Progress in ionic organic-inorganic composite membranes for fuelcell applications
R. K.
Nagarale†
*a,
Woonsup
Shin
a and
Pramod K.
Singh
b
aDepartment of Chemistry and Interdisciplinary Program of Integrated Biotechnology, Sogang University, Seoul, 121-742, Korea. E-mail: rnagarale@yahoo.com
bDepartment of Material Science and Research & Technology Development Centre, Sharda University, Greater Noida, 201 306, India
First published on 23rd December 2009
Abstract
Over the past decade, organic-inorganic composite ionic membranes have gained tremendous attention for use in medium temperature fuelcells. The addition of a functional or nonfunctional inorganic into the organic material is usual practice in proton exchange membranes to improve their thermal stability and performance. In the development process, perfluorosulfonated membranes and their composites with inorganic materials dominate and show better thermal, mechanical and chemical stability, and proton conductivities at intermediate temperatures with low fuel drag. Concurrently, composites of hydrocarbon polymers, such as poly(arylene ether ketone)s, poly(benzimidazole)s, poly(arylene sulfone)s, poly(vinyl alcohol) etc., have been developed as alternatives to perflurinated membranes. The present review addresses the basic route used to synthesize these composite membrane materials. The main emphasis has been given on the preparation procedures of inorganic materials, their modifications and composites with perfluorinated and hydrocarbon polymers. The properties are addressed with respect to fuelcell applications.
 R. K. Nagarale | R. K. Nagarale did his MSc in inorganic chemistry at Karnatak University Dharwad, Karnataka, India in 1999 and received Prof. G. K. Narayanareddy gold medal for standing first among the successful students. In 2006 he completed his PhD at Central Salt and Marine Chemicals Research Institute, Bhavnagar affiliated to Bhavnagar University, Bhavnagar, Gujarat, India. During his PhD course he was awarded a senior research fellowship from CSIR India. In 2006 he joined Prof. Woonsup Shin as a post-doctoral fellow, in the department of chemistry and Interdisciplinary program of integrated biotechnology, Sogang University, Seoul, Korea. In 2008 he became a research professor in the same department. He is presently visiting Prof. Adam Heller, in the department of chemical engineering, University of Texas at Austin, USA. His research interests include the ionic and functional ionic polymers for energy, biotechnology and separation/purification technologies. |
 Woonsup Shin | Woonsup Shin has been at Sogang University as a Professor of Chemistry from 1995 after receiving a PhD at Texas A&M University and postdoctoral work at Stanford University. He is currently Chair of Interdisciplinary Program of Integrated Biotechnology and Head of Inorganic and Bio-Material Center. He is a member of the Korean Chemical Society, Korean Electrochemical Society, International Society of Electrochemistry, The Electrochemical Society, American Chemical Society and serves as Vice-Chair of the Bioelectrochemistry Division of the International Society of Electrochemistry. |
 Pramod K. Singh | Pramod Kumar Singh received his MSc (Electronics) degree from the Purvanchal University, Jaunpur, India in 1994 and PhD degree from Banaras Hindu University in 2001. Dr Singh spent four years as a postdoc fellow with Professor Hee Woo Rhee’s group at Sogang University, Seoul, S. Korea working on dye sensitized solar cells. He is currently appointed as an Assistant Professor in Sharda University, Greater Noida, India. His research interests are focused mainly on dye sensitized solar cells using solid polymer electrolytes. |
1. Introduction
The production of composite membrane materials by bridging organic and inorganic chemistry at a molecular level is an extensive and fascinating field of investigation. A major benefit of such hybrid research activities is linked to synergetic effects of organic and inorganic matrix with desired and improved properties in comparison to own unique properties of each components (organic or inorganic).1 They expanded considerable applications in optoelectronic,2 ion-conduction,3 biology,4 catalysis5 and membranes.6Principally, ionic membrane materials have been developed for the medium temperature proton exchange membrane (PEM) fuelcell applications. Which have been identified as a nearly ideal solution to power the requirements for motor vehicle manufacturers, utility and nonutility generators, and portable electronic devices although efficiency is not up to the mark?7 Most commonly used PEMs are perfluorosulfonated membrane for example ‘Nafion’ developed by Dow Chemicals and is a bench mark for most of researchers. But these membranes have limitations such as high cost, use at low temperatures (lower than 100 °C because of low thermal stability) and the need for high humidity to achieve high proton conductivity and high methanol flux.8,9 But operation of proton exchange membranefuelcells (PEMFCs) at intermediate temperatures (100–200 °C) enhances performance by accelerating electrode reactions without CO poisoning of the Ptcatalyst. Alternatively, hydrocarbon based PEMs, which operates at intermediate temperatures have been well studied.3,10,11 They suffer from the disadvantage of dehydration at higher temperature and interaction of absorbed water with the acid group present in the membrane for generating proton conductivity. The ability of PEMs to absorb a large fraction of water increases the proton conductivity, fuel permeability and reduces the mechanical properties. Also, the nature of acid groups, i.e. sulfonic, phosphonic and carboxylic acid, and their distribution affects the fraction of water content in the membranes. Thus design of new membranes that can conduct protons with little or no water is perhaps the greatest challenge in the science community.
One promising strategy for improvement in performance of PEMs by water management is incorporation of nanometre sized particles of hygroscopic metal oxides (silica, titania, and zirconia) which act as a water reservoir.2,12 Furthermore, the methodology of introducing functional groups onto inorganic moieties has been successfully used to improve proton conductivity at higher temperature with low fuel flux and high mechanical and oxidation stability. This review gives an insight of literature available on possible approaches to develop organic-inorganic composite proton exchange membranes and their perspectives.
2. General consideration
2.1 Critical requirements from composite membrane materials
The critical requirement for the successful development of new ionic membrane materials are detailed in the following.(1) Ionic conductivity: high ionic conductivity is the critical requirement and main deciding factor for ionic membranes. Basically, it depends on the type of functional groups present i.e. strongly ionic groups such as sulfonics,13 phosphonic acids,14 and quaternary ammonium saltsetc.; and/or weakly ionic groups, for example, carboxylic acids,15 hydroxyls, and primary,16 secondary and tertiary aminegroupsetc. on the polymer backbone. The porosity of the membrane also affects the ionic conductivity. A highly porous membrane will have high ionic conductivity and vice versa. Thus in cases of organic-inorganic composite materials we can easily tune the pore structure of the membrane by appropriate organic or inorganic materials or synthetic methodology or even with ion-exchange groups. These are the big advantages for design of new generation ideal ionic polymers. Water content of the polymeric material has considerable effect on ionic conductivity. The higher the water content, higher is the ionic conductivity. But for wide number of applications, like fuelcell and electromechanical transducers low water content with high ionic conductivity is the primary requirement. Thus, in designing new types of ionic polymers one should keep in mind how one can lower down the water content by retaining good ionic conductivities.
(2) High permselectivity: the ionic polymer should have good permeability to counter ion but be impermeable to the co-ion. It basically depends on the type of ion-exchange groups present on the polymer backbone and its surface morphology, i.e. porosity of the membranes.
(3) Low electronic conductivity: the normal use of ionic membrane materials are in electro-membrane processes like electro-driven separation of ionic compounds, biologically important molecules, protein separation and fuelcelletc. In such cases, high electronic conductivity will decreases the open circuit potential of the system. But, exceptionally, in the case of electromechanical transducers, the presence of high surface electronic conductivity is essential for better actuation behavior of the polymers. Composites of metal nanoparticles and carbon nanotubes commonly have such requirements. Thus depending on the type of application, the requirements of electronic conductivity of the ionic polymers need to be tailored.
(4) Good chemical stability: this is another important deciding factor in the usefulness of the ionic polymers. For the wide range of applications the polymer should have good chemical stability, i.e. it should be resistant to a wide range of acids and alkalis over the complete pH range as well as stable in strong oxidizing, reducing and hydrolytic environments.
(5) Thermal stability: to render the applications in high temperature, the ionic polymers should have good thermal stability. Such requirement is challenging, but a demand of the future for high temperature fuelcell applications.
(6) Good mechanical and dimensional stability. Good mechanical and dimensional stability is required in all membrane based processes. The ionic membrane should not lose its mechanical strength both in dry and hydrated states. It should not change its dimensions when the process medium changes. For example when the membrane process is carried out in low and high ionic strength media, or the pH of the medium changes during the process. In fuelcell applications the detachment of the electrode from membrane due to dimensional changes (non uniform expansion/contraction) must not occur. Thus, the polymer must be compatible with the environmental fluctuations and show least variations in its dimensions.
(7) Low cost: for the successful commercialization and sustainable growth, the cost of the synthesized ionic membrane materials should be as low as possible and the materials used in the synthesis process should be readily available.
The parameters, as discussed above, determining the membrane properties often have opposing effects. For example, high ionic conductivity results from the high water content and low cross-linking but decreases the mechanical and dimensional stability of the membranes. Thus there is a compromise between these properties to develop good ionic membrane materials. Hence the progress towards the development of optimum quality ionic polymer has wide challenges and needs to be designed carefully.
2.2 Basic synthesis approach
Generally, two types of approaches have been used to synthesize ionic organic-inorganic composite membrane materials as shown in Fig. 1. The first one deals with the formation of macro-composite or nano-composites in which domain size of the inorganic phase is of about micrometre or nanometre range, leading to enormous interfacial areas. A simple way of synthesizing these types of membrane is the physical blending of two components in an appropriate solvent. During the synthesis process care should be taken to avoid the unwanted precipitation of the components. Furthermore, each component should be well dispersed and compatible to each other. In the second case, the hybrid structure is obtained at the molecular level with entirely different properties by co-polymerization of one monomer usually inorganic, in the presence of other, usually organic polymeric solution in the proper solvent and catalyst. The general methodology used to prepare these membranes is a sol–gel process. This process occurs at room temperature in liquid state with organometallic precursors (TMOS, TEOS, Zr(IV)-propoxide, Ti(IV)-butoxide, etc.) by hydrolysis-condensation in presence of suitable catalysts, acid or base. The process makes it possible at low temperature a relatively easy molecular level incorporation of pure inorganic phase into an organic matrix,17 which leads to vast applications. Nowadays, it is the fashion to use sol–gel to describe any low temperature preparation of organic-inorganic composite materials in which condensation polymerization of metal alkoxides takes place.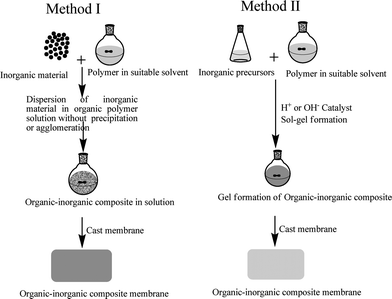 | ||
| Fig. 1 A schematic presentation of the basic preparation methodology for ionic organic-inorganic composite membranes. | ||
A sol is basically a dispersion of colloidal particles (size 1–100 nm) in a liquid, and a gel is an interconnected, rigid network with pores of sub micrometre dimensions and polymeric chains whose average length is greater than a micron. In the sol–gel process, the reaction of metal alkoxides and water in the presence of acid or base forms a one phase solution that goes through a solution-to-gel transition to form a rigid, two-phase system comprised of solid metal oxides and solvent filled pores. The physical and electrochemical properties of the resultant materials largely depend on the reaction conditions, i.e. the catalyst used. In the case of silica alkoxides,18–20 it was observed that the acid catalyst reactions results in linear polymers, which are weakly cross-linked. These polymers entangle and form additional branches, resulting in gelation. Whereas base-catalyzed reactions form highly branched clusters due to rapid hydrolysis condensation of alkoxide silanes.21Gelation occurs by linking of these clusters18–20 (Fig. 2). This difference in cluster formation is due to solubility of resultant metal oxides in reaction medium. The solubility of the silicon oxide is more in alkaline medium which favors the inter-linking of silicaclusters than acidic medium. The hydrolysis condensation of Zr(IV)-propoxide, Ti(IV)-butoxide usually occurs very fast with white precipitation in the reaction mixture and uneven distribution in the organic matrix. In such cases the sol–gel reaction should be carried out in organic solvent with little water content and in the presence of ethylacetoacetate.
 | ||
| Fig. 2 A cartoon showing linear weakly cross-linked and highly branched clusters, A: acid catalyzed B: base catalyzed hydrolysis. | ||
Both acid and base catalyzed reactions which occur in the sol–gel process are bimolecular nucleophilic substitution reactions. The fundamental steps involved are:
Hydrolysis of M(OR)4 (where M is Si, Ti, Zr)
| M(OR)4 + H2O → M(OR)3(OH) + ROH |
Intermediates due to the partial hydrolysis include molecules with M–OHgroups. Complete hydrolysis to form M(OH)4 is very difficult to achieve. Instead, condensation may occur between either two –OH or M–OHgroups and an alkoxygroup to form bridging oxygen and a water or alcohol molecule. An example of a condensation reaction between two –OH with the elimination of water is shown below.
| M(OR)4 + M(OR)3 (OH) → (RO)3MOM (OR)3(OH) + H2O |
Hydrolysis of (RO)3MOM(OR3)3 resulted in (RO)2 M(OH)OM(OR)3, which can undergo further polymerization reactions. The hydrolysis and polycondensation reactions are initiated at numerous sites and the kinetics of the reactions are therefore complex. When a sufficient number of interconnected M–O–M bonds are formed in a region, they interact cooperatively to form colloidal particles or a sol. With time the colloidal particles link together to form a three-dimensional network. At gelation, the viscosity of the solution increases dramatically and a solid object termed an alcogel results, a real sol–gel processes. A technologically important point is that alcogels can be formed in any shape or desired configuration. The possible mechanisms for both acid and base catalyzed reactions in case of silicon alkoxides is as shown in Fig. 3.
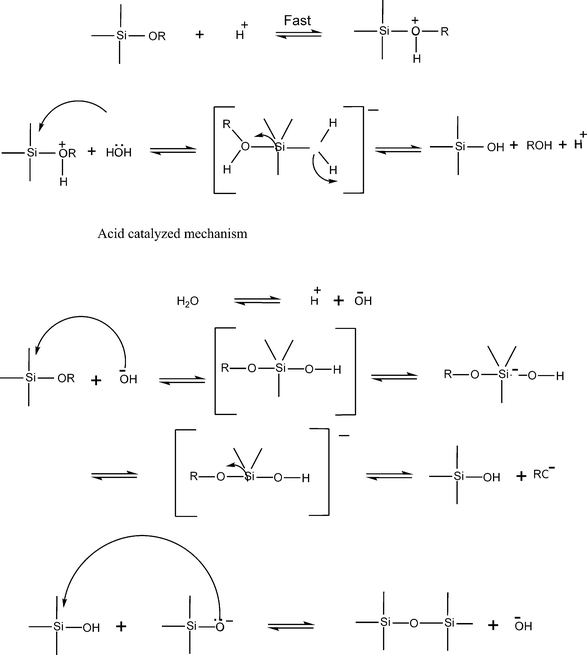 | ||
| Fig. 3 The reaction mechanisms for acid and base catalyzed hydrolysis condensation of alkoxy silane. | ||
3. Inorganic materials
3.1 Functionalized and non functionalized silica materials
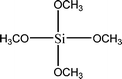
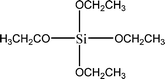
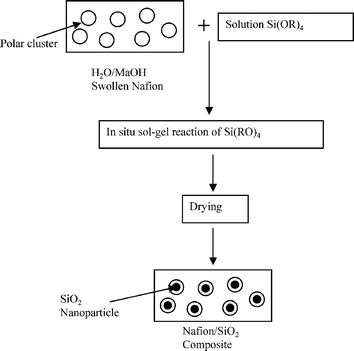
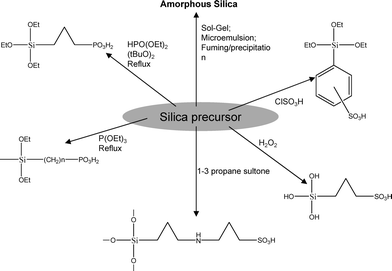
The name silica comprises a large family of products with general formula SiO2 or SiO2·xH2O. It is a naturally occurring material found in minerals, such as quartz and flints, and in plants such as bamboo, rice and barley. But silica used in chemical applications has a synthetic origin.22 There are two form of silica, crystalline and amorphous. The crystalline form involves a high degree of ordering. The active surface, which may participate in any chemical or physical interaction, is limited to the external surface of the crystalline particles. Amorphous silica occurs in various forms likes fibers, sheets, sols, gels and powders. It can be fabricated according to the application. A main feature of interest is the porosity of the amorphous silica forms. Porosity introduces a large surface area inside the silica particles. As interphase processes require a large surface/mass ratio. So, amorphous silica is far more interesting for chemical and physical applications than their crystalline counterparts. It is commonly used in modifying the properties of the ionic polymers.There are two techniques reported to synthesize silica particles used in modifying ionic polymers: the sol–gel method and the microemulsion method. The simple synthesis of monodisperse spherical silica particles is by means of hydrolysis of a dilute solution of TEOS in ethanol at high pH. Uniform amorphous silica spheres whose sizes ranged from 10 nm to 2 μm were obtained22 simply by changing the concentrations of the reactants. This method appears to be simplest and most effective route for synthesizing monodispersed silica spheres. The commercially available silicananoparticlesi.e.nanosilica powder is mainly produced by the fuming and precipitation method in industry. Fumed silica is a fine, white, odorless, and tasteless amorphous powder. It is manufactured by a high-temperature vapor process in which SiCl4 is hydrolyzed in a flame of oxygen–hydrogen. The silica has an extremely large surface area and smooth nonporous surface, which could promote strong physical contact between the filler and the polymer matrix. Precipitated silica is manufactured by a wet procedure by treating silicates with mineral acids to obtain fine hydrated silica particles in the course of precipitation. For the preparation of silica nanocomposites, fumed silica is commonly used and precipitated silica is rarely used since the precipitated one has more silanol (Si–OH) groups on the surface and consequently it is much easier to agglomerate than fumed one. The presence of hydroxylgroups on the surface holds individual silica particles together and aggregates remain intact even under the best mixing conditions if stronger filler-polymer interaction is not present. The dispersion of nanometre-sized particles in the polymer matrix has a significant impact on the properties of composites. A good dispersion may be achieved by surface chemical modification of the nanoparticles or physical methods such as a high-energy ball-milling process and ultrasonic treatment. The molecular level dispersion of silica can be achieved by in situhydrolysis condensation of silica alkooxide monomers in the polymeric solution. In the sol–gel processes choice of silica precursors, catalysis and solvent system is very important.13–16,23,24Table 1 shows the potential functional and non functional silica precursors. Using these precursors one can prepare weakly or strongly positive and negatively charged organic-inorganic composite membrane materials as per the desire applications.
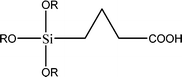

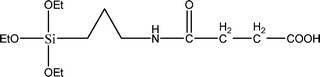
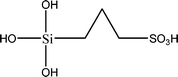


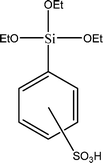
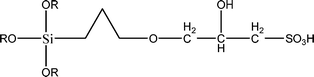
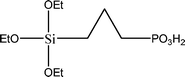
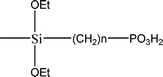
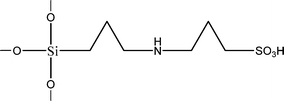
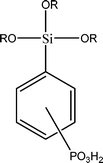

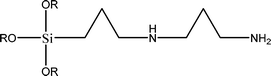
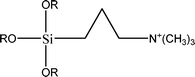
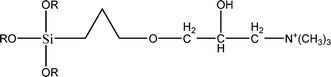
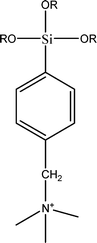
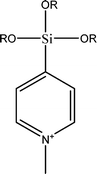
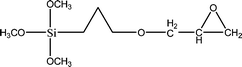

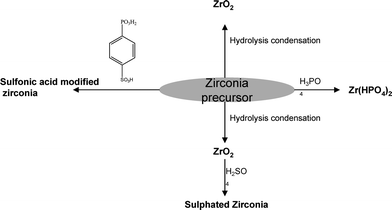
3.2 Zirconium phosphate
Zirconium phosphate is the most extensively studied inorganic material for proton exchange membranesfuelcells. Normal zirconium phosphate (PO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr = 2) is obtained as a gelatinous amorphous precipitate when an excess of phosphoric acid or soluble phosphate is added to a soluble zirconium salt. The insoluble precipitate is considered as zirconyl salt25 and the compound is represented as ZrO(H2PO)2.
:Zr = 2) is obtained as a gelatinous amorphous precipitate when an excess of phosphoric acid or soluble phosphate is added to a soluble zirconium salt. The insoluble precipitate is considered as zirconyl salt25 and the compound is represented as ZrO(H2PO)2.
Amphlett and his co-workers26 postulated that it consists of a network of zirconium atoms linked together by bridging oxygen atoms. The phosphate groups were thought to be bonded to the zirconium atoms and to contain replaceable hydrogen atoms. Washing normal zirconium phosphate with water removes phosphate groups by hydrolysis. Larsen and Donald27 exhaustively washed the normal phosphate with water and obtained a product in which PO4:Zr = 1.72. Baetslé and Pelsmaekers28 precipitated zirconium phosphates from solutions containing PO4:Zr ratios of 3, 2, and 1.75. The precipitates had different compositions but on washing, products of constant composition in which a PO4:Zr ratio of 1.66–1.68 was obtained. The washed zirconium phosphates exhibited weak X-ray diffraction patterns from which it was determined that the unit cell is cubic with a = 9.04 Å. From these observed X-ray and density data, they proposed structure for zirconium phosphate as shown in Fig. 4.
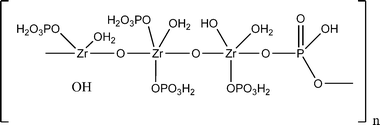 | ||
| Fig. 4 Larsen and Vissers’ proposed structural formula for zirconium phosphate. | ||
Crystalline zirconium phosphate was synthesized by Clearfield and Stynes29 for the first time in 1964. They initially prepared zirconium phosphate gels from hydrochloric acid solutions of zirconyl chloride using an excess of phosphoric acid as precipitant. Further, gelatinous precipitate was refluxed with phosphoric acid to give truly crystalline zirconium phosphate. They found that higher the concentration of refluxing phosphoric acid, faster the conversion rate into crystalline zirconium phosphate of formula Zr(HPO4)2·H2O. The ion-exchange capacity of the resulting material was found to be ∼2 meq. Na/g of the dried sample.
In a modified procedure, Alberti and Torracca30 found the best result by dissolving a suitable zirconium salt (oxychloride or nitrate) in hydrofluoric acid followed by adding concentrated phosphoric acid and allowing the solution to evaporate. In a typical procedure, 5.5 g of ZrOCI2·8H2O was dissolved in 80 ml of water; 4 ml of 40% hydrofluoric acid and 46 ml of 85% phosphoric acid was added ito the stirring solution. It is well known that hydrofluoric acid is a good complexing agent for zirconium; when the concentration of hydrofluoric acid is sufficiently high, precipitation by phosphoric acid is then inhibited. However, when the fluoride ion concentration is lowered (e.g. by evaporation of hydrofluoric acid) the fluozirconate complex gradually dissociates and zirconium phosphate begins to precipitate. The X-ray data and ion-exchange capacity were matched with that of Clearfield's data.
There are two types of predominate zirconiumhydrogen phosphate Zr(HPO4)2·H2O31 (henceforth ZrP) structures, α and γ (Fig. 5). Recent investigation has led to the discovery of two new structures synthesized via hydrothermal routes, designated as τ-ZrP and ψ-ZrP.31 In α-ZrP, the layers consist of ZrO6 octahedra in which zirconium atoms are located in same plane and bonded with one another by tetrahedral phosphate groups (HPO42−) lying above and below the plane composed of zirconium atoms. On the other hand, in γ-ZrP, ZrO6 is coordinated with two kinds of tetrahedron, H2PO4− and PO43−.32 The crystalline α-ZrP has composition Zr(HPO4)2·H2O with strictly stoichiometry and laminar structure.32 The selective sorption properties of ZrP gels are affected by the crystallinity and degree of dehydration. Also, the latter characteristic strongly affects the exchange kinetics. But for both structures the α and γ forms are best for proton transporting due to their pendent –OH groups. Generally α-ZrP has higher reactivity, ion-exchangeability and intercalation property, than the γ form due to its abundant hydroxylgroup,32 which extends into interlayer region and forms hydrogen bonded network with water. The hydroxylproton can react with sodium hydroxide or any alkyl amine. Upon reaction, the structure of α-ZrP remains unchanged, while interlayer distances increases to accommodate the guest ion or molecule. The transport mechanism in α-ZrP at room temperature is dominated by surface transport and it is four orders of magnitude higher than the bulk transport. Isoconductance measurements also indicate that the conductivity varies linearly with the number of surface phosphate groups. Additionally conductivity is highly dependent on degree of hydration, by varying two orders of magnitude of relative humidity; conductivity increases from 5 to 90%.31 Further, recent research has confirmed the dominance of surface transport by modifying P–OHgroups.31–35
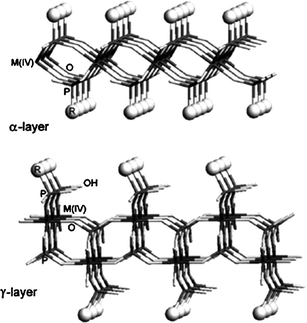 | ||
| Fig. 5 A schematic presentation of the α- and γ-layered structures of ZrP.35,36 | ||
Modification has been done with functionalized organic moieties or Brønsted bases.33 Significant improvement in conductivity was achieved with intercalation of strong acidic functional groups, –SO3H into the interlayer region (there was little improvement with the weak –COOH groups). Zirconium alkyl sulfophenylphosphonates or the variety of [Zr(O3PC6H4SO3H)0.85(O3PC2H5)1.15·nH2O; (Zr(O3PC6H4SO3H)x (O3PCH2OH)2−x·nH2O)] have been investigated for their conductivity under different temperature and relative humidity regimes.36–38 The best conductivity reached in the anhydrous state with ethylsulfophenyl phosphonate: 1.2 × 10−5 S cm−1 at 180 °C (as compared to 10−6 S cm−1 for microcrystalline α-ZrP). This value puts it amongst the best fully anhydrous proton conductors.36,38 In hydrated conditions at 20 °C the conductivity increased from 10−4 at 22% RH (relative humidity) to 1.6 × 10−2 S cm−1 at 90% RH while at 100 °C conductivities as high as 0.05 S cm−1 with 90% RH were reported. Unfortunately at less than 50% humidity, the conductivity of sulfophenylphosphonates showed increased dependence as compared to standard α-ZrP. However at 65% RH and 100 °C still they are capable of conducting at 0.01 S cm−1.39 The significant advantage of sulfophenylphosphonates is that; as temperature is increased from ambient conditions to 100 °C there is no drop in conductivity indicating hygroscopic nature has not been affected. More recent work34 has shown that the zirconium sulfoarylphosphonates also display high conductivity values, although it is strongly affected by humidity. The different zirconium compounds were synthesized with sulfonic acid attached to a phenyl, benzyl or to a fluorinated benzyl group. The highest conductivity reported for sulfophenylphosphonates is up to 5 × 10−2 S cm−1 at 100 °C to 2 × 10−2 S cm−1 at 150 °C with 100% RH.
Honma and co-workers35 developed a proton exchange membrane based on organically modified zirconia by a self-assemble sol–gel process. The monomers were covalently bonded at the zirconia interface to form macromolecular inorganic–organic networks. By altering the inorganic–organic molar ratios, a range of flexible and rigid films showed good chemical tolerance, mechanical property and thermal stability up to 325 °C. The proton conductance was obtained after treating membranes with 85% H3PO4 at 150 °C. The proton conductivities of phosphoric acids-incorporated inorganic–organic hybrids were found about 7 × 10−4 S cm−1 at 180 °C.
3.3 Heteropoly acids
Heteropoly acids have a very long history in basic and applied science. They are part of a large class of polyoxometalates (POMs) and are composed of a metal such as tungsten, molybdenum or vanadium; oxygen; an element from the p-block of the periodic table, such as silicon, phosphorous or arsenic and acidic hydrogen atoms. The conjugate anions of the heteropoly acids (HPAs) are known as polyoxometalates, having metal-oxygen octahedra as the basic structural unit.40 The first characterized and the best known of these is the Keggin heteropolyanion typically represented by the formula XM12O40x−8 where X is the central atom (Si4+, P5+, etc.), x is its oxidation state, and M is the metal ion (Mo6+ or W6+). The Keggin anion is composed of a central tetrahedron XO4 surrounded by 12 edge- and corner-sharing metal-oxygen octahedra MO6 (Fig. 6).40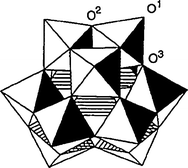 | ||
| Fig. 6 The Keggin structure of the XM12O40x−8 anion (R-isomer): 24 terminal (O1), edge-bridging (O2), and cornerbridging (O3) oxygen atoms. | ||
The octahedra are arranged in four M3O13groups. Each group is formed by three octahedra sharing edges and having a common oxygen atom which is also shared with the central tetrahedron XO4. Among a wide variety of HPAs, Keggin’s are the most stable and more easily available. Generally, solid HPAs form ionic crystals composed of heteropolyanions, countercations (H+, H3O+, H5O2+, etc.) and hydration water. The crystal structure depends on the amount of hydration water.40 This water can be easily removed on heating, whereby the acid strength is increased due to the dehydration of protons. This is a reversible process accompanied by changing the volume of crystal cell. Unlike the rigid network structure of zeolites, in HPA crystal the Keggin anions are quite mobile. Not only water but also a variety of polar organic molecules can enter and leave HPA crystal. HPAs (H3PW12O40, H4SiW12O40, H3PMo12O40) based on the Keggin structure have some of the highest reported acidity and solid state proton conductivities at room temperature. Their proton transport has been extensively studied and thus becoming an increasingly popular proton conducting additive in proton exchange composite membranes for use under dry and/or elevated temperature conditions of fuelcell.41–43
4. Organic materials
The first ever organic material used in a fuelcell was a styrene-divinyl benzenecopolymer followed by perfluorinated membranes in the 60s.10 To date perfluorinated membranes are dominant and subject of research to many groups worldwide due to their excellent chemical stability and proton conductivity. These are the bench mark for many researchers to find alternative membranes even though they have the disadvantage of thermal instability, high reactant flux and high cost. A large amount of literature is available8–10,12,44 on alternative membranes to perfluorinated materials which includes: (a) acid functionalized or doped poly(benzimidazole) (b) poly(arylene ketone) (c) poly(arylene sulfone) (d) poly(sulfides) (e) poly(phynelene oxides) (f) poly(phosphazenes) etc. Within this wide range of alternative membrane materials, no membranes are available for real-world applications. Now world research is progressing in the direction of organic-inorganic composite ionic membranes which are the only hope of meeting the requirements of fuelcells and other applications.5. Organic-inorganic composite materials
The use of composite membranes in fuelcell is very attractive because they exhibit high ionic conductivity and mechanical strength. Also they resist dehydration and exhibit fuel non-permeation. Table 2 gives brief information about the organic and inorganic components of the composite ionic membranes with highlighted properties. Basically two types of organic-inorganic composite membranes are being developed: (i) macro-composite membranes, which are a combination of polymer with an organic or inorganic structure of micrometer scale. These could be developed by simple physical mixing of inorganic into organic matrix with appropriate solvent and (ii) nano-composite membranes, which are a combination of the polymer with an organic or inorganic matrix of nanometer scale. These could be developed by in situpolymerization of inorganic monomer in the presence of an organic matrix with suitable solvent and catalyst.| Polymer | Inorganic material | Descriptions | References |
|---|---|---|---|
| Nafion | Tetraethylorthosilicate | ∼20% increase in water uptake compare to Nafion | 45,48 |
| Cross-linked network of 4,4′-methylenedianiline and 3-glycidoxypropyltrimethoxysilane | ∼σ = 3.4 × 10−2 S cm−1, ∼P = 1.1 × 10−8 cm2 s−1 | 50 | |
| Diphenyldichlorosilane and mercaptopropyltrimethoxysilane | ∼σ = 2.2–1.0 × 10−1 S cm−1, water content 16–21%, ∼P = 2–1 × 10−6 cm2 s−1 | 51–55 | |
| ZrO2 | 20–25% higher water uptake, σ = 2 × 10−2 S cm−1i.e. 8–10% higher, 30–40 mA cm−2 higher current at 0.5 V compared to Nafion® at 90–120 °C | 57,58 | |
| Sulfated- ZrO2 | ∼σ = 2.3 × 10−1 S cm−1 105–135 °C, single cell performance 1.35 W cm−2 | 60,61 | |
| Zr(HPO4)2 | ∼σ = 1.6–2.7 × 10−1 S cm−1; current density 0.25–1.5 A cm−2 at 0.45V | 63 | |
| Heteropolyacids | Water content 60–95%, ∼σ = 1.5–9.5 × 10−2 S cm−1; current density 0.695–0.940 A cm−2 | 12,68,69 | |
| SO3H-heteropolyacid-SiO2 | Single cell performance at 80–200 °C 33–44 mW cm2 | 70 | |
| Poly(oxypropylene) backboned quaternary ammonium salt modified Montmorillonite | Water content ∼28–32%, σ = 4–6 × 10−2 S cm−1, current density 0.56 A cm−2 and power density 0.13 W cm−2 at 0.2 V | 73 | |
| SO3H modified clay | Water content ∼70–87%, σ = 0.2–3.0 × 10−2 S cm−1, current density 7.20 A cm−2 and power density 4.30 W cm−2 at 0.6V and 80 °C | 74 | |
| PVDF | ZrO2·SO4 | σ = 1.2–5.2 × 10−2 S cm−1, single cell power 32 mW cm−2. | 56 |
| PVDF-HFP copolymer | Heteropolyacids | Current density 1.6 A cm−2 | 42 |
| SPEEK | SiO2 | σ ≥1.0 × 10−2 S cm−1 | 80,81 |
| SiO2–N-(-3-triethoxysilyl propyl)-4,5-dihydroimidazole | 40% reduction in water content and P = 13% reduction | 82 | |
| Montmorillonite | ∼50% water content, ∼σ = 1.2 × 10−2 S cm−1, ∼P = 10−8 cm2 s−1 | 86 | |
| Laponite | ∼50% water content, ∼σ = 0.3 × 10−2 S cm−1, current density = 3.70 Acm−2 at 0.6 V | 85 | |
| Heteropolyacid | σ = ∼1 × 10−1 S cm−1 at 100 °C | 88–91 | |
| TiO2 | ∼5% water content, σ = ∼0.2 × 10−2 S cm−1, ∼P = 0.5–2.0 × 10−6 cm2 s−1 | 97 | |
| BPO4 | ∼35% water content, σ = ∼2 × 10−2 S cm−1, ∼P = 0.5–2.0 × 10−6 cm2 s−1 | 98 | |
| SPAEEKK | SiO2 | σ = 2.0–9.7 × 10−2 S cm−1, ∼P = 1.4–6.9 × 10−7 cm2 s−1 | 79 |
| PPEK | SO3H–SiO2 | Water uptake 35%, 3.6 fold increase in σ | 83 |
| SPEK | SiO2–N-(-3-triethoxysilyl propyl)-4,5-dihydroimidazole | 40% reduction in water content and P = 13% reduction | 82 |
| Poly(ether sulfone) | SO3H–SiO2 | Water uptake ∼20% and σ= ∼1 × 10−2 S cm−1 at 90% humidity | 23 |
| Poly(ether sulfone) | PO3H2–SiO2 | Water uptake ∼28%, σ= ∼6.3 × 10−2 S cm−1, P = 5 × 10−7 cm2 s−1. | 105 |
| Zr(HPO4)2 | σ= ∼1.9 × 10−1 S cm−1 at 90 °C and 90% humidity | 106 | |
| Poly(biphenyl ether sulfone) | Ag–SiO2 | Better oxidative stability and long term cell performance with low limiting current density 10 mA. | 103 |
| Poly(ether imide) | SO3H–SiO2 | Water uptake ∼20% and σ = ∼1 × 10−2 S cm−1 at 90% humidity, good hydrolytic and oxidative stability | 23 |
| Poly(vinyl alcohol) | SiO2 | σ = ∼1 × 10−3 to 10−1 S cm−1, P = 1 × 10−7 to 10−8 cm2 s−1 | 107,108 |
| SO3H–SiO2 | Water uptake ∼32%, σ = ∼0.1–1.2 × 10−3 S cm−1. | 19 | |
| PO3H2–SiO2 | Water uptake ∼123%, σ = ∼5.5 × 10−2 S cm−1, P = ∼1.9 × 10−6 cm2 s−1. | 109 | |
| PEG | SiO2 | σ = ∼1 × 10−4 S cm−1 | 116,117 |
| Heteropolyacid | σ ≥ 1 × 10−2 S cm−1 | 118 | |
| Zr(HPO4)2 | σ = ∼1 × 10−4 S cm−1 at 80 °C | 35 | |
| — | Heteropolyacid, PDMS | σ = ∼7.7 × 10−2 S cm−1 high temperature stability. | 119 |
5.1. Inorganic composites of perfluorinated ionic membranes
The perfluorinated proton exchange membranes were developed by polymerization of monomers which can be converted into cations. These perfluorinated PEMs were commercialized in the trade name Nafion by DuPont, Flemion by Asahi glass and Aciplex by Dow Chemicals. The general structure is shown in Fig. 7. The properties of these long-side-chain perfluorinated PEMs (e.g.Nafion/Flemion/Aciplex) and the short-side-chain perfluorinated PEMs (e.g. Dow) are as follows: (a) EW range = 800–1500; (b) conductivity = 0.20–0.05 S cm−2 (for example, conductivity 1100 EW = 0.1 S cm−2 and conductivity 850 EW = 0.15 S cm−2).20 Morphologically they consist of clusters of long sulfonate-terminated side chains in a perfluorocarbon matrix that has a degree of ubiquitous crystallinity. These Quasi-order crystallinity gives d spacing around 35–50 Å, as evidenced by presence of single broad peak in small angle X-ray scattering (SAXS).45,46 This spacing forms the clusters of solvent molecules water and exhibit good conductivity at room temp. But as the temperature increases, the quasi-order crystallinity gets disturbed and dehydration results into lower conductivity. However, it is possible to retain quasi-order cryastallinity and hence conductivity by incorporation of crystalline materials such as silica, zirconia, titaniaetc. into the polymer matrix. This will act as a waterstorage medium enabling good proton conductivities even at 145 °C.47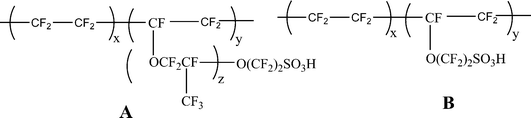
The proton exchange membranes with low methanol permeability were constructed by incorporating Nafion into a covalently cross-linked network composed of 4,4′-methylenedianiline (MDA) and 3-glycidoxypropyltrimethoxysilane (GPTMS).50 The hybrid membrane was prepared by acid hydrolysis of GPTMS in appropriate amount of ethanol. The robust framework with full of covalently bonded silica showed both good proton conductivity (i.e., σ = 3.4 × 10−2 S cm−1) and ultralow methanol permeability (i.e. P = 1.1 × 10−8 cm2 s−1) compare to bare Nafion-117 (σ = 4.5 × 10−2 S cm−1 and P = 2.2 × 10−6 cm2 s−1) under the same experimental conditions.
The hygroscopic silicananoparticles used in most of references are single functional i.e. hydrophilic even though nanoparticles that are both hydrophilic and proton conductive (i.e. bifunctional) as shown in Fig. 9 would have been preferred. Wang et al. and Liet al.51–53 made an attempt to developed functional silicananoparticles followed by its composite with Nafion (E.W. = 1100 g mol−1 SO3H). In the synthetic protocol, sulfonation of silica precursor, phenethyltrimethoxysilane (PETMS) was carried out with chlorosulfonic acid. A stoichiometric amount of chlorosulfonic acid was drop wise added into PETMS under rigorous stirring and kept at room temperature for 2 to 3 h to ensure complete sulfonation of phenethylgroups. After sulfonation, measured amounts of absolute ethanol and deionized (D.I.) water were added for the hydrolysis condensation of sulfonated PETMS. Hydrolysis of sulfonated PETMS was carried out under self-acid-catalysis of the sulfonic groups under stirring for 1 day. Then finally obtained sol was mixed to appropriate amount of Nafion solution in ethanol to give composite membranes with specific composition. The resulting membranes shows about 2.7–5.8 times higher conductivity than bare Nafionmembrane with good dimensional stability. Also these composite membranes shows better single cell performance and low methanol permeability in fuelcell at temperature rang 25–75 °C. The bifunctional silicananoparticles developed into pre-swelled Nafionmembrane by hydrolysis co-condensation of TEOS and γ-propyl mercaptotriethoxysilane (MPTMS) followed by oxidation of mercaptogroup enhances water sorption and proton conductivity than unmodified Nafion.54,55 A key parameter governing the performance of these Nafion/SiO2 hybrid membranes is the MPTMS/TEOS ratio used for the formation of the functionalized silica domains. The hybrid membrane obtained by a higher ratio MPTMS/TEOS exhibit intrinsically higher activation energy for the proton transport at high water activity and an enhanced proton conductivity in the range of temperature 40–70 °C at lower water activity (0.4–0.5).
 | ||
| Fig. 9 Common non functional and functional silica materials used in organic-inorganic composite proton exchange membrane preparation. | ||
A new approach for synthesizing low-cost perfluorinated organic-inorganic composite PEMs based on PVDF and silica with surface-anchored sulfonic acidgroups has been reported by Duvdevani et al.56 The membrane was thermally stable up to 250 °C. Preliminary tests on fuelcell showed cell resistance with a non-optimized membrane in the range of 3 Ω cm−2 and maximum power density of 32 mW cm−2 at 70 °C. The crossover-current density for a 100 μm thick membrane was measured in 1M methanol at 80 °C and found to be 110 mA cm−2.
 | ||
| Fig. 10 A schematic presentation of the modification of zirconia. | ||
Sulfated zirconia (S–ZrO2), a solid state super-acid, exhibits a Hammett acid strength H0 of −16.03, and was recognized as the strongest solid acid.59 Hara and Miyayama studied the proton conductivity of S–ZrO2 prepared by three different methods and found that it had a high conductivity of 2.3 × 10−1 S cm−1 at 105–135 °C.60 Datta and co-workers61 had synthesized ex situ and in situ S–ZrO2–NafionPEMs and evaluated their performance for higher temperature/lower RH operation. The promising potential of the S–ZrO2–Nafion composite showed improved hydration as well as conductivity at higher temperature and lower RH conditions. The polarization and power density curves of the single cells with Nafion 112, recast Nafionmembrane and 15 wt% S–ZrO2–Nafion composite membrane on H2/O2 at 80 °C (gases saturated at 80 °C, RH ≈ l) and 120 °C (gases saturated at 110 °C, RH ≈ 0.6) has been reported62 with the inlet pressure of PH2 = PO2 = 0.2 MPa. The result suggested that when operated at normal operational temperature, the cell performance with 15 wt% S–ZrO2/Nafion composite membrane reaches 1.35 W cm−2, which is superior to the single cell performance of Nafion 112 (1.22 W cm−2) and recast Nafion (1.28 W cm−2). They have the best performance of about 0.99 W cm−2 at 120 °C for S–ZrO2/Nafion composite membrane compare to bare Nafion-112 (0.75 W cm−2) and bare recast Nafionmembrane (0.72 W cm−2).
Zirconium phosphate (ZrP) is an inorganic proton conductor and highly hygroscopic insoluble solid63 and discussed in section 3.2. It has been shown, for example, that zirconium phosphate glasses prepared through a sol–gel route have a proton conductivity of 10−2 S cm−1 at room temperature under conditions of full humidification.64 It has been electrochemically65 and chemically66 precipitated in situ in the pores of a per-fluorinated ionomermembrane for PEFC applications. The composite membranes can be prepared by incorporating the zirconium phosphate into the membrane using the procedure described by Grot and Rajendra66 or by recasting from a Nafion solution with zirconium ions as described by Savadogo.12Zirconium phosphate was incorporated into Nafionvia an exchange reaction involving Zr+4 ions followed by precipitation of zirconium phosphate after immersion of membrane in a phosphoric acid solution. The usual preparation sequence given by Savadogo12 is as follows: (i) the membranes are swollen in a 1:1 vol methanol–water solution; (ii) dipped in zirconylchloride; (iii) rinsed and placed in a 1M phosphoric acid solution. This process leads to an insoluble zirconium phosphate in the nano-pores of the membrane. The properties of these membranes can be summarized as follows: (i) a composite membrane based on a commercial Nafion-115membrane exhibited a performance of 1 A cm−2 at 0.45 V, whereas an unmodified membrane exhibited a performance of 0.25 A cm−2 for an H2/O2PEMFC at 130 °C and a pressure of 3 bars. With the same operating conditions, the cell performance was 1.5 A cm−2 at 0.45 V for the composite recast membrane. The composite recast membranes showed a stable behavior over time when maintained at 130 °C, while non-composite membranes show irreversible degradation. The proton conductivity was found to be similar for pure Nafion and Nafion-ZrP. The activation energy associated with the proton conduction was found to be similar for pure Nafion (9.34 kJ mol−1) and composite Nafion-ZrP (9.82 kJ mol−1). This indicates that the presence of zirconium phosphate does not significantly change the proton conduction mechanism in a well-hydrated membrane. The slight improvement was attributable either to the hydroscopy of the zirconium phosphate or to the reduction in the number of free spaces in the nano-pores, promoting capillary condensation and thus waterretention and proton conductivity.63 Modified Nafion structures based on surface modification (surface cross-linking, etc.) and/or bulk modification (Nafion-inorganic hybrid structures or barrier layer laminated composite structures) are under development at DuPont12 for DMFC applications. The recasting process of the DuPont composite membrane is not known, but the above results on Nafion-ZrP composite membrane may induce some comments for further developments: (i) determination of the stability of the composite membrane with time, and a study of whether or not the dopant is retained in the composite membrane over an extended period; (ii) the stability of cell performance based on the composite membrane, in particular at cell temperatures higher than those of the humidifiers; (iii) determination of the fuel-cell exhaust, and the anode and cathode catalyst compositions, and of whether or not they are contaminated by the dopant; and (iv) monitoring of the conductivity of the composite membrane with time. In the other efforts Nafion-ZrP modified with room temperature ionic liquids has been used for the conductivity improvement but they have lower conductivity and have been explained in the review in ref. 12.
Zirconium sulfophenyl (ZrSPP) and zirconium alkyl sulfophenyl phosphonates36–38,67 showed increased conductivity dependent on relative humidity and discussed in section 3.2. They made efforts to make composites of these materials with the Nafion to increase the conductivity at high temperatures above 100 °C by dispersing the particles of ZrSPP synthesized from ZrOCl2 and m-sulfophenyl phosphonic acid. The composite membranes showed 700 mA cm−2 current density in fuelcell which was four times better than recast Nafion at 100 °C.
000 Pa) < Nafion-STA (14000 Pa) < Nafion-PMA (8000 Pa) < Nafion-PTA (3000 Pa), while their deformation (εmax) changes in the order of: Nafion-STA (45%) < Nafion-PMA (70%) < Nafion-PTA (170%) < Nafion-117 (384%). The voltage–current characteristics of polymer electrolyte fuelcells (PEFC) were determined.12 From the cell voltage–current plots, the current density at 0.600 V of PEMFCs based on various membranes varies in the order of: Nafion-117 (640 mA cm−2) < Nafion-STA (695 mA cm−2) < Nafion-PTA (810 mA cm−2) < Nafion-PMA (940 mAcm−2).
Sulfonic acid functionalized heteropolyacid-SiO2nanoparticles were synthesized by grafting and oxidizing of a thio-silane compound with a micro-emulsion technique.70Sodium bis(2-ethylhexyl)sulfosuccinate and heteropoly acid PTA was dissolved in water and mixed with cyclohexane to form a well-defined micro-emulsion phase. Tetraethoxysilane (TEOS) was dropped into the microemulsion phase and stirred at room temperature for 12 h. The resulted PTA-SiO2nanoparticles were collected by centrifugation and repeatedly rinsing with acetone. For the improvement of hydrophilic and proton conducting properties, sulfonic acidgroups were introduced onto the surface of the silica. It was carried by grafting and oxidizing of thio-silane compound.19,713-Mercaptopropyltrimethoxysilane (MPTMS) was dissolved in toluene and refluxed with PTA-SiO2nanoparticles for 24 h at 120 °C. After reflux, the nanoparticles were washed with dry toluene and dried for several hours at 120 °C. The oxidation of thiol group to sulfonic acid was carried out by treating the dried thiol-modified nanoparticles with aqueous H2O2 (30 wt%). The suspension was stirred at room temperature for 12 h. After the oxidation treatment, the resulting solution was centrifuged and washed separately with water and ethanol. The wet nanoparticles were suspended in 1 M H2SO4 solution for 4 h. Finally washed several times with water and ethanol and dried at 80 °C under vacuum. The atomic ratio of S/Si which was calculated by XPS measurement was found 0.105.70,71 The composite membranes were prepared by suspending functionalized PTA-SiO2 in 5 wt% Nafion solution and dried at 120 °C. The function of the sulfonic acid-functionalized PTA-SiO2nanoparticles was to provide a protoncarrier and act as a water reservoir in composite membrane at elevated temperature. The DMFC performance of the composite membrane was evaluated at temperatures from 80 to 200 °C. The power density was 33 mW cm−2 at 80 °C, 39 mW cm−2 at 160 °C and 44 mW cm−2 at 200 °C, respectively.
Malers et al.42 prepared composite membranes from polyvinylidenedifluoride-hexafluoropropylene (PVDF-HFP) and different heteropoly acid with weight ratio 1:1. For the first time they have presented complete polarization curves for a number of heteropoly acids (HPAs), H3PW12O40, H3P2W18O62, H6P2W21O71, and H6As2W21O69 as the only proton conducting component and hence showed promising candidates for solid acid fuelcell performance at >200 °C. The high proton conductivities reported at RT are demonstrated in fuelcells using HPA/PVDF-HFP composites with limiting current densities as high as 1.6 A cm−2 using dry O2 and H2. Moderate fuelcell activity was demonstrated for H3PW18O62 at 120 °C and 25% RH. Unfortunately all of the materials studied were somewhat porous and the open circuit potentials observed were somewhat low. They were also able to show that a HPAfuelcell could be shorted by reduction of the HPA to a heteropoly blue under exceptional circumstances. They have demonstrated higher proton conductivities than those reported in the literature and can be translated in to impressive currents in PEMfuelcells at RT with no external humidification. However, HPAs on their own do not have sufficient mobility of their protons for fuelcell temperature operation at 100 °C. Interestingly these materials may have application in high temperature PEMs at 200 °C. Although a small subset of the many structures available has been investigated. One HPA, H3P2W18O62, did show moderate activity at 120 °C with the application of some humidity. All of the membranes studied were certainly not optimized and suffered from porosity issues.
:methanol permeability).
Lin et al.73 studied clay composite membranes developed from montmorillonite (MMT) and Nafion. The modification of the montmorillonite with poly(oxyproplene)-backboned quaternary ammonium salts was utilized to improve the compatibility with Nafion. They evaluated the membrane performance in terms of water uptake, ion exchange capacity (IEC), methanol permeability, proton conductivity, and cell performance. They observed slight decrease in the water content, ion-exchange capacity and proton conductivity; but about 40% decrease in methanol permeability for the 5% MMT-POP-400-Nafion composite membrane. The same composite membrane shows the current density of 56 mA cm−2 and power density of 13.3 mW cm−2 in single cell performance at a potential of 0.2 V. The composite membrane from sulfonic acid modified clay: laponite and Nafion was studied by Bebin et al.74 The clay powder was modified by helium plasma under a pressure of 0.1 mbar using a 13.56 MHz frequency for 2 min. The activated clay was immediately immersed in a 3% (w/v) solution of p-styrene sulfonate in DMF and refluxed for 48 h. The grafted clay washed with DMF to remove unreacted monomers and oligomer moieties. The composite membranes were prepared by a recasting Nafion solution mixed with modified Laponite particles. The measured proton conductivity of the composite membrane was higher than that of the commercial Nafion in the temperature ranges 20–95 °C with a more significant difference at low relative humidity. At 80 °C and 0.6 V optimized composite membrane showed 720 mA cm−2 current density, which was corresponds to 20% improvement in power density (430 mW cm−2). At higher temperature, 120 °C with dilute cathode gas, air at lower pressure 3 bars; induce a drastic dehydration effect in the cell. The sensitivity of the membrane to humidity was easily observed with these cell conditions. With these fuelcell conditions Nafionmembrane could deliver 390 mA cm−2 of current at 0.6 V while the composite reach 500 mA cm−2, a 30% improvement in power density.
5.2. Inorganic composites of poly(arylene ether)s
Poly(arylene ether)s are well known engineering materials and thought to be an alternative candidate to perfluorinated polymers in high performance PEMs because of their availability, processability, options for chemical modification, and anticipated stability in a fuelcell environment. Specifically, poly(arylene ether) materials such as poly(arylene ether ether ketone) (PEEK), poly(arylene ether sulfone), and their derivatives are the focus of investigations. The synthesis of these materials has been widely reported.10,75 The poly(ether ketone) candidates from the polyarylene family differentiate through varying sequences of ether (E) and ketone (K) units to give ether rich: PEEK (Victrex® PEEK, GatoneTM PEEK, Gharda Chemicals) and PEEKK (Hostatec®), or ketone-rich semicrystalline thermoplastic polymers: PEK (Amoco Kadel®, FuMA-Tech) and PEKEKK, Ultrapek®, BASF)44 (Fig. 11). Oxidative and hydrolytic stability increase with increasing proportion of ketone segments as in case of PEKK (Declar®, Du Pont), and undergoes lower weight loss at ∼400 °C under water/oxygen than either PEKEKK or PEEK. In recent years, sulfonated poly(ether ketone) has been extensively studied than any other non-fluorinated system, with contributions from Jones, Kreuer, Kerres, Bauer, Rozière, and their co-workers and others,3,10–12,47,76,78 to studies ranging from modeling of the microstructure, proton transport properties, application in low- and medium-temperature PEMFC and DMFC, and as a component of polymer blend and hybrid inorganic–organic membranes.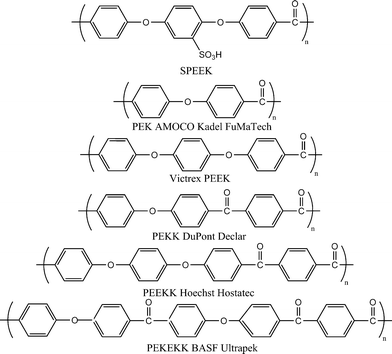 | ||
| Fig. 11 Structure of sulfonated poly(ether ether ketone) and representative membranes of poly(arylene ether ketone) family. | ||
Direct methanolfuelcell performance has been reported using hydrogen as fuel for a sulfonated PEEK membrane with an ion exchange capacity of 1.6 meq g−1 and 18 μm thickness.76 They have obtained 0.5 mA cm−2 current density with oxygen and air at voltages of 0.8 V and 0.72 V respectively, and temperature of 90 °C. With SPEEK membranes of 70 μm thickness they reported higher performance at 85 °C than Nafion-115 under the same test conditions. Patent reports claim 4000 h of functioning for SPEEK with an IEC of 1.47 meq g−1 at a modest cell temperature of 50 °C,77 giving a maximum power density of 0.386 W cm−2 at 0.52 V, whereas at 90 °C at constant current density of 0.5 mA cm−2SPEEK has attained 1000 h of functioning without failure.44 At higher temperatures, poly(arylene ether ketones) suffers from dehydration. It exhibits less pronounced hydrophobic/hydrophilic separation and different morphological behavior.78 This results in significant reduction in electro-osmotic drag and proton conductivity. Authors worldwide tried to over come these problems by making composites with charged and uncharged inorganic moieties.
Guiver and co-workers79 developed a composite membrane from sulfonated poly(arylene etheretherketoneketone) (SPAEEKK) with pendant sulfonic acidgroups. A series of hybrid membranes made from SPAEEKK incorporating different amounts of silicananoparticles were reported using the sol–gel reaction with tetraethyl orthosilicate (TEOS). The performances of hybrid membranes were investigated in doped and un-doped H3PO4 states. The H3PO4 doped SPAEEKK/silica hybrid membranes showed an improvement in membrane properties. The improvement is claimed on the basis of uniform distribution of nano-sized silica particles and H3PO4 into the membrane matrix. The silica has an effect on the state of water in the membranes. The ≡SiOHgroups of silica provide strong hydrogen bonding sites and increases contents of the bound to free water ratio into the membrane matrix. Further it retains water molecules at elevated temperatures and thus maintains proton conductivity. The proton conductivities of un-doped and doped H3PO4 hybrid membranes were in the range from 0.02 to 0.082 S cm−1 and from 0.024 to 0.097 S cm−1, respectively. The methanol permeabilities ranged between 1.4 × 10−7 and 6.9 × 10−7 cm2 s−1. In comparison with Nafion-117 (proton conductivity 0.1 S cm−1 and methanol permeability 7.7 × 10−6 cm2 s−1 at 80 °C), an H3PO4 doped SPAEEKK/silica hybrid membrane had 0.078 S cm−1proton conductivity and 6.06 × 10−7 cm2s−1 methanol permeability at 80 °C. The methanol permeability is 92.1% lower than Nafion-117. At 30 °C, the conductivity and permeability values of Nafion-117 were 0.078 S cm−1 and 2.52 × 10−6 cm2 s−1, respectively; while H3PO4 doped SPAEEKK/silica hybrid membrane had values of 0.032 S cm−1 and 2.02 × 10−7 cm2 s−1, with permeability being 92% lower. It shows H3PO4 doped SPAEEKK/silica as potential candidates for polymer electrolytes for DMFC and PEMFC applications.
The nanocomposites prepared from aminophenyl functionalized silica and sulfonated poly(ether etherketone) SPEEK showed conductivity higher than 0.01 S cm−1 at 25 °C and 100% RH. The improvement in proton transport was claimed to be caused by the presence of a membrane micro-structure and extensive silica-membrane interfacial regions.80 The existence of the interpenetration of the SPEEK polymer and functionalized aminophenyl silica networks with a domain size of about 4 nm indicates possible confinement of the polymer within the composite membrane. It was also claimed that nano-phase composite membranes up to 50 wt% silica can be obtained with SPEEK cast in dimethylsulfoxide. Composite membranes cast in lower dielectric constants such as dimethylformamide are translucent and more brittle.
Moeller and co-workers81 prepared composite polyelectrolytemembranes from solutions of sulfonated poly(ether ether ketone) (SPEEK) (DS 64%) and polyethoxysiloxane (PEOS) with N-(3-triethoxysilylpropyl)-4,5-dihydroimidazole as the compatiblizer in dimethylacetamide, following the concept of a semi-interpenetrating network. The in situ transformation of PEOS into SiO2 was done in a “water free” process. The morphology of the films obtained was controlled by a phase segregation process, determined by the rate of evaporation of the solvent and by the transformation of PEOS into SiO2-particles in the presence of suitable catalyst. By addition of N-(3-triethoxysilylpropyl)-4,5-dihydroimidazole they showed a dramatic reduction in the size of SiO2 particles. Proton conductivity of resultant composite membranes was lower than the Nafion-115. Nunes et al.82 showed a 13% reduction in methanol permeability and a 40% reduction in water flux with decrease in membrane conductance using composite membranes from SPEK (sulfonated poly(ether ketone)) and SPEEK. They prepared composites with N-(3-triethoxysilylpropyl)-4,5-dihydroimidazole and TEOS. Part of the membranes were prepared by first reacting the polymer with 1,1′-carbonyl-diimidazole (CDI) and aminopropyl triethoxysilane. Some other part of CDI was reacted with part of the sulfonic groups of the polymer, followed by addition of aminopropylsilane (APS), leading to alkoxy silanegroups covalently linked to the polymer.
It was claimed,83 that a composite membrane prepared from the derivative of poly(arylene ketone) family, i.e. poly(phthalazinone ether ketone) (PPEK) and SO3H-functionalized SiO2 showed 3.6 fold increase in conductivity and low methanol permeability compared to bare sulfonated PPEK. The acid functionlized silica was prepared from commercial available silica particles of 10–20 nm size and glycidyl phenyl ether (GPE).84
Well ordered silica materials such as Montmorillonite, Laponite and MCM-41, are familiar polymer filling materials. When polymers are mixed with these natural or synthetic inorganic materials, their properties are considerably improved. These inorganic materials have characteristics properties of improving mechanical strength, impact resistance and reducing permeability of gases and moisture.85 The PEMs composite membrane prepared from SPEEK and Montmorillonite by simple solution mixing and casting showed reduction in proton conductivity and methanol permeability. The organically modified montmorillonite showed a dramatic decrease in methanol permeability with retention of proton conductivity of 1.2 × 10−2 S cm−1 at 90 °C. This value is close to Nafion-115.86 A 370 mAcm−2 current density at 0.6 V for 10% Laponite/SPEEK composite membrane was reported. They performed fuelcell test for 100 h at 70 °C with same current density.85 Further increase in fuelcell performance was reported by modification of laponite with imidazole glycidoxypropyl triethoxysilane (IGPTES) and 3-2-imidazolin-1-yl-propyltrimethoxysilane (IPTMS) to decrease methanol permeability without losing membrane conductivity.87 The same type of results were shown for an MCM-41/SPEEK composite membrane by same author.
A series of composite membranes has been reported for incorporation of HPA: tungstophosphoric acid, its disodium salt and molybdophosphoric acid into a partially sulfonated PEEK and its derivatives.88–91 These membranes exhibited a rather high conductivity of 10−2 S cm−1 at ambient temperature, and up to a maximum of about 10−1 S cm−1 above 100 °C. It was also mentioned that there was an increase in thermal stability of composite membranes by DSC (differential scanning calorimeter) studies due to intermolecular interaction between SPEEK and HPA. The main disadvantage of these HPA-SPEEK composite membranes is the dissolution of HPA in the water formed by the electrochemical process of current generation with a consequent decay of cell performance.92
Many workers devoted research to overcoming the polyelectrolyte dissolution problem by entrapment of HPA into the host material, mostly silica oxide networks. Usually, the heteropolyacid was fixed through covalent or coulombic interactions. This involved the reaction between protons and SiO2, resulting in a decrease in the acid strength. The decrease in acid strength was described during investigations of 31P MAS NMR, which shows a chemical interaction between the protons of HPA and SiO2.92 There are two different approaches for covalent or coulombic entrapment of HPA into the polymeric matrix: the in situ generation of an oxide network by the sol–gel process from alkoxysilanes followed by covalent bonding of HPA with silica matrix and the modification of the anion structure of the heteropolyacid.93 The covalent bonding between HPA and inorganic phase was carried out using organosilyl derivatives of the divacant tungstosilicate [γ-SiW10O36]8− and GPTS (3-glycidoxypropyltrimethoxysilane). The presence of nucleophilic surface oxygen atoms at vacant site on Lacunary heteropolyacids [γ-SiW10O36]8− forms an effective organic-inorganic material, which allows covalent grafting of electrophilic groups. The organic functionality was linked to the polyanion surface by E–O–W bridges (E: Si(IV), P(V), Sn(IV), Ge(IV), or Ti(IV)), resulting in saturation of heteropolyanion surface. In this way organic-inorganic composite materials have been synthesized by addition of reactive organic groups (vinyl, allyl, methacryl, and styryl) to the lacunary heteropolyacids, [γ-SiW10O36]8− and [α-SiW11O396]8−.94 Nunes and co-workers92 introduced GPTS into the anion structure of lacunary heteropolyacid enabling its attachment to a host material, by an epoxy ring opening reaction with appropriate functional groups present in the surface of the host material, without involving the protons and therefore without affecting the acidity of the heteropolyacid. He got the membrane conductance to of the same order as the plane membrane with good bleeding stability in aqueous methanol solution. To avoid the problem of bleeding, a composite membrane of HPA loaded Y-zeolite and SPEEK has been reported95 and found to be suitable for PEM applications.
The conductivity of the SPEEK increases with increase in degree of sulfonation; its mechanical properties show a parallel progressive deterioration leading to a downfall of conductivity. To overcome this problem, TiO2 was used as filler in 90% degree of sulfonated PEEK. Because of rapid hydrolysis of Ti(OBu)4 in aqueous condition, the composites were prepared under non hydrolytic condition using two routes. In one route Ti(OBu)4 (1.11 mmol, 0.33 mL) and pyridine (12 mmol, 1 mL) were added. And in the second route, a sol was prepared by adding Ti(OBu)4 (1.11 mmol, 0.33 mL) and 2,4-pentandione (acac, 2.4 mmol, 0.22 mL) in DMAc (3 mL) at 0 °C. The resulting solutions were added to the SPEEK solution (1.68 g, 4.58 meq) and stirred at room temperature for 1 h. The solvent was evaporated to 5 mL at 120 °C, and then cast onto a Teflon plate and heated until dry. After cooling to room temperature, the resulting membranes were peeled off and dried at 100 °C for 10 h, then further dried under vacuum at 80 °C for 2 d for complete solvent removal. Before use, the membranes were treated with 5 M H2SO4. The reaction scheme is shown in Fig. 12. The prepared membranes showed a conductivity of about 5.8 × 10−2 S cm−1 at 120 °C in fully hydrated conditions.96,97 The methanol permeability of Nafion-115membrane is 2.32 × 10−6 cm2 s−1 at room temperature while it was between 0.5 × 10−6 and 2 × 10−6 cm2 s−1 for composite membrane with 65% sulfonated PEEK and TiO2,97 which is considerably smaller than that of Nafion-115membrane. The same membranes having 5wt% TiO2 have been reported the maximum fuelcell performance.
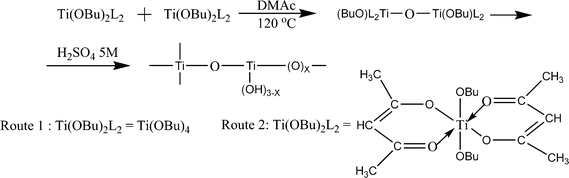 | ||
| Fig. 12 The reaction scheme for the preparation of TiO2. | ||
The SPEEK/BPO4 composite membranes were prepared from tripropylborate (C3H7O)3B and phosphoric acid (H3PO4), in equal molar proportion by a sol–gel method.98 The weight percentage of BPO4 in the composite membranes was determined by assuming total conversion of precursors to BPO4. Initially SPEEK was dissolved in N,N-dimethylacetamide (DMAc) at room temperature to prepare a 10 wt% solution and filtered. The requisite quantity of H3PO4 was added to the polymer solution and stirred with a magnetic stirrer for 10 min. Then, (C3H7O)3B was added and further stirred for 10 min. The resulting solution was degassed under vacuum and cast onto a flat-bottom Petri dish for membrane formation. The composite membranes showed six fold increases in conductivity with 30% loading of BPO4.
5.3. Inorganic composites of poly(arylene ether sulfone)s
Poly(arylene ether sulfone)s are another engineering material used in proton exchange membranesfuelcell. Los Alamos National Laboratory and Virginia Polytechnic Institute created the biphenol based poly(arylene ether sulfone) (PES) and a poly(arylene ether benzonitrile) (PEBN) membranes. It was reported99,100 that for these membraneswater uptake correlated proportionally with ion exchange capacity (IEC) and it is decreasing in the order of PES, PEBN and Nafion®. Nevertheless, methanol permeability, which is typically proportional to water uptake and IEC, was two times higher for Nafion® than the other two membranes.The MCO of PES and PEBN are nearly equal (81 × 10−8 and 87 × 10−8 cm2 cm−1) with 0.5 M methanol at T = 30 °C. At a slightly higher (10%) membrane resistance, the MCO is a factor of 2 lower than for Nafion® (167 × 10−8 cm2 cm−1). The combination of MCO and resistance properties results in a 50% improvement in selectivity, regardless of membrane thickness. The DMFC with this membrane has shown a performance of j = 200 mA cm−2 (compared to 150 mA cm−2 for Nafion®) at 0.5 V, T = 80 °C and ambient air pressure.101 Another prospective DMFCmembrane (originally developed for PEMFC) composed of modified poly(ether sulfone) (PES) is sulfonated poly(arylene thioether sulfone) (sPTES).102 This membrane was synthesized at the Air Force Research Laboratory (AFRL) and had the following advantages: high chemical and thermal stability; higher proton conductivity (>100 mS cm−1) than Nafion® 112 (80 mS cm−1) at T = 65 °C and RH = 85%; comparable performance to Nafion® in DMFC applications; specific area resistance (ASR) of 0.13 cm−2 at 80 °C and H2/air feed (compared to 0.18 S cm2 for Nafion®) retains large amounts of water over a wide temperature range; cheaper than Nafion®
There are reports of better performances of poly(arylene ether sulfone)s membrane in fuelcell, however their chemical stability is always questionable because of oxidative degradation of nonfluorinated backbone.103 The generation of hydroxyl and hydroperoxyl radicals in either anode or cathode after a 2-electron reduction reaction between H2 and O2 cross over the PEM to opposite electrode responsible for chemical stability of membranes.104 In the perfluorinated backbone, C–F bond energy is high (485 kJ mol−1). The fluorine atoms wrapped the C–C main chain closely to avoid attack from oxidative free radicals. This is the cause of that the film has good oxidation resistance. However, in the non-fluoridePEMs, the C–H bond dissociation enthalpy is lower and that oxidative degradation occurred easily. For the non-fluorideproton exchange membrane, oxidative degradation problem has become more prominent.
Xing et al.103 developed composite membranes from a simple dispersion of Ag–SiO2 particles in sulfonated poly (biphenylethersulfone) (SPSU-BP) to increase chemical stability against oxidative degradation by catalyticdecomposition of generated H2O2. He observed that the initial limiting currents of the two membranes, with Ag–SiO2 and without Ag–SiO2, were very similar, about 5 mA. But after 22 h the former membrane showed high limiting current of about 70 mA. Whereas, the latter membrane, even after 40 h, showed a lower limiting current of about 10 mA.
The composites of the bifunctional silicas are always preferred in fuel call applications.23 The composite membranes prepared by the methylphosphorylation of aminopropyl silane and sulfonated poly(ether sulfone) showed an increase in proton conductivity and decrease in methanol permeability compared to Nafion-117.105 It was also reported that the current–voltage polarization curves were in good agreement with Nafion-117.
The chemical and hydrolytic stability of amorphous silica during fuelcell operation is still questionable.106 The incorporation of zirconia and zirconium phosphate into hydrocarbon membranes are, in practice, composite membranes prepared from zirconium phosphate and sulfonated poly(ether sulfone) which showed an increase in proton conductivity at higher temperatures. But there are no such reports available on the fuelcell performance of these composite membranes and hence it is the subject of intensive study.
5.4 Inorganic composites of polybenzimidazole
Polybenzimidazole (PBI) based proton exchange membranes are successful hydrocarbon membranes for fuelcell applications. PBI is an aromatic heterocyclic basic polymer (pKa = 5.5) which can be complexed with strong acids or very strong bases. Bare PBI is an electronic and ionic insulator but becomes a very good ionic conductor after doped with acids in the proper conditions and remains an electronic insulator.12,99 The composite of PBI–phosphoric acid has been extensively studied at Case Western University, USA for high temperature DMFC applications.12,99 It was observed that the in situ casted PBI/phosphoric acid composite membrane exhibited better performance in DMFC than the composite membrane prepared by immersing pre-formed PBI film into the phosphoric acid solution. Depending on the preparation method of PBI, the properties of the composite membranes varies. But in general it has high proton conductivity, low electro-osmotic drag, capable of operating at high temperature (T = 200 °C) and low gas humidification, low MCO and low cost in comparison with Nafion/perfluorinated ionomers. The major disadvantage is leaching of the low molecular weight acid in hot methanol solutions. These problems were solved by the addition of high molecular weight phosphotungstic acid as a replacement of the low molecular weight acid. At T > 130 °C the conductivity of the doped PBImembrane is similar to Nafion® (30 mS cm−1 at 130 °C and 80 mS cm−1 at 200 °C).5.5 Inorganic composites of poly(vinyl alcohol)
Poly(vinyl alcohol) (PVA) is a synthetic polymer having excellent film forming, emulsifying and adhesive properties accompanied with good resistant to oil, grease and organic solvents. It has high tensile strength and flexibility. Its melting point, 180–230 °C depending on the degree of hydrolysis makes it a promising candidate in DMFC applications. The presence of reactive –OH group further adds options of chemical modification to tailor its properties.The sulfonic acid functionalized organic–inorganic hybrids based on (PVA)/SiO2/sulfosuccinic (SSA) acid were reported.107 In the hybrid membranes, sulfosuccinic acid served as both sulfonating as well as cross-linking agent. The proton conductivity and methanol permeability of the hybrid membranes were studied with changing SSA content from 5 to 25 wt% and found very much depends on SSA contain They found proton conductivities in the range of 10−3 to 10−2 S cm−1, and the methanol permeability 10−8 and 10−7 cm2 s−1 range. They claimed the decrease in methanol permeability in the presence of silica particles. Similarly, composite membranes of PVA/SiO2 with copolymer of 2-acrylamido-2-methyl-propanesulfonic acid (AMPS) and 2-hydroxyethyl methacrylate (HEMA) crosslinked by poly(ethylene glycol) dimethacrylate (PEGDMA) has been reported.108 They found proton conductivities of 0.02–0.11 S cm−1 with significantly lower fuel permeabilities than that of Nafion. Sulfonic acid functionalized silica/PVA composite membranes have been reported, created by a sol–gel method, where –SO3H groups were introduced by oxidation of –SH group present in mercaptopropylmethyldimethoxysilane (MPDMS).19 The resulting membrane had good thermal and electrochemical properties. The phosphonic acid functionalized silica/PVA composite proton exchange membrane has been reported. The phosphonic acid functionalisation was carried by phosphomethylation of aminopropyltriethoxysilane in aqueous medium.109 The resulting composite membrane had an excellent methanol barrier capability with good hydrophilicity and proton conductivity at higher temperatures. From selectivity parameter, they found, 50% silica loaded PVA composite with 3 h of phosphorylation resulted best proton-exchange membrane. It was about 20% more suitability in comparison to Nafion 117 membrane for direct methanolfuelcell applications. Apart from the silica composites, heteropolyacid and zirconium phosphate composite membranes have been reported with good proton conductivity and lower methanol permeability.110,111
5.6 Miscellaneous organic-inorganic composite ionic materials
The classic approach for developing new types of organic-inorganic composite PEM materials is sol–gel chemistry. The commonly used precursors in sol–gel chemistry are tetramethoxysilane (TMOS) and Tetraethoxysilane (TEOS).112–114 The introduction of organic functional groups into these inorganic precursors leads to organically modified sol–gel materials, called ormocers or ormosils or ceramers. The main advantages to have organic groups in sol–gel materials are: (i) during the preparation of purely inorganic materials, they can control the reaction rates of the reactants, the rheology of the sols, or the homogeneity and microstructure of the derived gels. (ii) The organic groups can be modified with different functional groups; acid functionalization is very common in fuelcell applications. The resulting final materials are composed of inorganic (oxidic) structures cross-linked or substituted by functionalized organic groups. For fuelcell applications, these hybrid materials can be prepared by two approaches. The first one is the physical mixing of acid functionalized molecules with an ormosil matrix and second one is the covalent linking of acid functionalized molecules with an ormosil matrix. The latter approach requires precursors in which the organic group is bonded to the oxide-forming element in a hydrolytically stable way.115Honma et al.116,117 did pioneering work in organic-inorganic composite proton exchange membrane development. In his initial approach the poly(ethylene oxide) (PEO) was end capped with triethoxysilane. PEOs of different molecular weights were mixed with silica precursors such as 3-isocyanatopropyltriethoxy silane in N2 atmosphere at 70 °C for 5 days to give PEO with end capped urethane linked silica as shown in Fig. 13. By this method, they have produced a flexible, free standing membrane. The proton conductivity was obtained by dispersing monododecylphosphate into these ormosil matrixes. The obtained membrane showed good proton conductivity of about 10−4 S cm−1 at higher temperatures. In other efforts117 he controlled the hydrophobic/hydrophilic characteristic of the membrane by organically bridged tri-alkoxysilane (bridged polysilsesquioxanes) precursors. They were used temperature tolerant polymers such as polyethers, alkylenes and aromatics to bridge alkoxysilane functional groups for organic-inorganic hybrid monomers. The proton conductivity was obtained by addition of heteropolyacids such as PWA into the sol. Fig. 14 shows the molecular structure of the organically bridged polysilsesquioxane monomers used in their work, where bifunctional polyalkylenes (ethane, hexane, octane, decaneetc., (CH2)n: n = 2 to 14) were terminated by alkoxysilanes. The bridged polysilsesquioxanes were synthesized using the three most common methodologies, (1) metallization of aryl, alkyl, or alkynyl precursors, followed by reaction with a tetrafunctional silane, (2) hydrosilylation of dienes (or polyenes) or less commonly diynes, and (3) reaction of a bifunctional organic group with an organotrialkoxysilane bearing a reactive functional group. The bridged polysilsesquioxanes they obtained either by synthesis using hydrosilylation reactions or from commercially (Gelest) available monomers.118 The resulting membranes exhibited large proton conductivities (>10 −2 S cm−1) at high temperatures of up to 160 °C, where the conductivity of the hybrid membrane can be tailored by varying the processing conditions, such as the amount of water in the sol solution and the equivalent weight of PWA incorporated in the macromolecules. The fast ionic conduction in the resulting materials may be based on the synthetic mechanism of amphiphilic macromolecules in the solution. The bridged octane hybrid membrane showed the humidity dependence for the conductivity from 120 to 160 °C, while the conductivity decreases exponentially with the water vapor pressure. The conductivity exceeds 2 × 10 −3 S cm−1 level even at 20% R.H. at 120 °C. Proton conductivity was stable above 100 °C with a small dependence on the R.H. The flexible polymer electrolyte membranes with zirconia, titania and polydimethylsiloxane (PDMS) of different molecular mass: 4500 and 600 have been reported.119 The membranes showed thermal stability and flexibility up to 300 °C proton conductivity of about 7.7 × 10−2 S cm−1 by addition of 12-phosphotungstic acid (PWA).
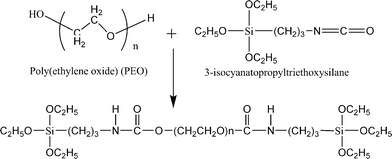 | ||
| Fig. 13 A schematic presentation of the end capped silica.116 | ||
 | ||
| Fig. 14 Structures of different bridged polysilsesquioxanes. | ||
These above discussed ionic inorganic material dispersed membranes have the drawback of leaching of the ionic conducting materials (heteropolyacids) with time. This could be prevented by forming covalent bonds between the ionic conducting material and the inorganic precursors. In the first effort, Honma and co-workers35 developed zirconiamembranes with trimethylene glycol, octamethylene glycol and hexadecanediol by the sol–gel method. The self-assembled hybrid organic-inorganic membranes showed high flexibility, chemical tolerance, and thermal stability, and good mechanical properties up to 325 °C. The maximum proton conductivity of about 7 × 10−4 S cm−1 at 180 °C was obtained after putting the hybrid membrane into 85% H3PO4 at room temperature or boiling at 150 °C. This is due to the formation of a zirconium phosphate type texture in the polymer matrix.
Proton conductive glasses are another type potential ionic materials used in DMFCs.120Protons are highly mobile when they are strongly hydrogen bonded in glasses, and their conductivity is primarily determined by the degree of hydrogen bonding and the concentration of mobile protons in the glass structure. In the case of silicate and phosphate glasses, protons are bound with oxygens to form hydroxylgroups attaching to network forming cations, such as Si4+ and P5+ ions. Comparing silicate glasses with phosphate glasses, the hydroxylgroups in phosphate glasses are strongly hydrogen bonded.121–123 The existence of P2O5 in the glass composition plays a significant role in providing high protonic conduction. The content of protons in glasses decreases with an increase in the sintering temperature of the glasses. Therefore, it is desirable to prepare a glass with a large amount of protons at low temperature, like in a sol–gel process. By a sol–gel process, Wang and Nogami et al.123–125 have independently prepared plates and films of hydrated phosphor–silicate glasses with proton conductivities of about 10−3 S cm−1. However, the gelation time of the glass membrane in their sol–gel process is about 1–6 months. Tung and Hwang120 reported a short time synthetic methodology for inorganic proton conductive electrolytes based on hydrated phosphor–silicate glass by an accelerated sol–gel process with water/vapor management. Due to the formation of the P2O5 and SiO2 network structure, the hydrated phosphor–silicate glass membranes show good thermal stability. They observed two or three kinds of pore sizes in the synthesized glass membranes. Increasing the P2O5 content of the glass membrane leads to a decrease of its major pore size and an increase of its porosity. However, it was observed that the pore size of the glass membrane becomes larger when its P2O5 content was higher than 40%. They also observed conductivity and methanol permeability increase with increasing the content of the P2O5, and interestingly, a maximum selectivity (the ratio of the conductivity to permeability) observed at the 30% P2O5 and 70% SiO2 in glass membrane. From the above discussion it is clear that the membranes with desired properties can be prepared by appropriate rationing of P2O5 and SiO2. But, the main drawback of these proton conducting glasses is their fragile nature, which prevents them from use in a wide range of applications.
6. Concluding remarks and perspective
From the above discussion it is evident that the addition of inorganic materials improves the membrane's thermal, mechanical and chemical stability, ionic conductivity at intermediate temperature with low fuel drag. The improvement in these properties is due to the combined effect of organic and inorganic phases, which are usually bound together with hydrogen bonding and ionic interactions. The addition of acid functionalized and non-functionalized silica material is usual practice for improving fuelcell performance. But the literature is lacking in systematic and comparative research on the effects of degree of crystallinity, size, orientation, and distribution of added silica particles into the membranes. The effect of other metal oxides such as TiO2, ZrO2, etc.has not been extensively studied and needs to explore in details. In composite with metal phosphonates; only zirconium phosphate has been well studied by Alberti and Casciola11 and other metals phosphonates need to be investigated.In the investigation processes, inorganic material should be accommodated in the organic matrix in such a way that it should permit the utilization of maximum elementary functions in a small volume, and hence optimize complementary possibilities and properties of the inorganic and organic components. A very interesting and appealing approach has been proposed by Sanchez and co-workers.126 They prepared transparent hierarchical hybrid functionalized membranes using in situ generation of mesostructured hybrid phases inside a non-porogenic hydrophobic polymeric host matrix by surfactant template method. The formation of hierarchically ordered domains was favored by the hydrophobic character of the non-porogenic polymer, allowing the surfactant molecules to organize in micelles together with the hydrolysis–condensation of inorganic (M(OR)n,M = Si, Ti,…) or hybrid (R–Si(OR)3) precursors. They proposed two different polymers, one highly hydrophobic containing perfluorinated chains, poly(vinylidene fluoride) (PVDF), and a second less hydrophobic, poly(vinyl butyral), as a host organic matrix associated with two different templates, an ionic surfactant, cetyltrimethylammonium bromide (CTAB), and a non-ionic surfactant, C12H25(EO)4 (BRIJ 30). To avoid segregation and sedimentation of the inorganic network during the concentration step, the synthesis requires an adequate solvent to be selected with specific physico-chemical properties (solubility, hydrophilicity, volatility). Among organic solvents, tetrahydrofuran (THF) has the most suitable behavior for the synthesis of these mesostructured hybrid materials. In addition to the polymer–solvent choice, functionalization of the inorganic phase was possible by means of this one-pot synthesis using a selected precursor carrying an active organic group R. So, direct functionalization of the silica network was performed using organosilane precursors (R–Si[OR]3) (R being any function such as mercaptopropyl, phenylethyl, methyl, and so on) co-condensed with Si(OR)4 as the silica precursor (OR being OEt). They further extended this approach to ionic polymer composites, such as composites with Nafion and found considerable increase in their performance.
This synthetic pathway looks appealing with regards to the formation of hierarchically ordered ionic particles into the host organic matrix with a homogeneous distribution. But an important drawback is the orientation of hierarchically ordered particles. Usually, in the casting method, orientation of the inorganic particles favors parallel arrangement to the membrane surfaces. This parallel orientation increases the length of the transport pathway concomitant lowering of the conductivity in ionic polymers. So for the ideal ionic membrane, hierarchically ordered ionic channels/particles should orient vertical to the membrane surface. This concept was proved by Azzaroni et al.127,128 He proposed a new approach to the facile large-scale fabrication of robust siliconmembranes with artificial, ordered two dimensional ion-conducting channels from ordered two-dimensional macroporous silicon and poly(sulfo-propyl methacrylate) hybrid materials. Ordered two-dimensional macroporous silicon was rendered proton conducting by growing a thick uniform polyelectrolyte brush using surface-initiated atom transfer radical polymerization throughout the porous matrix. This method proves the concept of orientation of ion-conducting channels vertical to the membrane surface and may find good applications in some specific areas. But, lack of membrane flexibility renders wide utilization.
Thus ionic membranes with ordered two dimensional ion-conducting channels vertically aligned to the membrane surface with excellent mechanical, chemical and dimensional stability and flexibility is an urgent need of advanced energy and biotechnology applications.
References
- C. Sanchez, B. Julian, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC
.
- Y. Lu, Y. Yang, A. Sellinger, M. Lu, J. Huang, H. Fan, R. Haddad, G. Lopez, A. R. Burns, D. Y. Sasaki, J. Shelnutt and C. J. Brinker, Nature, 2001, 410, 913 CrossRef CAS
- D. J. Jones and J. Rozière, Adv. Polym. Sci., 2008, 215, 219 CAS
- H. K. Baca, C. Ashley, E. Carnes, D. Lopez, J. Flemming, D. Dunphy, S. Singh, Z. Chen, N. Liu, H. Fan, G. P. Lopez, S. M. Brozik, M. W. Washburne and J. Brinker, Science, 2006, 313, 337 CrossRef CAS
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403 CrossRef CAS
- T. C. Merkel, B. D. Freeman, R. J. Spontak, Z. He, I. Pinnau, P. Meakin and A. J. Hill, Science, 2002, 296, 519 CrossRef CAS
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef CAS
- R. Souzy and B. Ameduri, Prog. Polym. Sci., 2005, 30, 644 CrossRef CAS
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587 CrossRef CAS
- G. Alberti and M. Casciola, Annu. Rev. Mater. Res., 2003, 33, 129 CrossRef CAS
- O. Savadogo, J. Power Sources, 2004, 127, 135 CrossRef
- R. Rodriguez, R. Herrera, L. A. Archer and E. P. Giannelis, Adv. Mater., 2008, 20, 1
- H. Steininger, M. Schuster, K. D. Kreuer and J. Maier, Solid State Ionics, 2006, 177, 2457 CrossRef CAS
- E. Besson, A. Mehdi, H. Chollet, C. Reye, R. Guilard and R. J. P. Corriu, J. Mater. Chem., 2008, 18, 1193 RSC
- T. Tezuka, K. Tadanaga, A. Hayashi and M. Tatsumisago, J. Am. Chem. Soc., 2006, 128, 16470 CrossRef CAS
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33 CrossRef CAS
- R. K. Nagarale, V. K. Shahi and R. Rangarajan, J. Membr. Sci., 2005, 248, 37 CrossRef CAS
- R. K. Nagarale, G. S. Gohil, V. K. Shahi and R. Rangarajan, Macromolecules, 2004, 37, 10023 CrossRef CAS
- R. K. Nagarale, G. S. Gohil and V. K. Shahi, Adv. Colloid Interface Sci., 2006, 119, 97 CrossRef CAS
-
G. W. Klemperer, V. V. Mainz, S. D. Ramamurthi and S. F. Rosenberg, in Better Ceramics Through Chemistry III, ed. E. D. Brinker, E. D. Clark, R. D. Ulrich, Materials Research Society, Pittsbuegh, PA, 1988, vol. 121, p. 15 Search PubMed
- H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893 CrossRef CAS
- K. Miyatake, T. Tombe, Y. Chikashige, H. Uchida and M. Watanabe, Angew. Chem., Int. Ed., 2007, 46, 6646 CrossRef CAS
- M. Aparicio, E. Lecoq, Y. Castro and A. Duran, J. Sol-Gel Sci. Technol., 2005, 34, 233 CrossRef CAS
- G. Hevesy and K. Kimura, J. Am. Chem. Soc., 1925, 47, 2540 CrossRef CAS
- C. B. Amphlett, L. A. McDonald and M. J. Redman, J. Inorg. Nucl. Chem., 1958, 6, 220 CrossRef CAS
- Edwin M. Larsen and R. Donald, J. Phys. Chem., 1960, 64, 1732 CAS
- L. Baetslé and J. Pelsmaekers, J. Inorg. Nucl. Chem., 1961, 21, 124 CrossRef CAS
- A. Clearfield and J. A. Stynes, J. Inorg. Nucl. Chem., 1964, 26, 117 CrossRef CAS
- G. Alberti and E. Torracca, J. Inorg. Nucl. Chem., 1968, 30, 317 CrossRef CAS
- W. H. J. Hogarth, J. C. Diniz da Costa and G. Q. Lu (Max), J. Power Sources, 2005, 142, 223 CrossRef CAS
- A. Clearfield, Annu. Rev. Mater. Sci., 1984, 14, 205 CrossRef CAS
- G. Alberti, M. Casciola, U. Constantino and R. Vivani, Adv. Mater., 1996, 8, 291 CAS
- G. Alberti, M. Casciola and R. Palombari, J. Membr. Sci., 2000, 172, 233 CrossRef CAS
- J. D. Kim, T. Mori and I. Honma, J. Membr. Sci., 2006, 281, 735 CrossRef CAS
- G. Alberti, M. Casciola, U. Costantino, A. Peraio and E. Montoneri, Solid State Ionics, 1992, 50, 315 CrossRef CAS
- E. W. Stein Sr., A. Clearfield and M. A. Subramanian, Solid State Ionics, 1996, 83, 113 CrossRef
- G. Alberti, L. Boccali, M. Casciola, L. Massinelli and E. Montoneri, Solid State Ionics, 1996, 84, 97 CrossRef CAS
- G. Alberti, M. Casciola, R. Palombari and A. Peraio, Solid State Ionics, 1992, 58, 339 CrossRef CAS
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS
- A. M. Herring, Polym. Rev., 2006, 46, 245 Search PubMed
- J. L. Malers, M. A. Sweikart, J. L. Horan, J. A. Turner and A. H. Herring, J. Power Sources, 2007, 172, 83 CrossRef CAS
- I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2007, 262, 86 CrossRef CAS
- J. Roziere and D. J. Jones, Annu. Rev. Mater. Res., 2003, 33, 503 CrossRef CAS
- K. A. Mauritz, Mater. Sci. Eng., C, 1998, 6, 121 CrossRef
- S. J. Paddison, Annu. Rev. Mater. Res., 2003, 33, 289 CrossRef CAS
- J. A. Kerres, J. Membr. Sci., 2001, 185, 3 CrossRef CAS
- L. C. Klein, Y. Daiko, M. Aparicio and F. Damay, Polymer, 2005, 46, 4504 CrossRef CAS
- Y. Daiko, L. C. Klein, T. Kasuga and M. Nogamia, J. Membr. Sci., 2006, 281, 619 CrossRef CAS
- W. F. Chen and P. L. Kuo, Macromolecules, 2007, 40, 1987 CrossRef CAS
- H. Wang, B. A. Holmberg, L. Huang, Z. Wang, A. Mitra, J. M. Norbeck and Y. Yan, J. Mater. Chem., 2002, 12, 834 RSC
- C. Li, G. Sun, S. Ren, J. Liu, Q. Wang, Z. Wu, H. Sun and W. Jin, J. Membr. Sci., 2006, 272, 50 CrossRef CAS
- Y. F. Lin, C. Y. Yen, C. C. M. Ma, S. H. Liao, C. H. Lee, Y. H. Hsiao and H. P. Lin, J. Power Sources, 2007, 171, 388 CrossRef CAS
- M. Lavorgna, L. Mascia, G. Mensitieri, M. Gilbert, G. Scherillo and B. Palomba, J. Membr. Sci., 2007, 294, 159 CrossRef CAS
- B. P. Ladewig, R. B. Knott, D. J. Martin, J. C. D. Costa da and G. Q. Lu, Electrochem. Commun., 2007, 9, 781 CrossRef CAS
- T. Duvdevani, M. Philosoph, M. Rakhman, D. Golodnitsky and E. Peled, J. Power Sources, 2006, 161, 1069 CrossRef CAS
- W. Apichatachutapan, R. B. Moore and K. A. Mauritz, J. Appl. Polym. Sci., 1996, 62, 417 CrossRef
- N. H. Jalani, K. Dunn and R. Datta, Electrochim. Acta, 2005, 51, 553 CrossRef CAS
- G. D. Yadav and J. J. Nair, Microporous Mesoporous Mater., 1999, 33, 1 CrossRef CAS
- S. Hara and M. Miyayama, Solid State Ionics, 2004, 168, 111 CrossRef CAS
- T. M. Thampan, N. H. Jalani, P. Choi and R. Datta, J. Electrochem. Soc., 2005, 152, A316 CrossRef CAS
- Y. Zhai, H. Zhang, J. Hu and B. Yi, J. Membr. Sci., 2006, 280, 148 CrossRef CAS
- P. Costamagna, C. Yang, A. B. Bocarsly and S. Srinivasan, Electrochim. Acta, 2002, 47, 1023 CrossRef CAS
- Y. Abe, G. Li, M. Nogami and T. Kasuga, J. Electrochem. Soc., 1996, 143, 144 CAS
-
R. P. Hamlen, US Patent, US 5849428, 1998 Search PubMed
-
W. G. Grot and G. Rajendran, US Patent, US 5919583, 1999 Search PubMed
- Y. T. Kim, K. H. Kim, M. K. Song and H. W. Rhee, Curr. Appl. Phys., 2006, 6, 612 CrossRef
- V. Ramani, H. R. Kunz and J. M. Fenton, Electrochim. Acta, 2005, 50, 1181 CrossRef CAS
- V. Ramani, H. R. Kunz and J. M. Fenton, J. Membr. Sci., 2006, 279, 506 CrossRef CAS
- H. J. Kim, Y. G. Shul and H. Hana, J. Power Sources, 2006, 158, 137 CrossRef CAS
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448 CrossRef CAS
- E. N. Gribov, E. V. Parkhomchuk, I. M. Krivobokov, J. A. Darr and A. G. Okunev, J. Membr. Sci., 2007, 297, 1 CrossRef CAS
- Y. F. Lin, C. Y. Yen, C. C. M. Ma, S. H. Liao, C. H. Hung and Y. H. Hsiao, J. Power Sources, 2007, 165, 692 CrossRef CAS
- P. Bebin, M. Caravanier and H. Galiano, J. Membr. Sci., 2006, 278, 35 CrossRef CAS
-
S. Wang and J. E. McGrath, Synthesis of Poly(arylene ether)s, Synthetic Methods in Step-Growth Polymers, Wiley, New York, 2003, p. 327 Search PubMed
- B. Bauer, D. J. Jones, J. Roziere, L. Tchicaya, G. Alberti, M. Casciola, L. Massinelli, A. Peraio, S. Besse and E. Ramunni, J. New Mater. Electrochem. Syst., 2000, 3, 93 Search PubMed
-
T. S. Guth, G. Frank, J. Baurmeister, J. Pawlik and R. Knauf, US Patent, US 6632847, 2003 Search PubMed
- K. D. Kreuer, Solid State Ionics, 2000, 136–137, 149 CrossRef CAS
- D. S. Kim, B. Liu and M. D. Guiver, Polymer, 2006, 47, 7871 CrossRef CAS
-
J. Roziere, D. J. Jones, T. L. Bouckary and B. Bauer, European Patent, EP01/07774, 2002 Search PubMed
- I. Colicchio, H. Keul, D. Sanders, U. Simon, T. E. Weirich and M. Moeller, Fuel Cells, 2006, 6, 225 CrossRef CAS
- S. P. Nunes, B. Ruffmann, E. Rikowski, S. Vetter and K. Richau, J. Membr. Sci., 2002, 203, 215 CrossRef CAS
- Y. H. Su, Y. L. Liu, Y. M. Sun, J. Y. Lai, D. M. Wang, Y. Gao, B. Liu and M. D. Guiver, J. Membr. Sci., 2007, 296, 21 CrossRef CAS
- Y. L. Liu, C. Y. Hsu, Y. H. Su and J. Y. Lai, Biomacromolecules, 2005, 6, 368 CrossRef CAS
- J. H. Chang, J. H. Park, G. G. Park, C. S. Kim and O. O. Park, J. Power Sources, 2003, 124, 18 CrossRef CAS
- Z. Gaowen and Z. Zhentao, J. Membr. Sci., 2005, 261, 107 CrossRef
- C. S. Karthikeyan, S. P. Nunes, L. A. S. A. Prado, M. L. Ponce, H. Silva, B. Ruffmann and K. Schulte, J. Membr. Sci., 2005, 254, 139 CrossRef CAS
- S. M. J. Zaidi, S. D. Mikhailenko, G. P. Robertson, M. D. Guiver and S. Kaliaguine, J. Membr. Sci., 2000, 173, 17 CrossRef CAS
- X. Li, D. Xu, G. Zhang, Z. Wang, C. Zhao and H. Na, J. Appl. Polym. Sci., 2007, 103, 4020 CrossRef CAS
- H. Zhang, J. H. Pang, D. Wang, A. Li, X. Li and Z. Jiang, J. Membr. Sci., 2005, 264, 56 CrossRef CAS
- D. E. Katsoulis, Chem. Rev., 1998, 98, 359 CrossRef CAS
- M. L. Ponce, L. A. S. A. Prado, V. Silva and S. P. Nunes, Desalination, 2004, 162, 383 CrossRef CAS
- M. L. Ponce, L. Prado, B. Ruffman, K. Richau, R. Mohr and S. P. Nunes, J. Membr. Sci., 2003, 217, 5 CrossRef CAS
- C. R. Mayer, I. Foumier and R. Thouvenot, Chem.–Eur. J., 2000, 6, 105 CrossRef CAS
- M. I. Ahmad, S. M. J. Zaidi and S. U. Rahman, Desalination, 2006, 193, 387 CrossRef CAS
- M. L. D. Vona, Z. Ahmed, S. Bellitto, A. Lenci, E. Traversa and S. Licoccia, J. Membr. Sci., 2007, 296, 156 CrossRef
- P. Kalappa and J. H. Lee, Polym. Int., 2007, 56, 371 CrossRef CAS
- P. Krishnan, J. S. Park and C. S. Kim, J. Membr. Sci., 2006, 279, 220 CrossRef CAS
- V. Neburchilov, J. Martin, H. Wang and J. Zhang, J. Power Sources, 2007, 169, 221 CrossRef CAS
- Y. S. Kim, M.
J. Summer, W. L. Harrison, J. S. Siffle, J. E. McGrath and B. S. Pivovar, J. Electrochem. Soc., 2004, 151, A2150 CrossRef CAS
- P. Piela, C. Eickes, E. Brosha, F. Garzon and P. Zelenay, J. Electrochem. Soc., 2004, 151, A2053 CrossRef CAS
- F. Lufrano, G. Squadrito, A. Patti and E. Passalacqua, J. Appl. Polym. Sci., 2000, 77, 1250 CrossRef CAS
- D. Xing, H. Zhang, L. Wang, Y. Zhai and B. Yi, J. Membr. Sci., 2007, 296, 9 CrossRef CAS
-
A. B. LaConti, M. Hamdan and R. C. McDonald, Handbook of Fule Cells: Fundamentals, Technology, and Applications, ed. W. Vielstich, A. Lamm, and H. A. Gasteiger, Wiley, New York, 2003, vol. 3, p. 647 Search PubMed
- V. K. Shahi, Solid State Ionics, 2007, 177, 3395 CrossRef CAS
- G. M. Anilkumar, S. Nakazawa, T. Okubo and T. Yamaguchi, Electrochem. Commun., 2006, 8, 133 CrossRef CAS
- D. S. Kim, H. B. Park, J. W. Rhim and Y. M. Lee, J. Membr. Sci., 2004, 240, 37 CrossRef CAS
- R. Q. Fu, L. Hong and J. Y. Lee, Fuel Cells, 2008, 8, 52 CrossRef CAS
- V. V. Binsu, R. K. Nagarale and V. K. Shahi, J. Mater. Chem., 2005, 15, 4823 RSC
- Y. Jin, J. C. D. Costa and G. Q. Lu, Solid State Ionics, 2007, 178, 937 CrossRef CAS
- M. Helen, B. Viswanathan and S. S. Murthy, J. Power Sources, 2006, 163, 433 CrossRef CAS
- V. S. Tripathi, V. B. Kandimalla and H. Ju, Sens. Actuators, B, 2006, 114, 1071 CrossRef
- V. D. Noto and M. Vittadello, Electrochim. Acta, 2005, 50, 3998 CrossRef
- U. Schubert, N. Husing and A. Lorenz, Chem. Mater., 1995, 7, 2010 CrossRef CAS
- Y. I. Park and M. Nagai, Solid State Ionics, 2001, 145, 149 CrossRef CAS
- I. Honma, S. Hirakawa, K. Yamada and J. M. Bae, Solid State Ionics, 1999, 118, 29 CrossRef CAS
- I. Honma, H. Nakajima, O. Nishikawa, T. Sugimoto and S. Nomura, Solid State Ionics, 2003, 162–163, 237 CrossRef CAS
- S. Li, Z. Zhou, H. Abernathy, M. Liu, W. Li, J. Ukai, K. Hase and M. Nakanishi, J. Mater. Chem., 2006, 16, 858 RSC
- J. D. Kim and I. Honma, Electrochim. Acta, 2004, 49, 3429 CrossRef CAS
- S. P. Tung and B. J. Hwang, J. Mater. Chem., 2005, 15, 3532 RSC
- M. Nogami, K. Miyamura and Y. Abe, J. Electrochem. Soc., 1997, 144, 2175 CAS
- M. Nogami and Y. Abe, Phys. Rev. B: Condens. Matter, 1997, 55, 12108 CrossRef CAS
- C. Wang, M. Nogami and Y. Abe, J. Sol-Gel Sci. Technol., 1999, 14, 273 CrossRef CAS
- C. Wang and M. Nogami, Mater. Lett., 2000, 42, 225 CrossRef CAS
- M. Nogami, H. Matsushita, Y. Goto and T. Kasuga, Adv. Mater., 2000, 12, 1370 CrossRef CAS
- K. Valle, P. Belleville, F. Pereira and C. Sanchez, Nat. Mater., 2006, 5, 107 CrossRef CAS
- B. Yameen, A. Kaltbeitzel, A. Langner, H. Duran, F. Muller, U. Gosele, O. Azzaroni and W. Knoll, J. Am. Chem. Soc., 2008, 130, 13140 CrossRef CAS
- B. Yameen, A. Kaltbeitzel, A. Langer, F. Muller, U. Gosele, W. Knoll and O. Azzaroni, Angew. Chem., Int. Ed., 2009, 48, 3124 CrossRef CAS
Footnote |
| † Present address: Department of Chemical Engineering, The University of Texas at Austin, 1 University Station C0400, Austin, Texas 78712, USA. Tel: +1 512 471 1287 |
| This journal is © The Royal Society of Chemistry 2010 |