrounding molecules. Notably, this mechanism occurs without affecting the oxidation state of the Mn ions because the absorption spectra of sML1 and sML2, and Mn2O3 are almost identical, as evidenced by their comparison presented in Fig. 6 (upper panel).
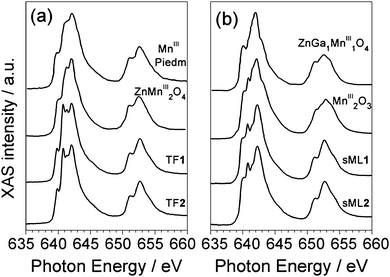 | ||
| Fig. 5 TF1 and TF2 XAS spectra compared with those of Piedmontite and ZnMn2O4 (a), and sML1 and sML2 spectra compared with those of MnIII2O3 and ZnGa1Mn1O4 (b).29 | ||
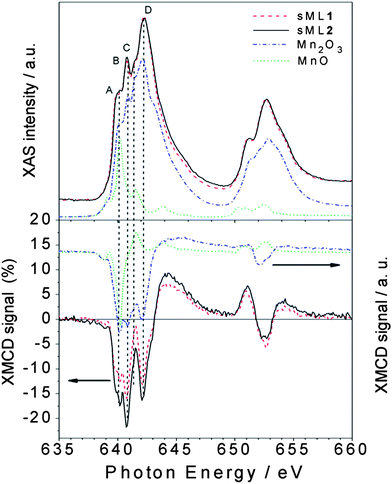 | ||
| Fig. 6 Normalized XAS (top panel) and percentage XMCD (bottom panel) spectra of sML1 and sML2 compared with those of MnIII2O3 (dash dotted curve) and MnIIO (dotted curve). The different A (640 eV), B (640.8 eV), C (641.5 eV) and D (642.2 eV) features observed in the sMLs and in the Mn2O3 are indicated (see the text). | ||
If the modifications in the XAS spectrum lead to different structural and electronic properties, those in the XMCD reflect the deviations of the magnetic properties. Comparison of the XMCD spectra (Fig. 6, bottom panel) highlights that sML1 and sML2 are very similar to Mn2O3 and once again rather different from MnO—in magnetic behaviour also. Since no significant MnII component is visible in sML1 and sML2 we can conclude that a large fraction of Mn6 molecules have been deposited on Au surface preserving the MnIII oxidation state; unlike that observed for Mn12ac where a systematic reduction of the magnetic core occurs after the adsorption.15,16,31,32 The sharp feature B of the reference compound Mn2O3 is observed in TF1 and TF2 as well as in sML1 and sML2 but only with different intensities between TFs and sMLs.
The application of the XMCD spin moment sum rule for MnIII is unfeasible because (a) the L2,3 edges overlap due to strong spin–orbit coupling and (b) of the complications arising from the determination of the magnetic dipole operator (Tz).33–36 Conversely, the orbital moment sum rule still holds and by definition it is proportional to the integral of the XMCD signal through the L2,3 edges.34 The XMCD integral curves reported in Fig. 4 vanish for TF1, sML1 as well as TF2(Fig. 2) and sML2 (not reported) giving evidence for quenched orbital moment.33 Hence, the total magnetic moment possesses only spin contribution. Since the orbital moment is also quenched for the Mn6benz compounds, we conclude that neither the tpc functionalization nor the deposition on gold surface affects the degree of quenching of the orbital momentum at the Mn sites. VTVH XMCD signals (%) at the L3 edge for TF1, TF2, sML1 and sML2 are plotted in Fig. 7. The uncertainty is estimated to be 20% of the corresponding maximum values. The following observations can be made: (i) as expected the dichroic signal of TF2 is more intense with respect to TF1 in agreement with the nominal values of their spin; (ii) the dichroic signal for the TFs and sMLs decreases with increasing temperature from 10 to 25 K due to the increased thermal fluctuation of the spin magnetic moments; (iii) upon increasing temperature the dichroic signal in the sMLs decreases with respect to the TFs, from 30% at 10 K to 50% at 25 K. Because the error bars of the TF and sML isothermal curves do not overlap, the MnIII spin magnetic moment shows an effective decrease. In accordance with the results of XAS measurements (Fig. 4) this can be explained in terms of structural changes of the Mn local environment when they are grafted on gold surface. In fact such changes may lead to different relative distances and bonding angles between Mn ions and to the modification of the Mn–Mn exchange couplings with respect to the corresponding TFs. The origin of such structural changes can be ascribed to the molecule–gold surface interactions and/or to the lack of isotropic interactions with the surrounding molecules. In fact, in the TFs the molecules are packed and, unless in the very first layer, the molecules probed by XMCD in TEY mode are surrounded by others. Hence isotropic molecule–molecule interactions are expected, while interactions with the surface do not play any role. This is because the TFs are deposited on the non-reactive HOPG surface, and because of the small sampling depth of the TEY mode (2–3 molecular layers) with respect to the TF thickness. In the sMLs the molecules are surrounded by the surface on the bottom, free space on the top and molecules in the plane (Fig. 1). Local and molecular symmetries are driven by anisotropic molecule–molecule and molecule–surface interactions. Other effects such as preferential orientation of the molecular easy axis of the magnetisation in the plane of the surface, and high-spin to low-spin transitions driven by a competition between the interatomic exchange couplings and the crystal field strength must be considered. If the molecular easy axis lies preferentially in the plane of the surface, then the variable field XMCD signal is expected to be lower than the relative TFs because the magnetic field is applied perpendicular to the surface and along the molecular hard axis. This can be proven only with further studies based on grazing incident XMCD experiments, although STM studies favour a preferential molecular axis perpendicular to the surface.24 The transition of MnIII ions from high-spin to low-spin can be also disregarded because the XAS lineshapes for TF1, TF2, sML1 and sML2 have a well-defined multiplet structure, which is a clear fingerprint of a localized 3d orbital with a high-spin state. Conversely, itinerant systems with a low-spin state are characterized by a smooth XAS spectral lineshape.37,38
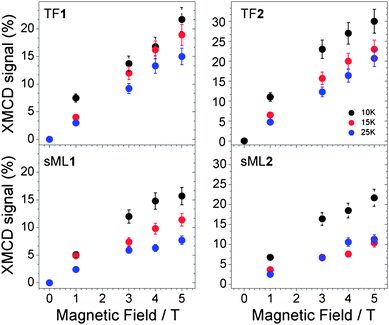 | ||
| Fig. 7 Variable temperature and variable field XMCD signal (%) measured for the samples TF1, TF2, sML1 and sML2. | ||
Conclusions
The electronic and magnetic properties of two functionalised derivatives 1 and 2 belonging to a Mn6 SMM family have been investigated by XAS and XMCD techniques. The surface effects of a gold surface on deposited Mn6 derivatives have been studied by comparing the variable temperature and variable field XMCD spectra of the Mn6 sub-monolayers with those of relative thick films. The results show the average MnIII spin magnetic moment of the sub-monolayers samples to be decreased with respect to the relative thick films in both derivatives. The main effect responsible for this result has been ascribed to local distortions of Mn environments followed by modified Mn–Mn exchange couplings when Mn6 clusters are deposited on gold surface. We argue that the origin of these local distortions might be due to the molecule–gold surface covalent bond interactions and/or to the lack of isotropic interactions with all surrounding molecules. In addition we have proved that (i) the tpc functionalization of 1 and 2 does not affect the main electronic and magnetic properties of the Mn6 magnetic core, and (ii) the MnIII oxidation state is preserved even after the deposition of Mn61 and 2 on gold surface. In perspective, we suggest that Mn6 magnetic core is reasonably robust to make this class of SMMs suitable for the synthesis of new derivatives with different sulfur-based functionalization which may drive the deposition of ordered arrays of Mn6 SMMs on gold surface. In addition, the electronic stability of the Mn6 core encourages the use of MnIII ions having a large magnetic anisotropy as building blocks for SMMs with enhanced anisotropy energy barriers in view of their potential application as molecular unit of data storage devices.Acknowledgements
We are grateful to Ivan Marri for stimulating and helpful discussions. This work has been supported by FP7-ICT FET Open “MolSpinQIP” project, contract no. 211284. Marco Evangelisti acknowledges funding from the Spanish Ministry for Science and Innovation through grants MAT2009-13977-C03 and CSD2007-00010. Euan K. Brechin thanks The Leverhulme Trust for funding. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities.References
- G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66–71 CAS.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS.
- N. Ishikawa, M. Sugita, N. Tanaka, T. Ishikawa, S. Y. Koshihara and Y. Kaizu, Inorg. Chem., 2004, 43, 5498–5500 CrossRef CAS.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., Int. Ed., 2005, 44, 2931–2935 CrossRef CAS.
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541–548 CrossRef CAS.
- O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS.
- F. Meier, J. Levy and D. Loss, Phys. Rev. Lett., 2003, 90, 47901 CrossRef.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- T. Lis, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2042–2046 CrossRef.
- J. R. Friedman, M. P. Sarachik, J. Tejada and R. Ziolo, Phys. Rev. Lett., 1996, 76, 3830–3833 CrossRef CAS.
- L. Thomas, F. Lionti, R. Ballou, D. Gatteschi, R. Sessoli and B. Barbara, Nature, 1996, 383, 145–147 CrossRef CAS.
- M. Cavallini, J. Gomez-Segura, D. Ruiz-Molina, M. Massi, C. Albonetti, C. Rovira, J. Veciana and F. Biscarini, Angew. Chem., Int. Ed., 2005, 44, 888–892 CrossRef CAS.
- L. Bogani, L. Cavigli, M. Gurioli, R. L. Novak, M. Mannini, A. Caneschi, F. Pineider, R. Sessoli, M. Clemente-Leon, E. Coronado, A. Cornia and D. Gatteschi, Adv. Mater., 2007, 19, 3906–3911 CrossRef CAS.
- M. Mannini, P. Sainctavit, R. Sessoli, C. C. D. Moulin, F. Pineider, M. A. Arrio, A. Cornia and D. Gatteschi, Chem.–Eur. J., 2008, 14, 7530–7535 CrossRef CAS.
- S. Voss, M. Fonin, U. Rudiger, M. Burgert, U. Groth and Y. S. Dedkov, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 45102 CrossRef.
- C. J. Milios, J. Constantinos, A. Vinslava, P. A. Wood, S. Parsons, W. Wernsdorfer, G. Christou, S. P. Perlepes and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 8–9 CrossRef CAS.
- C. J. Milios, R. Inglis, A. Vinslava, R. Bagai, W. Wernsdorfer, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 12505–12511 CrossRef CAS.
- C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 2754–2755 CrossRef CAS.
- C. J. Milios, A. Vinslava, A. G. Whittaker, S. Parsons, W. Wernsdorfer, G. Christou, S. P. Perlepes and E. K. Brechin, Inorg. Chem., 2006, 45, 5272–5274 CrossRef CAS.
- C. J. Milios, M. Manoli, G. Rajaraman, A. Mishra, L. E. Budd, F. White, S. Parsons, W. Wernsdorfer, G. Christou and E. K. Brechin, Inorg. Chem., 2006, 45, 6782–6793 CrossRef.
- C. J. Milios, C. P. Raptopoulou, A. Terzis, R. Vicente, A. Escuer and S. P. Perlepes, Inorg. Chem. Commun., 2003, 6, 1056–1060 CrossRef CAS.
- C. J. Milios, A. Vinslava, W. Wernsdorfer, A. Prescimone, P. A. Wood, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 6547–6561 CrossRef CAS.
- F. Moro, V. Corradini, M. Evangelisti, V. D. Renzi, R. Biagi, U. del Pennino, C. J. Milios, L. F. Jones and E. K. Brechin, J. Phys. Chem. B, 2008, 112, 9729–9735 CrossRef CAS.
- U. del Pennino, V. Corradini, R. Biagi, V. De Renzi, F. Moro, D. W. Boukhvalov, G. Panaccione, M. Hochstrasser, C. Carbone, C. J. Milios and E. K. Brechin, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 85419 CrossRef.
- V. Corradini, F. Moro, R. Biagi, U. del Pennino, V. De Renzi, S. Carretta, P. Santini, M. Affronte, J. C. Cezar, G. Timco and R. E. P. Winpenny, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 14402 CrossRef.
- A. Ghirri, G. Lorusso, F. Moro, F. Troiani, V. Corradini, C. Muryn, F. Tuna, G. Timco, R. E. P. Winpenny and M. Affronte, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 224430 CrossRef.
- J. Stohr, J. Electron Spectrosc. Relat. Phenom., 1995, 75, 253–272 CrossRef.
- H. J. Noh, S. Yeo, J. S. Kang, C. L. Zhang, S. W. Cheong, S. J. Oh and P. D. Johnson, Appl. Phys. Lett., 2006, 88, 485208.
- S. P. Cramer, F. M. F. Degroot, Y. Ma, C. T. Chen, F. Sette, C. A. Kipke, D. M. Eichhorn, M. K. Chan, W. H. Armstrong, E. Libby, G. Christou, S. Brooker, V. McKee, O. C. Mullins and J. C. Fuggle, J. Am. Chem. Soc., 1991, 113, 7937–7940 CrossRef CAS.
- N. Grumbach, A. Barla, L. Joly, B. Donnio, G. Rogez, E. Terazzi, J. P. Kappler and J. L. Gallani, Eur. Phys. J. B, 2010, 73, 103–108 CrossRef CAS.
- A. Naitabdi, J. P. Bucher, P. Gerbier, P. Rabu and M. Drillon, Adv. Mater., 2005, 17, 1612–1616 CrossRef CAS.
- P. Ghigna, A. Campana, A. Lascialfari, A. Caneschi, D. Gatteschi, A. Tagliaferri and F. Borgatti, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 132413 CrossRef.
- P. Carra, B. T. Thole, M. Altarelli and X. D. Wang, Phys. Rev. Lett., 1993, 70, 694–697 CrossRef CAS.
- J. P. Crocombette, B. T. Thole and F. Jollet, J. Phys.: Condens. Matter, 1996, 8, 4095–4105 CrossRef CAS.
- E. Goering, Philos. Mag., 2005, 85, 2895–2911 CrossRef CAS.
- H. A. Durr, G. van der Laan, D. Spanke, F. U. Hillebrecht and N. B. Brookes, Phys. Rev. B: Condens. Matter, 1997, 56, 8156–8162 CrossRef CAS.
- G. van der Laan, B. T. Thole, G. A. Sawatzky and M. Verdaguer, Phys. Rev. B: Condens. Matter, 1988, 37, 6587–6589 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |