Programming nanostructures of polymer brushes by dip-pen nanodisplacement lithography (DNL)†
Xuqing
Liu
,
Yi
Li
and
Zijian
Zheng
*
Nanotechnology Center, Institute of Textiles and Clothing, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China. E-mail: tczzheng@inet.polyu.edu.hk; Fax: +852-27731432; Tel: +852-27666441
First published on 19th October 2010
Abstract
We report a facile and versatile scanning probe based approach—dip-pen nanodisplacement lithography (DNL)—for manipulating nanostructures of polymer brushes. Nanostructured polymer brushes with sizes as small as 25 nm are made by DNL patterning of the initiator molecules and subsequent surface-initiated polymerization. Nanoconfinement effects including chain collapsing and spreading are observed in the nanopatterned polymer brushes. In addition to chemical structure, size, topography and shape, our approach can also readily program the grafting density, chain configuration, hierarchical structure and multiplexing of the polymer brushes, which allows for the realization of complex chemical surfaces.
Introduction
Polymer brushes, polymers with one end tethered on a surface, are of great interest in the past decade because of their abundant diversity in chemical and mechanical properties.1–7 The patterning of polymer brushes with well-defined molecular architecture, chemical functionality, lateral size and topographical structure are critical to the realization of functional surfaces, which can find a wide variety of applications in cell biology, tissue engineering, medical science, actuation, optical and electronic devices.8–17 Patterning ultrafine (size less than 100 nm) polymer brushes also opens up the opportunity towards the fundamental understanding of polymer chain dynamics and transitions.18In general, patterned polymer brushes are mostly fabricated by surface-initiated polymerization from patterned initiator molecules. Recently, several lithography techniques have been developed for making nanopatterns of functional polymer brushes, including electron-beam lithography,18–20 nanoimprint lithography,21–24 microcontact printing,25–27 and various scanning probe lithography (SPL) methods.28–32 Among the many advances, SPL methods are particularly attractive due to the combinatorial attributes of chemical flexibility, low-cost, high resolution and high registration; and they offer highly scalable throughput with the use of tip arrays.33–39 Dip-pen nanolithography (DPN) represents the constructive SPL approach for making polymer nanostructures. The process consists of DPN-tip direct-writing of initiator molecules, and subsequent growth of polymer brushes from the initiator-patterned areas. Although the patterning process is simple and straightforward, the diffusion of the initiator limits the application of this technique because: (1) the diffusion rate is difficult to control, and varies a lot between different initiator molecules; and (2) volatile initiator molecules can diffuse onto random areas of the surface through air, and thus are not ideal for DPN patterning. On the other hand, a destructive SPL approach such as nanoshaving relies on selective removal of an inert self-assembled monolayer (SAM) which protects the surface from contamination, and subsequent solution backfilling of initiator onto the uncovered areas. Because the initiator is assembled on a chemically confined surface, the pattern size is not affected by molecular diffusion. Rather, the dynamic exchange between initiator and inert SAM molecules associated with the multiple solution processes may induce many defects on the pattern surfaces. The need for an additional liquid reaction cell is also regarded as a drawback.
Herein, we report the fabrication of programmable polymer brushes by a novel, facile, and versatile scanning-probe-based approach called Dip-Pen Nanodisplacement Lithography (DNL). Inspired by DPN and nanoshaving, DNL combines the attributes of both techniques to yield a high resolution, high registration, solution-free and diffusion-limited tool for delivering initiator molecules onto a surface in air and ambient conditions. Complex nanostructures of polymer brushes with not only controllable chemical structure, topography, size and shape, but also grafting density, polymer chain configuration, hierarchical structure, and multiplexing can be readily made by this method.
Results and discussion
The DNL process is schematically illustrated in Fig. 1A. First, an atomic force microscope (AFM) tip inked with initiator molecules is brought into contact with a surface modified with a dense SAM. Under high load (>10 nN), the SAM molecules are mechanically cleaved away by the AFM tip, and the initiator molecules simultaneously self-assemble onto the uncovered areas of the surface to achieve the “nanodisplacement”. The SAM acts as a resist layer to protect the other areas of the surface from contamination by the diffusion of ink molecules. As a result, the feature size is equal to the cleaving area and is independent of environmental conditions. Polymer brushes are then grown from the initiated areas by surface-initiated polymerization. At low load (1–3 nN), the AFM tip does not cleave the SAM so that it can be used for reading, allowing for high registration patterning. We demonstrate herein patterning of the initiator, ω-mercaptoundecyl bromoisobutyrate (MUDBr), on 16-mercaptohexadecanoic acid (MHA) passivated gold substrate by DNL, and then grow polymer brushes by surface-initiated atomic transfer radical polymerization (SI-ATRP).13,40 | ||
| Fig. 1 (A) Schematic illustration of the fabrication of polymer brushes by DNL. (B) LFM image of a MUDBr square written by DNL. (C) MUDBr nanodots made by DNL at constant tip–substrate contact force (1000 nN), but different tip–substrate contact time. Each dot was made by indenting the tip onto the MHA-Au one at a time. | ||
We first study the DNL writing of MUDBr. In a typical experiment, a contact mode AFM tip (Vistaprobes) was first inked with MUDBr by immersion in an ethanol solution of 1 mM MUDBr for 10 s and drying in the air. This will produce a large amount of ink molecules on the tip which can typically last for the duration of a one-day DNL experiment. The MUDBr inked tip was then loaded onto an XE-100 AFM (Park System) and used for DNL patterning onto a gold substrate that was previously coated with MHA. The movement of the tip was precisely controlled by programming the x–y–z piezo of the AFM with XE-100 lithography software. As proof-of-concept, a 2 μm × 2 μm MUDBr square was first written by scanning the same area at high tip–substrate contact force (1000 nN, 4 μm s−1), and imaged by lateral force microscopy (LFM) mode with the same tip at 1 nN (Fig. 1B). Lower lateral friction, which arises from the more-hydrophobic bromoisobutyrate terminal groups of MUDBr, was indeed observed only at the high force scanned area, indicating that the displacement of MHA by MUDBr occurred site-selectively, critical for high resolution patterning.
Similar to the microstructure, nanodots and nanolines of MUDBr can be made by indenting or shaving the MHA-Au surface with the MUDBr-inked tip at high force, respectively. Again, lower lateral friction arising from the MUDBr was observed only at the “nanodisplacement” positions. The resolution (defined here as smallest lateral feature size) of the MUDBr feature is mainly controlled by the radius of curvature of the writing tip, and is independent from the tip–substrate contact time. Fig. 1C shows a 5 × 5 array of MUDBr dots made with constant indentation force (1000 nN), but different indentation time spanning from 0.1 s to 10 s. Each dot was generated by a single indentation so as to generate the smallest feature size. The size of the 25 dots measured by LFM appeared to be uniform. The average feature size calculated by measuring the radius with the XE-100 software (assuming circular features) is 26 ± 4 nm. This is because that the expansion of the MUDBr feature on gold by surface diffusion is prohibited by the MHA resist layer. Note that the shapes of the MUDBr features are not perfectly circular, which can be attributed to the grain boundary of polycrystalline gold and the shape of the writing tip. The resolution can be potentially improved by using a shaper tip and flatter gold substrate (such as <111> Au), and vice versa. Nanolines produced by single shaving (1000 nN) but different scanning speed have a similar line width resolution of 27 ± 4 nm (Fig. S1 in the ESI†).
Note that MUDBr is volatile and can self-assemble onto an uncovered gold surface through random diffusion in the air. Fortunately, the DNL approach is particularly suitable for volatile inks because the MHA layer can effectively passivate the gold surface, i.e., no bare gold surface is available for MUDBr unless it is uncovered by the DNL cleaving. Furthermore, DNL can make good use of the volatile nature of MUDBr for high speed patterning without humidity control as a consequence of the fact that the assembly of MUDBr does not necessarily depend on the water meniscus. For example, we could write the 2 μm × 2 μm MUDBr square at 100 μm s−1 and 20% relative humidity (Fig. S2, ESI†). In contrast, direct-write SPL techniques such as DPN typically operate with a lower speed (0.1–4 μm s−1) because the formation of a water meniscus, which is inevitable to the writing process, is interrupted at high scan speed. Such volatile molecules are not ideal to be patterned by DPN because the non-patterned gold surface can be easily contaminated by the ink (Fig. S3, ESI†). Compared with destructive SPL techniques such as nanoshaving and nanografting, DNL is simpler and faster since it does not require additional solution backfilling process and the liquid cell. The characteristics of DPN, nanografting, and DNL are briefly summarized in Table S1 in the ESI.†
We then study the growth of polymer brushes from the MUDBr pre-patterns made by DNL. Very importantly, combined with SI-ATRP, DNL offers the unique capability for programming the chemical structure, topography, size, shape, grafting density, chain configuration and multiplexing components of polymer brush nanostructures on a surface. As proof-of-concept, we patterned poly[2-(methacryloyloxy)ethyltrimethylammonium chloride] (PMETAC) brushes.15,17,21,25 First of all, uniformed nanostructures of PMETAC brushes with <10% size variation in all dimensions can be achieved by SI-ATRP from MUDBr features previously made by DNL at the same conditions. For example, Fig. 2A shows a tapping mode AFM image of a 5 × 5 nanodot array of PMETAC with 99 ± 9 nm in width, and 7 ± 5 nm in height. No growth of polymer brushes was observed at the MHA-passivated areas, further confirming that the previous DNL process was highly site-selective.
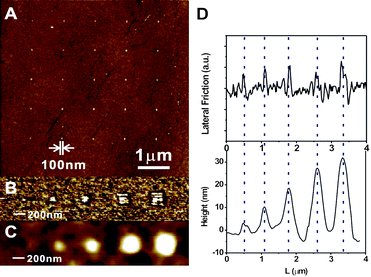 | ||
| Fig. 2 (A) AFM topographic image of 5 × 5 array of PMETAC dots. (B) LFM image of MUDBr of different feature sizes made by DNL. (C) AFM topographic image of PMETAC brushes grown from the MUDBr features in B. (D) Cross sectional profiles of B (upper) and C (lower). | ||
The size of PMETAC can be tailored by changing the size of the corresponding MUDBr feature. We made a series of MUDBr dots with different scanning size at 1 μm s−1 and 1000 nN. The MUDBr dots measured by LFM are 68 nm, 93 nm, 136 nm, 199 nm, 272 nm (Fig. 2B). PMETAC brushes grown from the dot series under the same conditions are 210 nm, 226 nm, 274 nm, 355 nm, 379 nm in width, and 4 nm, 11 nm, 20 nm, 28 nm, 33 nm in height (Fig. 2C). Notably, the size the PMETAC feature, D, is larger than that of the corresponding MUDBr footprint, d (Fig. 2D). The D/d ratio decreases with increasing d (Fig. 3A). On the other hand, the height of the PMETAC feature, h, also increases with increasing d, although the polymer brushes were grown at the same solution. Such a phenomenon can be explained by a recent theoretical model by Jonas et al.,41 which takes into account both of the chain entropy and wetting energy of nanoscale patterned polymer brushes. In the present system, the AFM images show that PMETAC nanodots form a droplet-like structure (Fig. 3B). The chains at the periphery of the droplet try to spread on the surface because the quaternary ammonium groups of PMETAC have an affinity for the carboxylic background. The relaxation of the chains at the rim of the features creates extra room for neighboring chains further inside the nanobrushes, which relax by tilting away from the normal to the surface and by decreasing the distance between their ends. That results in a decrease in the maximum height of the brush, and an increase in the lateral dimension of the brush compared with that of the initiator footprint. Such nanoconfinement phenomenon is more pronounced for smaller diameter. In the above AFM measurements, although tip broadening effect will overestimate the feature size, the trend of size variation should still be true and parallel.
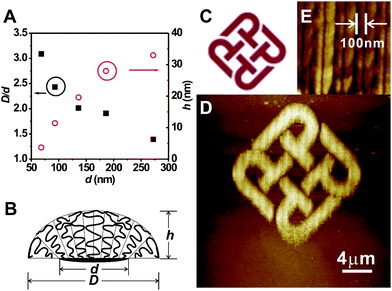 | ||
| Fig. 3 (A) The ratio of PMETAC size to corresponding MUDBr footprint, D/d (solid square), and height of polymer brushes, h (empty circle), versus the MUDBr footprint, d. (B) A model of the nanobrush droplet. The chains at the periphery of the droplet try to spread on the surface, and the relaxation of the chains at the rim of the features creates extra room for neighboring chains further inside the nanobrushes, which relax by tilting away from the normal to the surface and by decreasing the distance between their ends. (C) “PolyU Logo” of the Hong Kong Polytechnic University. (D) AFM topographic image of a PMETAC pattern of PolyU Logo made by DNL and SI-ATRP. (E) A zoomed-in AFM topographic image of D, showing the center-to-center distance of the nanolines is 100 nm, while the width of the lines is ∼75 nm. | ||
Arbitrary structures of polymer brushes can be made by programming the initiator pattern through the XE-100 lithography software. To demonstrate an example, we took a bitmap of the “PolyU Logo” of the Hong Kong Polytechnic University (Fig. 3C) and converted the colored areas into a pattern consisting of various lengths of vertically parallel line segments with 100 nm center-to-center spacing at the x axis. MUDBr was written according to this pattern by DNL at 10 μm s−1 and 1000 nN, and then PMETAC was grown. Fig. 3D shows the tapping mode AFM image of the as-made PMETAC hierarchical arbitrary structure. The zoomed-in AFM image confirms that the line spacing is indeed 100 nm, while the width of the nanoline is ∼75 nm (Fig. 3E). The height and width of the nanolines seem to be uniform over the pattern.
Very interestingly, and importantly, we found that the grafting density of the polymer brushes was dependent on the tip–substrate contact force in the DNL process. A series of 2 μm × 2 μm MUDBr squares were written by DNL on the same substrate at 4 μm s−1, but with a different tip–substrate contact force, ranging from 5 nN to 1000 nN. PMETAC were then grown in the same polymerization solution from all the MUDBr squares simultaneously, and imaged by tapping mode AFM. We observed a mono-increase in the thickness of the polymer brushes with an increasing force (Fig. 4A). For example, PMETAC brush grown from MUDBr written at 1000 nN is ∼16 nm thick, but only ∼2 nm thick for the 100 nN counterpart. We attribute this effect to the increase in grafting density at higher contact force. With a harder applied force, more MHA resist was removed so that greater amount of MUDBr was assembled, leading to a higher grafting density of the polymer brushes. Compared with traditional methods that use an initiator/inert molecule mixture to adjust the grafting density,42 our approach opens up the opportunities for high resolution and site-specific control of the grafting density, and allows for the fabrication of a hierarchical structure with different grafting densities and chain configuration. Notably, such mutli-level chemical structures are critical to the realization of many functional surfaces.4,9,26,43–45 As a proof-of-concept experiment, we patterned a three-layer hierarchical initiator structure consisting of three co-centered square features of different grafting densities. First, a 30 μm × 30 μm bottom layer of loose MUDBr square was patterned at 4 μm s−1 and 100 nN. Second, a middle 11 μm × 11 μm square was patterned on top of the bottom layer at 500 nN. Third, a third 1 μm × 1 μm central square was patterned at 1000 nN. Finally, PMETAC brushes were grown. Indeed, tapping mode AFM reveals a three-step-thickness topography of the as-made PMETAC hierarchical structure (Fig. 4B), while the phase image shows a very low contrast, indicating that the surface is covered with the same chemicals (Fig. 4C). The bottom polymer layer has the least thickness because the polymer chains collapse into a “mushroom” morphology as a result of very low grafting density. The central layer has the highest thickness because the polymer chains are closely packed and stretched up into a “brush” morphology (Fig. 4D).
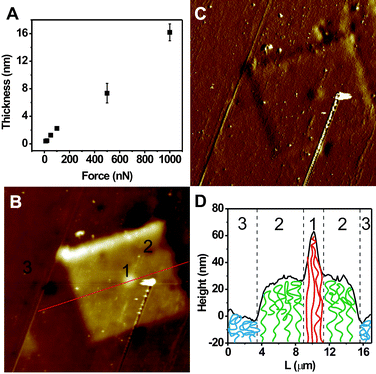 | ||
| Fig. 4 (A) Thickness of PMETAC brushes versus the tip–substrate contact force used for DNL writing of the underlined MUDBr. (B) AFM topographic image of a hierarchical PMETAC structures made by three-step DNL with different grafting densities (scan size: 60 μm × 60 μm). (C) AFM phase image of B (scan size: 60 μm × 60 μm). (D) Cross sectional profile of B, and a scheme showing that the chain configuration can be controlled by varying the grafting density, leading to the thickness difference. | ||
Lastly, we demonstrate the fabrication of multiplexed polymer brushes towards a multifunctional surface. A 100 μm × 100 μm pattern consisting of line segments of PMETAC was first made by DNL and SI-ATRP, as stated above. NaN3 was used to ensure termination of the ATPR initiator after the first growth. Previously, we have proven that NaN3 could effectively terminate the Br initiator.25 The non-displaced MHA areas were still available for second DNL patterning. Because the DNL tip can be used for reading, we could readily relocate the tip to the first pattern area. A second polymer brush, poly (N-isopropylacrylamide) (PNIMPAm), was then patterned (shorter line segments) across the first PMETAC pattern. Again, the result nanostructures are highly uniform and site-selective (Fig. 5). In principle, one can make as many polymer components as possible once there is still spare MHA area available.
 | ||
| Fig. 5 (A) AFM topographic image of two multiplexed polymer brushes made by multi-cycle DNL and SI-ATRP. The scan size is 60 μm × 60 μm. (B) Zoomed-in image of A. | ||
Conclusions
In summary, we have reported the fabrication of highly programmable nanostructures of polymer brushes enabled by DNL. The DNL method has several important attributes. (1) The nanodisplacement process is a combination of constructive and destructive procedures. (2) The method is diffusion-limited. The MHA resist prevents the gold surface from undesirable surface and air diffusion, so that the resolution of this method is mainly defined by the radius curvature of the tip. In the present work, the resolution is ∼25 nm using a 20-nm-sharp tip. (3) DNL operates in air and ambient conditions, without the need for liquid cell or careful control over the environment humidity. (4) DNL is ideal for patterning volatile molecules in the air. To the best of our knowledge, this is the first reported SPL technique aiming at patterning volatile molecules in the air. (5) The grafting density of patterned molecules is force-dependent, which allows for precise site-selective tailoring of a surface at the molecular-level. (6) The writing tip can be used for reading at low force, enabling high resolution and high registration patterning of multiple functional polymers. In principle, one can create surfaces with wide-ranging combinations of chemical structure, topography, size, shape, grafting density and chain configuration of various polymer brushes by controlling the DNL parameters, the choice of initiators and monomers and the polymerization conditions. This will benefit a large number of applications and fundamental researches, especially those in materials science, biotechnology and life science.Experimental
Materials: NaN3, 11-mercapto-1-undecanol, 16-mercaptohexadecanoic acid (MHA), N-isopropylacrylamide (NIPAm), 2-(methacryloyloxy)ethyltrimethylammonium chloride (METAC), N,N,N,N,N-pentamethyldiethylenetriamine (PMDETA), 2,2-dipyridyl, Cu(I)Br, and Cu(II)Br2, triethylamine, dimethylformamide, dichloromethane, alcohol and ethanol were purchased from Aldrich and used without further purification. ω-Mercaptoundecyl bromoisobutyrate (MUDBr), for ATRP was synthesized from 11-mercapto-1-undecanol and 2-bromo-2-methylpropionyl bromide by using a modified procedure according to ref. 13.Preparation of MHA-Au: Self-assembled MHA monolayers were formed on the surface of gold films by immersion of the films into 5 mM MHA solutions at room temperature for 48 h. The substrates were rinsed sequentially in ethanol and hexane, and then dried under a stream of nitrogen. Note: immersion for short time such as 1 h may resulted in less-dense MHA monolayers, which may lead to contamination in DNL.
SI-ATRP: Polymer brush growth was achieved by placing the patterned gold substrates in Schlenk tubes under an N2 atmosphere and adding degassed solutions. The solution to prepare PMETAC composed of 9.2 g METAC, 10 mL of MeOH, 2,2-dipyridyl (0.48 g), Cu(I)Br (0.12 g), and Cu(II)Br2 (0.019 g) and the polymerization time was 10 h at room temperature. After taking out the substrates from the polymerization solution, they were rinsed extensively with H2O and MeOH.
Two-component SI-ATRP: The terminal Br of the PMETAC brush was passivated by immersing it into a 0.12 M NaN3/dimethylformamide (DMF) solution for 50 h. It was then ultrasonically cleaned with de-ionized water and dried under a stream of nitrogen. A second MUDBr pattern was written onto this substrate. Then, the substrate was immerged in Schlenk tubes again under an N2 atmosphere and degassed solutions. The solution for preparation of the PNIPAm was composed of NIPAm (12.6 g), CuBr (0.16 g), and PMDETA (0.70 mL) in 6.3 mL H2O and 6.3 mL MeOH. The polymerization was allowed to proceed for 5 min at room temperature. The polymerization mixture was poured into ice-cold MeOH. The substrates were also finally rinsed with H2O and MeOH, and dried.
Acknowledgements
This work is supported by Research Grant Council of Hong Kong (PolyU 5234/08E), Niche Area (J/BB6Q), and The Hong Kong Polytechnic University (A-PK09 and 1-ZV5Z).Notes and references
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14 RSC.
- E. P. K. Currie, W. Norde and M. A. C. Stuart, Adv. Colloid Interface Sci., 2003, 100, 205–265 CrossRef.
- W. Senaratne, L. Andruzzi and C. K. Ober, Biomacromolecules, 2005, 6, 2427 CrossRef CAS.
- L. Leger, E. Raphael and H. Hervet, Poly. Confined Environments, 1999, 138, 185 Search PubMed.
- B. Yu, Z. J. Zheng, Y. Li and F. Zhou, Text. Bioeng. Inform. Symp. Proc., 2009, 1–2, 73 Search PubMed.
- J. L. Zhang and Y. C. Han, Chem. Soc. Rev., 2009, 39, 676 Search PubMed.
- C. Y. Hong, Y. Z. You and C. Y. Pan, Chem. Mater., 2005, 17, 2247 CrossRef CAS.
- X. M. Li, J. Huskens and D. N. Reinhoudt, J. Mater. Chem., 2004, 14, 2954 RSC.
- R. Ducker, A. Garcia, J. M. Zhang, T. Chen and S. Zauscher, Soft Matter, 2008, 4, 1774 RSC.
- W. T. S. Huck, Mater. Today, 2008, 11, 24 CrossRef CAS.
- Z. H. Nie and E. Kumacheva, Nat. Mater., 2008, 7, 277 CrossRef CAS.
- U. Schmelmer, A. Paul, A. Kuller, M. Steenackers, A. Ulman, M. Grunze, A. Golzhauser and R. Jordan, Small, 2007, 3, 459 CrossRef CAS.
- R. R. Shah, D. Merreceyes, M. Husemann, I. Rees, N. L. Abbott, C. J. Hawker and J. L. Hedrick, Macromolecules, 2000, 33, 597 CrossRef CAS.
- M. Husemann, M. Morrison, D. Benoit, K. J. Frommer, C. M. Mate, W. D. Hinsberg, J. L. Hedrick and C. J. Hawker, J. Am. Chem. Soc., 2000, 122, 1844 CrossRef CAS.
- O. Azzaroni, Z. J. Zheng, Z. Q. Yang and W. T. S. Huck, Langmuir, 2006, 22, 6730 CrossRef CAS.
- O. Azzaroni, S. E. Moya, A. A. Brown, Z. J. Zheng, E. Donath and W. T. S. Huck, Adv. Funct. Mater., 2006, 16, 1037 CrossRef CAS.
- X. Q. Liu, H. X. Chang, Y. Li, W. T. S. Huck and Z. J. Zheng, ACS Appl. Mater. Interfaces, 2010, 2, 529 Search PubMed.
- A. M. Jonas, Z. J. Hu, K. Glinel and W. T. S. Huck, Nano Lett., 2008, 8, 3819 CrossRef CAS.
- S. J. Ahn, M. Kaholek, W. K. Lee, B. LaMattina, T. H. LaBean and S. Zauscher, Adv. Mater., 2004, 16, 2141 CrossRef CAS.
- A. Rastogi, M. Y. Paik, M. Tanaka and C. K. Ober, ACS Nano, 2010, 4, 771 CrossRef CAS.
- Z. L. Liu, H. Y. Hu, B. Yu, M. A. Chen, Z. J. Zheng and F. Zhou, Electrochem. Commun., 2009, 11, 492 CrossRef CAS.
- S. B. Jhaveri, M. Beinhoff, C. J. Hawker, K. R. Carter and D. Y. Sogah, ACS Nano, 2008, 2, 719 CrossRef.
- M. Beinhoff, A. T. Appapillai, L. D. Underwood, J. E. Frommer and K. R. Carter, Langmuir, 2006, 22, 2411 CrossRef CAS.
- A. Genua, J. A. Alduncin, J. A. Pomposo, H. Grande, N. Kehagias, V. Reboud, C. Sotomayor, I. Mondragon and D. Mecerreyes, Nanotechnology, 2007, 18, 7.
- F. Zhou, Z. J. Zheng, B. Yu, W. M. Liu and W. T. S. Huck, J. Am. Chem. Soc., 2006, 128, 16253 CrossRef CAS.
- M. Wang, J. E. Comrie, Y. P. Bai, X. M. He, S. Y. Guo and W. T. S. Huck, Adv. Funct. Mater., 2009, 19, 2236 CrossRef CAS.
- M. L. van Poll, F. Zhou, M. Ramstedt, L. Hu and W. T. S. Huck, Angew. Chem., Int. Ed., 2007, 46, 6634 CrossRef CAS.
- H. W. Ma, J. H. Hyun, P. Stiller and A. Chilkoti, Adv. Mater., 2004, 16, 338 CrossRef CAS.
- X. G. Liu, S. W. Guo and C. A. Mirkin, Angew. Chem., Int. Ed., 2003, 42, 4785 CrossRef CAS.
- W. K. Lee, L. J. Whitman, J. Lee, W. P. King and P. E. Sheehan, Soft Matter, 2008, 4, 1844 RSC.
- W. K. Lee and P. E. Sheehan, Scanning, 2008, 30, 172 CrossRef CAS.
- W. K. Lee, K. C. Caster, J. Kim and S. Zauscher, Small, 2006, 2, 848 CrossRef CAS.
- D. S. Ginger, H. Zhang and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 30 CrossRef.
- K. Salaita, Y. H. Wang and C. A. Mirkin, Nat. Nanotechnol., 2007, 2, 145 CrossRef CAS.
- F. W. Huo, Z. J. Zheng, G. F. Zheng, L. R. Giam, H. Zhang and C. A. Mirkin, Science, 2008, 321, 1658 CrossRef CAS.
- L. G. Rosa and J. Liang, J. Phys.: Condens. Matter, 2009, 21, 18.
- N. A. Amro, S. Xu and G.-y. Liu, Langmuir, 2000, 16, 3006 CrossRef CAS.
- M. Liu, N. A. Amro and G.-y. Liu, Annu. Rev. Phys. Chem., 2008, 59, 367 CrossRef CAS.
- M. Kaholek, W. K. Lee, S. J. Ahn, H. W. Ma, K. C. Caster, B. LaMattina and S. Zauscher, Chem. Mater., 2004, 16, 3688 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337 CrossRef CAS.
- A. M. Jonas, Z. J. Hu, K. Glinel and W. T. S. Huck, Macromolecules, 2008, 41, 6859 CrossRef CAS.
- D. M. Jones, A. A. Brown and W. T. S. Huck, Langmuir, 2002, 18, 1265 CrossRef CAS.
- S. J. Sofia, V. Premnath and E. W. Merrill, Macromolecules, 1998, 31, 5059 CrossRef CAS.
- R. R. Bhat, B. N. Chaney, J. Rowley, A. Liebmann-Vinson and J. Genzer, Adv. Mater., 2005, 17, 2802 CrossRef CAS.
- I. Luzinov, S. Minko and V. V. Tsukruk, Soft Matter, 2008, 4, 714 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: LFM images of MUDBr patterns; digital image of irregular polymer brushes grown on gold caused by random diffusion of MUDBr through the air. See DOI: 10.1039/c0nr00565g |
| This journal is © The Royal Society of Chemistry 2010 |