Self-healing polymers prepared via living radical polymerisation†
Jay A.
Syrett
a,
Giuseppe
Mantovani
b,
William R. S.
Barton
c,
David
Price
c and
David M.
Haddleton
*a
aDepartment of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: d.m.haddleton@warwick.ac.uk
bSchool of Pharmacy, University of Nottingham, Nottingham, UK NG7 24D. E-mail: giuseppe.mantovani@nottingham.ac.uk
cLubrizol Corporation, Hazelwood Research Centre, Derby, UK DE56 1QN
First published on 15th January 2010
Abstract
Diels–Alder chemistry has been used to synthesise polymerisation initiators and a dimethacrylic cross-linker that leads to efficient cleavage and reformation; self-healing. The initiators were prepared using 1-(2-hydroxyethyl)-1H-pyrrole-2,5-dione (3) as an intermediate, and reacting this with furfuryl alcohol to afford 2-bromo-2-methyl-propionic acid 2-[1-(2-bromo-2-methyl-propionyloxymethyl)-3,5-dioxo-10-oxa-4-aza-tricyclo[5.2.1.02,6]dec-8-en-4-yl]-ethyl ester (7) and 9-anthracenemethanol to yield initiator (8). The former exhibited polymers with excellent cleavage properties (Mn 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 to 6500 g mol−1) with reformation at efficiency of 50% (Mn 8900 g mol−1). The initiator from the anthracene derivative initially indicated that retro-Diels–Alder was not occurring with no change in the NMR or GPC data. An excess of a rhodamine-based dienophile was added to capture any cleaved anthracene-terminated polymer, with results indicating that the polymer is cleaving and reforming upon the cooling cycle. Similar results were observed with arm first stars, made using a Diels–Alder based dimethacrylate cross-linker. Little cleavage was observed initially, however, addition of the tag gave a significant reduction in Mw (7080 g mol−1 to 5300 g mol−1) and Mw/Mn (1.78 to 1.26).
000 g mol−1 to 6500 g mol−1) with reformation at efficiency of 50% (Mn 8900 g mol−1). The initiator from the anthracene derivative initially indicated that retro-Diels–Alder was not occurring with no change in the NMR or GPC data. An excess of a rhodamine-based dienophile was added to capture any cleaved anthracene-terminated polymer, with results indicating that the polymer is cleaving and reforming upon the cooling cycle. Similar results were observed with arm first stars, made using a Diels–Alder based dimethacrylate cross-linker. Little cleavage was observed initially, however, addition of the tag gave a significant reduction in Mw (7080 g mol−1 to 5300 g mol−1) and Mw/Mn (1.78 to 1.26).
1. Introduction
Self-healing materials are inspired by biological systems where damage leads to an automatic healing response,1 for example, the self-repair of cuts and bruises on the human body. The application of this philosophy to polymers and materials is relatively new but gaining in interest for a number of potential applications.2,3 Synthetic polymers often have a limited lifespan with a tendency to damage or degrade over time due to constant stresses and strains. This is driving the development of polymeric materials that can self-heal, either as an effect of an external stimuli or ideally by an auto-response.3 In conventional materials damage first occurs at the microscopic level and repair at this level is essential to restore the full physical/mechanical properties, as opposed to macroscopic repair with adhesives, etc. For polymers this has been achieved by both irreversible repair, i.e. by incorporating microcapsules containing reactive monomer into a thermoset matrix2 or by including an element of reversibility, either covalent or non-covalent, into cross-linked polymers.3Diels–Alder (DA) cycloaddition is a convenient route for the formation of carbon–carbon bonds via a facile reaction under undemanding conditions that fulfills the requirements of a “click” reaction.4–8 Diels–Alder cycloadditions are thermoreversible reactions9 and this feature has been exploited in the preparation of self-healing polymers carrying functionalities, either as the polymer chain-end or in the repeating units.3 Furthermore, the reverse DA reaction does not involve free radicals, thus avoiding many side reactions that might prevent reformation.10 Diels–Alder moieties were first incorporated into polymers by Stevens and Jenkins.11 Subsequently, Saegusa and co-workers developed a thermally “mendable” polymeric DA network.12 A Diels–Alder cross-linked polymer synthesised for the specific use of self-healing was prepared by Wudl et al. in 2003, using furan- and maleimide-based monomers cross-linking along the polymer backbone.9 In this study it was shown that fractured polymer materials, heated to 120 °C, exhibited 83% recovery of the polymers original strength. Crucially, this fracture/repair cycle could also be repeated, making this the first real DA polymeric system exhibiting a thermally responsive self-healing behaviour.
This route to self-healing polymers is applicable to composite materials and large cross-linked networks. Advances in controlled radical polymerisation (CRP) allow for the synthesis of functional polymers with excellent control over molecular weight, molecular weight distribution, architecture and incorporation of functionality. The introduction of a range of techniques, ATRP,13–15 RAFT16–18 and NMRP,19 gives the polymer chemist a great deal of control of polymer properties. Herein, we report the use of a living radical polymerisation to synthesise a self-healing polymer. The chemistry employed is efficient and relatively simple and has allowed for the design of cleavable linkers that contain polymerisation initiators. Additionally, a DA based cross-linker that can be employed in the synthesis of arm first stars is reported.
In this present study we report the synthesis of novel well-defined linear and star methyl methacrylate (MMA) polymers bearing DA adducts within their macromolecular backbone and a preliminary evaluation of their ability to cleave and reform under external thermal stimuli.
2. Results and discussion
Synthesis of initiators and cross-linking agents
The synthetic protocol developed for the synthesis of the initiators DA1 and DA2 is shown in Scheme 1. The Diels–Alder adduct (1) was prepared by heating a suspension of maleic anhydride in furan and toluene at 120 °C for 24 h to give, after crystallisation, exclusively the exo isomer. A solution of 1 in methanol and triethylamine (TEA) was subsequently treated with monoethanolamine at 0 °C, and slowly heated to 67 °C, as described by Hung et al.,20 affording, after crystallisation, the alcohol (2) in 72% yield. This was then subjected to a retro-Diels–Alder process in refluxing toluene for 24 h, to give the maleimide-containing alcohol (3), which served as a versatile intermediate for the synthesis of both initiators DA1 (7) and DA2 (8). Initiator DA1 was obtained by treatment of 3 with furfuryl alcohol to give diol (4), which was esterified with 2-bromoisobutyryl bromide in THF–TEA, to give, after flash chromatography, the difunctional initiator DA1 (7) in 78% yield. DA2 (8) was prepared in an analogous way, by treating 3 with 9-anthracenemethanol followed by esterification of the resulting diol (5) with 2-bromoisobutyryl bromide in THF–TEA, to give DA2 (8) in 81% yield. The diacrylate (6), employed later in this study as a thermoresponsive cross-linking agent, was prepared in a 75% yield, again from 4, using acryloyl chloride and TEA in anhydrous dichloromethane.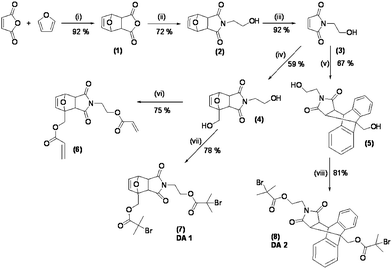 | ||
| Scheme 1 Reagents and conditions: (i) toluene, 80 °C; (ii) ethanolamine, Et3N, MeOH, 0 °C to 70 °C; (iii) toluene, reflux, 24 h; (iv) furfuryl alcohol, benzene, reflux; (v) anthracenemethanol, toluene, reflux; (vi) acryloyl chloride, dichloromethane, 0–25 °C; (vii and viii) 2-bromo isobutyl bromide, Et3N, 0–25 °C, THF. | ||
Polymerisation
Linear poly(methyl methacrylate) (PMMA) was prepared by polymerisation, using DA1 (7) or DA2 (8) as the initiator with Cu(I)Br–pyridine imine ligand as catalyst.21 Polymerisation in relatively non-polar/coordinating solvents is typically carried out between 70 °C and 90 °C; in the present case the tendency of the DA1 difunctional initiator to dissociate on heating (DSC analysis of DA1 at 5 °C min−1, suggested retro-Diels–Alder dissociation occurring at ca. 120 °C, see ESI†) prompted us to lower the polymerisation temperature to 50 °C. All polymerisations proceeded as expected, showing relatively linear first-order kinetic plots and linear increase of Mn with conversion (Fig. 1), affording well-defined polymers of tunable chain size and narrow molecular weight distributions determined by GPC analysis using chloroform–TEA 95 : 5 as the eluent (Table 1).![(i) Comparison of rates for polymers 10 (50 °C) and 12 (70 °C). (ii) Mnvs. conversion for the polymers 10 and 12. [Cu(i)Br]–[ligand]–[initiator]0–[MMA]0 2 : 4 : 1 : 100.](/image/article/2010/PY/b9py00316a/b9py00316a-f1.gif) | ||
| Fig. 1 (i) Comparison of rates for polymers 10 (50 °C) and 12 (70 °C). (ii) Mnvs. conversion for the polymers 10 and 12. [Cu(I)Br]–[ligand]–[initiator]0–[MMA]0 2 : 4 : 1 : 100. | ||
The ability of polymers 9–12 to respond to thermal stimuli was investigated. The 1H NMR signals for the Diels–Alder derived polymer feature a very distinctive pattern of signals, which can be easily distinguished from the dissociated polymer (Fig. 2).
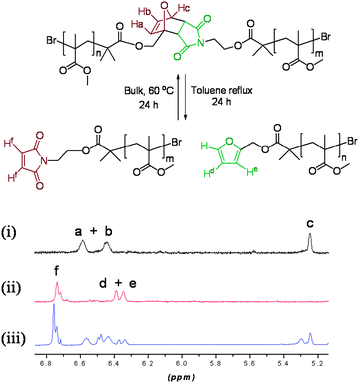 | ||
| Fig. 2 1H NMR of polymer 10 (i) prior to heating, (ii) cleaved polymer following heating, and (iii) reformed polymer. | ||
Following thermal treatment of polymers 9–11 under reflux in toluene over 24 h a completely different pattern of peaks in the 1H NMR was observed and each signal was easily identified, indicating that a retro-Diels–Alder process occurred efficiently under these conditions, to give N-maleimide (6.89 ppm) and furfuryl ester (6.36 and 6.39 ppm)-terminated polymers. After removal of the solvent under reduced pressure, the reaction mixture was heated at 60 °C for 24 h. NMR analysis of the resulting reaction mixture revealed that approximately 50% of the Diels–Alder linker had reformed under these conditions (5.22, 6.45 and 6.57 ppm). This value is comparable to the 59% yield observed for the analogous cycloaddition in which the maleimide alcohol (3) and furfuryl alcohol were converted into the adduct (4). It is noted that previous reports have often reported healing efficiencies of <50%, which were found to be sufficient for significant recovery of mechanical properties.22 In the 1H NMR spectrum two new signals at 6.49 and 5.3 ppm, not previously seen either in the spectra of the DA polymers (9–11) or in the maleimide- and furan-terminated polymers, were observed after the retro-Diels–Alder process (Fig. 2). NMR investigation and comparison with data reported in the literature23 indicated that the reformed polymer was a mixture of endo and exo isomeric Diels–Alder linkers.
GPC traces confirmed a significant proportion of polymer reformation (Fig. 3), although a quantitative assessment of the yields for this Diels–Alder process was not extrapolated from these data due to partial overlapping of the peaks of the cleaved and non-cleaved polymers. As expected, the molecular weight of the former was found to be approximately half that of the original polymer, whilst, upon reformation, the molecular weight increases, as expected. The PDI's also increased upon Diels–Alder linker reformation, which was due to partial peak overlap, Tables 1 and 2.
| Polymer | M n/kDaa | M w/kDaa | M w/Mna | [η]b |
|---|---|---|---|---|
| a Determined by GPC analysis using THF–TEA 95 : 5 (v/v) as the mobile phase and PMMA as calibrants. b Intrinsic viscosities were measured for polymer 10 and its cleaved/reformed derivatives. | ||||
| 9 | 5.40 | 5.99 | 1.11 | — |
| Cleaved | 2.76 | 3.25 | 1.18 | — |
| Reformed | 4.10 | 4.96 | 1.21 | — |
| 10 | 12.0 | 13.5 | 1.12 | 0.072 |
| Cleaved | 6.5 | 7.81 | 1.21 | 0.041 |
| Reformed | 8.9 | 11.0 | 1.24 | 0.052 |
| 11 | 54.7 | 62.9 | 1.15 | — |
| Cleaved | 28.4 | 34. 7 | 1.22 | — |
| Reformed | 42.0 | 52.0 | 1.24 | — |
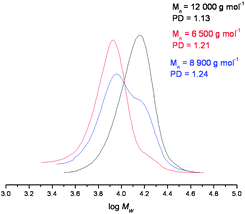 | ||
| Fig. 3 GPC data of (i) polymer 10 (black), (ii) cleaved polymer (red), and (iii) the reformed polymer (blue), under the conditions described in Fig. 2. | ||
Polymers prepared from the difunctional initiator DA1 (7) showed excellent cleavage/re-healing properties. Polymer 12, prepared from the difunctional initiator DA2 (8), was introduced as maleimide–anthracene molecules which are known to undergo Diels–Alder process in a facile manner. Aydan et al. showed that star polymers could be synthesised via a Diels–Alder click reaction.24 An arm first core was grown first with an anthracene-based initiator. Subsequently, a protected maleimide functional polymer was introduced, and a highly efficient deprotection followed by click reaction to the anthracene core in one pot occurred.24 However, subsequent deprotection of these polymers was not reported. This suggested either that the retro-Diels–Alder did not occur or that the process did occur to a certain extent, but the Diels–Alder linkers subsequently did reform, upon cooling or directly at the temperature employed for the deprotection, which is common in self-healing systems.9 The polymers formed from DA1 (7) did not exhibit reformation upon cooling.
In order to investigate whether the anthracene–maleimide linker was cleaving and then reforming upon cooling, an excess of a dienophile was added to capture any cleaved anthracene-terminated polymer (Fig. 4). For this purpose, we synthesised a maleimide-containing rhodamine B probe (Fig. 5). Retro-Diels–Alder cleavage was attempted in the presence of 50-fold molar excess of the rhodamine tag, in refluxing DMSO. The tag was introduced as its furan adduct which released the desired maleimide-containing fluorophore in situ, upon heating. Complete cleavage, as in the case of polymers 9–11, should halve the Mn. In this case the Mn decreased by only 20%, suggesting that approximately a fifth of the linker had effectively cleaved throughout the process. Evidence that exchange has taken place came from GPC with UV-Vis detection at λ = 550 nm, the resulting material was now clearly seen (Fig. 6). The starting material (12) has no absorption at this wavelength. This suggests that the anthracene–maleimide linker in 12 has a higher thermal stability than the furan–maleimide present in 9–11, and that any cleaved materials formed upon heating 12 at 200 °C were able to undergo Diels–Alder cycloaddition upon slow cooling back to ambient temperature.
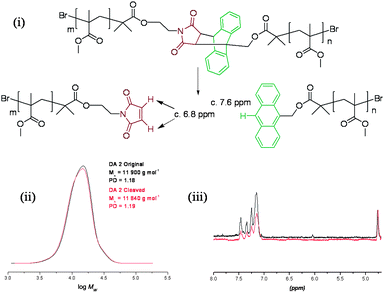 | ||
| Fig. 4 Attempted deprotection of 12 in DMSO at 200 °C: (i) reaction scheme, (ii) GPC data, and (iii) partial 1H NMR spectra before and after the attempted retro-Diels–Alder polymer cleavage. | ||
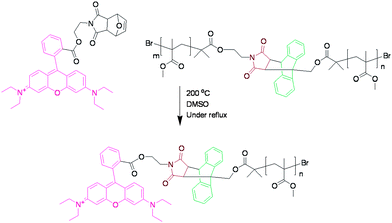 | ||
| Fig. 5 The expected product 12 cleaved during the heating process. This product could then be detected by UV GPC. | ||
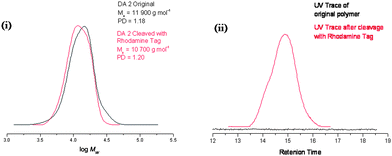 | ||
| Fig. 6 Cleavage of DA2 polymer monitored by GPC with (i) DRI detection and (ii) UV detection showing no absorbance in the original polymer, and a peak following cleavage and addition to the rhodamine tag, retention time is given for the UV trace as this detector was not calibrated. | ||
In order to investigate the applicability of the approach the study was extended to star polymers. A difunctional cross-linker monomer containing a substituted furan–maleimide Diels–Alder adduct was synthesised for this purpose. Due to structural similarities with polymers 9–11 obtained from the initiator DA1 (7) we expected the retro-Diels–Alder cleavage of the cross-linker to occur to a relatively large extent.
Refluxing 13 (Fig. 7) in toluene for 24 h suggested that a small amount of cleavage takes place with a shoulder corresponding to a lower molecular weight material detected in the GPC chromatogram, Fig. 8. As polymers 9–11 were easily cleaved under analogous reaction conditions, it was suspected that a significant proportion of the star cleaves and reforms within the cooling cycle. In this system complete cleavage of the star was expected to afford lower molecular weight materials with narrower PDI's. One possible explanation for this behaviour was that in a cross-linked network a less than quantitative retro-Diels–Alder process would caused the diene/dienophile cleaved moieties to remain spatially close to each other which would favour the reformation of the Diels–Alder linkers.
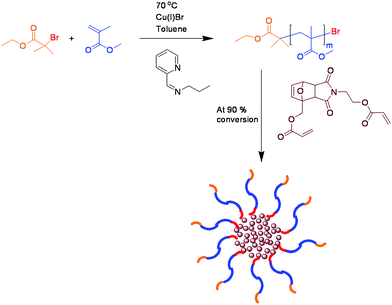 | ||
| Fig. 7 Synthesis of star polymer (13). | ||
 | ||
| Fig. 8 GPC data before and after attempted cleavage of the star polymer. Evidence that some cleavage has occurred is seen from the appearance of low molecular shoulder in the cleaved product. | ||
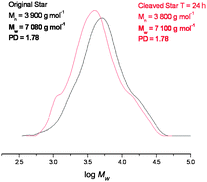
In order to investigate this the maleimide functionalised rhodamine trapping agent was added in the reaction mixture. In this case the Mn and Mw of the star polymer were reduced dramatically, and the PDI decreased from 1.78 to 1.26 indicating that the majority of the cross-linker units was effectively cleaving under these conditions (Fig. 9).
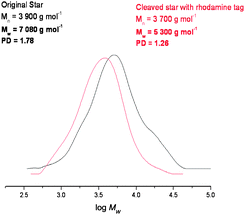 | ||
| Fig. 9 GPC data before and after cleavage with the rhodamine tag, showing a vast reduction in the polydispersity and Mw data. | ||
At this stage it is still not clear whether this system did reheal upon cooling or whether the Diels–Alder linkers were constantly cleaving and reforming. The cleaving and reforming throughout the heating cycle were confirmed by high temperature GPC at 140 °C, with 1,2,4-trichlorobenzene as eluent, which showed little change from the original polymer (see ESI†).
3. Summary
Two polymerisation initiators and one cross-linker were prepared using DA chemistry. DA1 exhibited exceptional cleavage properties, with 50% reformation occuring upon the reheating cycle. However, the facile nature in which this linker cleaved can be a problem for practical applications. Changing the alcohol to 9-anthracenemethanol gave the Diels–Alder linkage higher thermal stability. The forward DA reaction is highly efficient when using 9-anthracenemethanol, and the small amount of cleavage occuring is reforming in the cooling cycle. In order to investigate different polymer architectures, the same principle was applied to arm first stars, however, this time it was the shear quantity of linkages that enabled the Diels–Alder bond to constantly cleave and reform throughout the heating processAcknowledgements
The work was funded by EPSRC and Lubrizol Ltd under an EPSRC CASE award. We also thank the M. W. Jones and J. Burns for their help and advice. The SEC equipment used in this research was obtained through Birmingham Science City (AM2), with support from Advantage West Midlands and part funded by the European Regional Development Fund (ERDF).Notes and references
- N. Sottos, S. White and I. Bond, J. R. Soc. Interface, 2007, 4, 347–348 CrossRef.
- W. Jean-Luc and P. S. Rint, Angew. Chem., Int. Ed., 2008, 47, 8161–8163 CrossRef.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630–5639 CrossRef CAS.
- E. Gungor, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3409–3418 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, New J. Chem., 2007, 31, 718–724 RSC.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- F. R. Kersey, D. M. Loveless and S. L. Craig, J. R. Soc. Interface, 2007, 4, 373–380 CrossRef CAS.
- M. P. Stevens and A. D. Jenkins, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 3675–3685 Search PubMed.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2636–2641 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- D. M. Haddleton, C. B. Jasieczek, M. J. Hannon and A. J. Shooter, Macromolecules, 1997, 30, 2190–2193 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- C. C. Hung, M. Sai, G. M. Sulzer and R. Venkataram, WO2007044169 (A1), 2007.
- D. M. Haddleton, M. C. Crossman, B. H. Dana, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, Macromolecules, 1999, 32, 2110–2119 CrossRef CAS.
- T. C. Mauldin, J. D. Rule, N. R. Sottos, S. R. White and J. S. Moore, J. R. Soc. Interface, 2007, 4, 389–393 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906–1912 CrossRef CAS.
- D. Aydan, D. Hakan, T. Umit and H. Gurkan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 178–187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and additional characterisation data. See DOI: 10.1039/b9py00316a |
| This journal is © The Royal Society of Chemistry 2010 |