Mass spectrometry in polymer chemistry: a state-of-the-art up-date†
Till
Gruendling
ab,
Steffen
Weidner
c,
Jana
Falkenhagen
*c and
Christopher
Barner-Kowollik
*a
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Fax: +49 721 608 5740; Tel: +49 721 608 5641
bBioanalytical Mass Spectrometry Facility, UNSW Analytical Centre, The University of New South Wales, Sydney, New South Wales 2052, Australia. E-mail: till.gruendling@gmx.de
cFederal Institute for Materials Research and Testing (BAM), Richard-Willstaetter-Strasse 11, D-12489, Berlin, Germany. E-mail: jana.falkenhagen@bam.de; Fax: +49 30 8104 1137; Tel: +49 30 8104 3398
First published on 26th January 2010
Abstract
Two decades after the introduction of matrix assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI), soft ionization mass spectrometry represents a powerful toolset for the structural investigation of synthetic polymers. The present review highlights the current state-of-the-art, covering the latest developments of novel techniques, enabling instrumentation as well as the important applications of soft ionization MS from the beginning of 2008. Special attention is paid to the role that soft ionization MS has played in the mechanistic investigation of radical polymerization processes since 2005.
 Till Gruendling | Till Gruendling studied chemistry at the University of Marburg, until 2007. He is currently a PhD student at the University of New South Wales (UNSW)/Karlsruhe Institute of Technology (KIT) under the supervision of Prof. Barner-Kowollik and Prof. Brynn Hibbert. Till Gruendling has authored 9 publications and has presented his work at international conferences. He currently holds a University International Postgraduate Award. His research involves the application of liquid chromatography – mass spectrometry for the characterization of synthetic polymers and the development of novel quantitative methods in liquid chromatography – mass spectrometry, with focus on the elucidation of free radical polymerization processes. |
 Steffen Weidner | Steffen Weidner received a Diplom degree in chemistry from the Berlin Humboldt-University and a PhD in chemistry from the Technical University of Berlin. He is head of the Polymer Analysis group at the Federal Institute for Materials Research and Testing and a member of the German Society for Mass Spectrometry. He is author of numerous scientific publications including a book chapter. His research is driven from a regulatory perspective having strong ties to academia and industry. His particular interest is focused on the application of MALDI-TOF-MS to synthetic polymers by developing suitable hyphenated chromatography approaches for efficient reduction of complexity. |
 Jana Falkenhagen | Jana Falkenhagen studied chemistry at the Martin-Luther-University Halle-Wittenberg, and completed her studies with the Diplom degree. She received a PhD from the Technical University Berlin. Her main field of research is the characterization of complex polymers by hyphenated coupling techniques. She was strongly involved in the development of two-dimensional chromatography (LC × SEC) of polymers and its introduction in industry. Her key activity at the Federal Institute for Materials Research and Testing (BAM) is the development of hyphenated chromatographic and spectroscopic techniques with a main focus on LC/MALDI and LC/ESI-TOF-MS. She is author of numerous publications, book chapters and holds one patent. |
 Christopher Barner-Kowollik | Christopher Barner-Kowollik studied chemistry at the Universities of Konstanz and Göttingen. He completed a PhD at the University of Göttingen, before leading a research group as Full Professor at the Centre for Advanced Macromolecular Design/University of New South Wales until mid-2008. He is currently Full Professor of Preparative Macromolecular Chemistry at the Karlsruhe Institute of Technology, and has published over 195 papers, several patents and 9 book chapters. His research interests range from the synthesis of macromolecular architectures via living/controlled polymerization, the development of novel polymer conjugation methods, to high resolution mass spectrometry on polymer systems coupled with multi-detector chromatographic techniques. |
Introduction and scope
The properties of a polymer are governed by the topology of the polymer, the composition, the degree and sites of functionalization as well as the shape of the molecular mass distribution. The tools available today for controlling the polymer architecture to a fine degree are multifold. In particular, the advent of versatile radical polymerization processes featuring living characteristics has enormously simplified access to complex architecture polymers. Recently, the ability to create intricate macromolecular designs has been further enhanced by the introduction of modular and highly efficient synthetic concepts summarized by the term click chemistry. Concomitantly to the recent advances in macromolecular synthesis, the methodologies for the characterization of macromolecules have become more refined, as conventional analytical techniques, such as conventional size exclusion chromatography (SEC) coupled to refractive index or UV/Vis detectors only provides very limited or indeed no chemical information about the analyte. Yet, the determination of the degree of functionalization and the chemical constitution of macromolecules is of paramount importance for the optimization and design of well-defined macromolecules. Mass spectrometry (MS) has gained prominence in the field of polymer synthesis and the characterization of macromolecules, particularly since the advent of so-called soft-ionization protocols, including electrospray ionization (ESI) and matrix assisted laser desorption/ionization (MALDI). Inspection of the literature indicates that progress in these areas is tremendous, both in the area of method development as well as the range of polymer systems that have been analyzed. Writing a review is a process of selection, discarding some of the wealth of literature that is present and prioritizing others. Thus, in the current review we have limited ourselves to the – in our opinion – most significant instrumental innovations and novel analytical procedures, and among these mainly those employing the soft ionization techniques MALDI and ESI, published between the start of 2008 and October 2009. Furthermore, we demonstrate how these instrumental innovations have aided in the elucidation of (radical) polymerization mechanisms focusing on articles published between 2005 and today. An earlier feature article1 covers reports on the application of soft ionization mass spectrometry to radical polymerisation before this date. The reader is advised that due to space limitations, mechanistic investigations of non-radical chain- and step-growth polymerizations – although numerous – have not been included in the current manuscript, as have been the many analyses where MS has merely served as a tool to confirm polymer and endgroup identity in functional polymer synthesis.Existing reviews and books
Early 2010 will see the publication of ‘MALDI Mass Spectrometry for Synthetic Polymer Analysis’.2 Anticipated as a reference guide to the field, the basics of polymer analysis by mass spectrometry will be discussed as well as hands-on procedures for sample preparation, analysis and data treatment. A number of reviews have been published in the covered time-frame, paying tribute to the on-going attention of the polymer community in mass spectrometry and highlighting a number of new and promising trends in the field. In the biennial series “Mass Spectrometry of Synthetic Polymers” Weidner and Trimpin gave a comprehensive overview of the literature published in 2006 and 2007.3 Batoy et al. focussed on MALDI, giving a short account of the current understanding of the mechanism of ionization.4 Attention was subsequently drawn to the critical role that proper matrix selection and sample preparation plays in synthetic polymer MALDI-MS. Literature on tandem ESI- and MALDI-MS/MS of synthetic polymers published after 2006 was covered by Crecelius et al.5 These authors noted the potential benefits of tandem mass spectrometry (MS/MS) in that structural information on homo- and copolymers is gained from the characteristic fragmentation patterns in MS/MS, complementary to that from mass spectrometry alone. Two publications highlighted the increasing awareness of the utility of secondary ion mass spectrometry (SIMS) in the characterization of soft surfaces and films. In a feature article, Walker argued that the advent of commercially available cluster ion sources has lead to a renaissance in SIMS during the recent years.6 Cluster ion sources now provide excellent ion yields and decreased sample damage. The sub-micrometre lateral resolution of SIMS as well as the ability of depth profiling make this technique a powerful chemical analysis tool for the 2D and 3D imaging of soft surfaces. Walker concluded that regretfully there seem to be some dated preconceptions which are currently hindering faster proliferation of SIMS in the chemical community. Arriving at similar conclusions about the advances of the method, Mahoney provided a very detailed review on cluster SIMS of polymeric materials.7 The author discussed in detail the intricacies and advantages of using cluster primary ions, as well as a number of applications of the technique in synthetic polymer analysis and materials science. The reader is also advised of a recent review article by Rial-Otero et al., which provides a concise overview of the contemporary fields of application of pyrolysis gas chromatography mass spectrometry (Py-GC-MS) and recent important developments of the technique.8Soft ionization techniques
Transfer of the solid or dissolved polymer molecule into the gas phase and simultaneous ionization with negligible fragmentation stands at the basis of a holistic analysis of polymer structure by mass spectrometry. Laser desorption ionization (LDI) and – to a lesser extent – electrospray ionization have become the workhorse ionization techniques in this regard. In the recent years, a number of long standing issues, such as the soft desorption ionization of polymers that are insoluble or weakly soluble as well as of non-polar species have found potential solutions and a number of matrix free LDI techniques have been presented which will be discussed here.Atmospheric pressure photo-ionization (APPI) has recently been introduced for the ionization of low-polarity compounds such as polyaromatic hydrocarbons, lipids and steroids (see for example ref. 9) that are difficult or impossible to ionize by either of the contemporary soft ionization techniques. Kéki and co-workers applied this technique to the ionization of two non-polar polymers, namely polyisobutylenes10,11 and polyethylene waxes.12 Spectra of the intact end-group-functional polymer adduct ions [M + Cl]− were obtained in negative mode by dopant-assisted APPI using toluene as well as chlorinated hydrocarbon solvents as dopants. A general kinetic model was proposed for the formation process of the Cl− quasimolecular ions.10
Surface assisted laser desorption/ionization (SALDI) is a new matrix-free laser desorption/ionization technique, in which (metal-oxide) nanoparticles act as the energy transfer medium to promote ion formation. SALDI was successfully applied by Watanabe et al. to study poly(ethylene oxide) (PEO), polystyrene (PS) and poly(methyl methacrylate) (PMMA).13 These authors used anisotropic ZnO nanoparticles to achieve virtually the same ionization efficiency for PEO as with a traditional matrix (dihydroxybenzoic acid, DHB) (see Fig. 1). A great advantage of matrix-free ionization is realized immediately from the spectrum in Fig. 1(b), i.e. the total lack of any low molecular weight matrix ions. Surface assisted ionization using TiO2 nanoparticles led to significant fragmentation of the polymer, due the strong photocatalytic nature of TiO2 [Fig. 1(c)]. Although not a main concern of their study, Gorman et al. used a very similar technique, desorption/ionization on silicon (DIOS), in which a porous silicon surface acts as the energy transfer medium.14
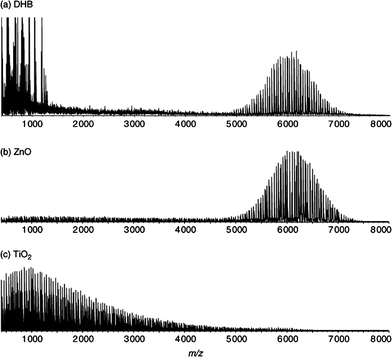 | ||
| Fig. 1 Comparison of ion intensities of polyethylene oxide 6000 (100 pmol) by laser desorption/ionization MS using (a) DHB (b) ZnO nanoparticles and (c) TiO2 nanoparticles. The image is taken from ref. 13 with kind permission of Wiley and Sons. | ||
An unconventional technique, termed electrospray droplet impact (EDI)/secondary ion mass spectrometry (SIMS) was applied to the analysis of PS and PEO samples by Asakawa et al.15 In this technique, which is principally related to cluster SIMS, ionization of polymer samples with molecular weight of up to 6 kg mol−1 was achieved. The apparent lack of significant fragmentation is notable, which is usually rather pronounced in cluster SIMS.
In the analysis of functional polymers by soft ionization techniques, loss of the end-group also means a loss of chemical information. The softness of the ionization depends on the polarity of the polymer, with less polar polymers requiring harsher ionization conditions. A number of recent studies therefore investigated the ability to attain soft ionization of the important but rather difficult to ionize polystyrene (PS), carrying labile functional groups.
Gruendling et al. used silver tetrafluoroborate as dopant to demonstrate that ESI of dithioester-functional PS from reversible addition fragmentation chain transfer (RAFT) polymerization is possible without loss of the sensitive end-group.16 In another study, the same authors applied statistical design of experiment (DoE) to optimize source conditions in ESI-MS of labile, bromide functional PMMA.17 Later, Ladavière and colleagues compared in a comprehensive manner the ability of ESI and MALDI with different doping salts to ionize polystyrene carrying labile dithioester-, nitroxide- and halide-end-groups from living/controlled radical polymerization.18 These authors noted that using ESI-MS with sodium doping, significantly more intact end-groups were detected than with MALDI-MS and silver trifluoroacetate as dopant. However, strong fragmentation was observed with both ionization techniques, for chloride-, bromide- and iodide-functional PS and in ESI, the doping salt (Na+vs. Ag+) was found to play a key-role in determining the extent of fragmentation. From earlier studies by MALDI it is also known that hydrosilane (–SiHMe2) functional polystyrenes are oxidized by Ag+ in the presence of dithranol so that only the siloxane (–SiOHMe2) units are observed in the spectrum.19 Dempwolf et al. analyzed functional polystyrenes produced by nitroxide mediated polymerization (NMP) by MALDI-MS.20 Using dihydroxybenzoic acid (DHB) as matrix, the intact nitroxide-capped PS were observed as protonated species, whereas use of dithranol/AgTFA as matrix lead to chain-end degradation.
Direct ionization techniques
Direct or ‘ambient’ ionization techniques are those allowing ionization of an analyte in its native state, usually as a solid material, without requiring any sample preparation or pre-treatment steps. These techniques are advantageous in many applications such as quality assurance, high throughput synthesis or field testing, because they are inherently fast and have little requirements on the laboratory infrastructure. Harris et al. have given a brief overview of the many currently employed ‘ambient’ ionization sources, of which some are available commercially.21Williams and Scrivens used desorption electrospray ionization (DESI) for the direct analysis of – amongst others – PEO excipients in drug formulations.22 These authors demonstrated the advantages of using gas-phase ion mobility separations in conjunction with direct ionization to improve spectral interpretability. Direct analysis in real time (DART) is another ‘ambient’ ionization technique. Ionization in DART is mainly achieved by reaction of the desorbed analyte with protonated atmospheric water-clusters.21 Rothenbacher and Schwack used this technique to analyze the phthalic acid ester content of toys made of poly(vinyl chloride) in a rapid and non-destructive manner.23
Wesdemiotis and co-workers introduced direct-probe atmospheric pressure chemical ionization (DP-APCI),24 which they used to analyze the thermal desorption/degradation products of several amphiphilic copolymers and networks based on N,N-dimethylacrylamide and dimethylsiloxane. As an intermediate technique between in-vacuum direct probe ionization and APCI, DP-APCI allows structural and compositional characterization of synthetic polymers too large or too complex for in-vacuum techniques.
Flowing afterglow atmospheric pressure glow discharge mass spectrometry (FA-APGD-MS) is a hard ionization technique, that was used by Jecklin et al. to fingerprint a number of synthetic and biopolymers, including polyisoprene, PEO, poly(ethylene terephthalate) and polysaccharides.25 Using statistical data treatment (principal component analysis) these authors were able to rapidly differentiate between pectin, amylopectin and cellulose, three chemically very similar polysaccharides.
MALDI sample preparation
Sample spot preparation is an important part of MALDI-MS analysis. It involves mixing the polymer sample with a matrix, which can be achieved by various methods. One of the most common procedures, known as the “dried droplet” method, involves the deposition of dissolved matrix and analyte either successively to form a “sandwich” layer or together in one droplet. Rings of dried matrix and analyte are often formed after evaporation. This type of preparation cannot be regarded as homogeneous. Often so-called “hot” spots are found, where intense signals can be recorded. Hot spot formation might not be important if the structure of repeating units of a polymer or its end-groups are to be investigated. However, for the analysis of molecular masses and mass distributions homogeneous preparations are indispensable. MALDI and SIMS imaging was used by different groups to demonstrate the homogeneity of sample spots.26,27Depending on the solvents used for the spot preparation, significant lateral molecular mass differences accompanied by segregation of sample and matrix, and of the polymer homologues is observed (see Fig. 2). The transport of sample and/or matrix towards the periphery of the sample spot is caused by capillary flow.28 The segregation of polymers is under investigation and a possible reason is not yet clear. Several processes are imaginable, including solubility or diffusion effects during solvent evaporation.
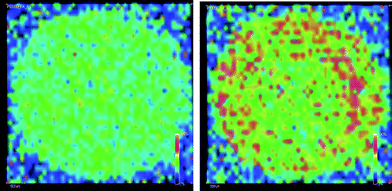 | ||
| Fig. 2 MALDI-imaging of a polystyrene spot (M ≈ 5000 kg mol−1) prepared using the “dried droplet” method (THF, all-trans retinoic acid/AgTFA), ion intensities of PS42 (left) and PS58 (right) showing accumulation of higher masses at the outer periphery of the spot (unpublished results of the authors). | ||
To overcome this problem, several other sample preparation methods have been reported. Since the separation phenomena in a drying droplet are caused during evaporation of the solvent, a straight-forward way to avoid this separation is to avoid the use of a solvent. The solvent-free sample preparation method for the cationization of linear polyethers with alkali metals was reported by Hortal et al.29 A similar method was used by Mazarin to promote the protonation of PEOs with labile end-groups in MALDI.30 Continuous developments of the solvent-free method have led to a multi-sample vortex approach which enables the simultaneous preparation of 384 spots using tiny stainless steel balls for the mixing of sample and matrix, followed by their simultaneous deposition on a MALDI target. Hanton and Stets used microscopy images and MALDI mass spectra to show that remarkably short mixing times (10 s) are required by this vortex method to produce high quality mass spectra.31 The evaporation-grinding method represents an alternative method to prepare MALDI sample spots.32 By means of this procedure, based on a grinding of the dissolved matrix and non-dissolved polymer, the degradation of polysulfones was investigated.32
Another approach to avoid inhomogeneities in solvent-based sample preparation are spray methods. Electrospray deposition has been shown to form homogeneous layers although one has to consider that the molecular mass distribution can change at higher voltages due to fragmentation. Erb and Owens presented a dual-spray electrospray deposition system.33 The homogeneity of sample spots was comparable to that obtained by a conventional single-spray electrospray system. However, the dual-spray system allowed for intimate mixing of separately prepared sample components and resulted in improved quantitative results. The development of this device also allows the mixing of sample components prepared in different solvents without the need to be concerned with solvent miscibility. Air-spraying of the chromatographically separated polymers onto MALDI plates (either pre-mixed with the matrix or directly onto matrix coated targets) results in homogeneous sample traces. This has also been demonstrated by MALDI imaging.27 Recently, an innovative approach for preparing MALDI spots was presented – the electric field enhanced sample preparation via induction based fluidics (IBF). Combined with a new contact-free, small droplet deposition method to dispense nanolitre drops the signal intensity and sensitivity achieved by MALDI-MS was improved remarkably.34
Miniaturization of MALDI spots seems to be a promising approach in order to avoid sample/matrix inhomogeneities and to save sample material. A practical overview on this topic is given by Tu and Gross35
The choice of suitable matrices for MALDI still represents a challenge. Various isomers of dihydroxybenzoic acid (DHB) – a common MALDI matrix – were investigated with respect to the analysis of PEOs using both solvent-free and solvent-based preparations.36 The authors revealed that best results could be obtained with 2,6-DHB. Wurm et al. demonstrated the applicability of pencil lead for the investigation of dimethylsilyl-terminated PS synthesized in a microstructured reactor.37 The ionic liquids (N,N-diisopropylethylammonium alpha-cyano-4-hydroxycinnamate [DEA-CHCA] and aniline/alpha-cyano-4-hydroxycinnamic acid) were used as new matrices, showing their suitability to assist in the ionization of polar biodegradable polymers.38 A general overview to aspects of matrix choice for MALDI was given by Batoy et al.4
Different ultrasonic devices, including an ultrasonic bath with dual frequency, a sono-reactor and an ultrasonic probe, were tested for their ability in the MALDI sample treatment using three polymer standards and dithranol as the matrix.39 The Mn and Mw values obtained for one ultrasonic device were similar to those acquired with vortex mixing and were in concordance with the manufacturers' values.
Ion mobility spectrometry
Although MS techniques are extremely informative, they may be regarded as limited in the sense that they measure ion intensity and mass-to-charge ratios only. A novel and high-level approach for synthetic polymer characterization is the dynamic combination of ion mobility spectrometry and mass spectrometry (IMS-MS) as well as more sophisticated techniques such as IMS with collision-induced dissociation (CID) MS/MS. Ion mobility provides an additional dimension of separation based on structure, similar to chromatography. Ions are separated as they drift through a gas (typically He, N2, or air, depending on the application) under the influence of an electric field. Typical IMS spectra consist of 2D graphs composed of drift time vs. m/z ratios. The drift times depend on the conformational state, the mass and the charge of the molecules, which enables analyte ions to be separated by size and shape. Thus, compact ions traverse the drift tube more quickly than elongated ions (for a given mass and charge state). IMS enables conformational studies, reducing sample complexity. IMS spectrometry is available with common ionization modes such as ESI, APPI, APCI and MALDI.Ion mobility separation enables components previously invisible to traditional ESI or MALDI analyses to be revealed, allowing species of interest within complex polymer samples to be separated from isobaric co-eluting compounds, MALDI matrix ions, or background interferences. For example, peaks from branched polymers are potentially isobaric. The separation of branched and linear species in aramid fibers applying MALDI-IM/TOF-MS was reported by Gies et al.40 MALDI-IM/TOF-MS combined with the evaporation-grinding (E–G) sample preparation (see also chapter “sample preparation”) resulted in 2D plots of drift time vs. m/z showing clusters of peaks, that indicate a clear separation of branched and linear species.
ESI-IM-MS/MS was employed by Hilton et al.41 to separate and differentiate between polyether oligomers with identical nominal mass-to-charge ratios (m/z), but differing structures. For these experiments PEO 1000 and PEO mono-oleates with different EO concentration in isobaric oligomers, and PEO mono-stearates and PEO bis(2-ethyl hexanoate) with isobaric oligomers having a constant number of EO were investigated. Due to the inherent limitations in precursor ion resolution of the used quadrupole analyzer, ESI-MS/MS alone was not able to resolve these isobaric species unequivocally. IM-MS/MS in contrast was able to aid the identification of the backbone and end-groups of the pairs of isobaric polyethers.
ESI of PEOs and PEGylated proteins usually leads to complicated mass spectra due to overlapping charge state distributions. In a study by Bagal et al., gas phase base-mediated charge-stripping of protonated PEO in the atmospheric pressure source region significantly reduced the number of charge states present.42 According to the authors this feature can be easily implemented into commercial instrumentation. By a combination of this method with IMS and TOF mass spectrometry it was possible to further enhance detection sensitivity and accuracy.
Trimpin and Clemmer gave a comprehensive overview of the possible applications of a newly developed high resolution ESI-IM/IM-MS cell demonstrating a fast and comprehensive analysis of a number of synthetic polymers (e.g. ternary blends of PEMA, PtBMA and PnBMA; PEO/PPO blends and random EO–PO copolymers).43 Due to the pre-separation of the different charge states before MS the dynamic range and sensitivity of MS was enhanced. Additionally, the end-group sensitive separation of blends of functional homopolymers and copolymers was demonstrated. Size-to-charge induced folding transitions of molecules that are highly shape sensitive resulted in different drift times. Furthermore, a high throughput analysis with automated sample introduction (<10 min/sample) should be possible. A prospective possibility of a fast interpretation of recorded spectra for quality control by means of automated image analysis was predicted.
Liquid chromatograpy/mass spectrometry (LC/MS)
For the past decade two-dimensional chromatographic polymer characterization has been used for a simultaneous analysis of molar masses and chemical heterogeneity (e.g. end-groups, copolymer composition etc.). The principle is based on a coupling of two different chromatographic modes. In the first dimension molecules are separated according to their chemical heterogeneity by liquid adsorption chromatography at critical conditions (LACCC). In the second dimension fractions are analyzed with regard to their molar mass distribution (MMD) by means of SEC. Frequently, appropriate standards for a calibration of the SEC are not available. MALDI- and ESI-MS are increasingly applied as a proxy for the SEC dimension. MS allows simultaneous access to MMDs and structural information. Concomitantly, in the past two years the LC/MALDI- or ESI-MS coupling was predominantly applied for identification and quantification of pharmaceuticals and biopolymers. Only a few articles concerning synthetic polymers have been published, although the performance of these techniques was already demonstrated.Structure-performance relationships of solid epoxy resins were studied in detail by Julka et al.44 Components were characterized qualitatively and quantitatively by the classical approach of coupling a comprehensive multidimensional liquid chromatography system to ESI-MS. The modes of orthogonal separation applied were SEC in the first dimension and LACCC in the second dimension. Several classes of polymers with different functional groups were separated and identified, including epoxy-epoxy and epoxy-alpha-glycol functional oligomers, and their individual molar mass values were determined. The obtained repeatability was found to range from 0.5% for the main product to 21% for oligomers at the 0.4% concentration level.
Copolymers consisting of neopentyl glycol (NPG), adipic acid (AA) and hexane diol (HD) were investigated by LC/MALDI-MS regarding their composition and end-group distribution.45 The deficiency of MALDI-MS, which was not able to distinguish between the number of NPG-AA and HD-AA units and/or different end-groups of the same chemical structure (mass difference of 14 Da in any case resulting in overlapping signals), was overcome by applying ESI-qTOF-MS. This method offers a very high mass resolution especially in the lower mass regions. Yet another way of solving this problem is to use tandem mass spectrometry. The same copolymers were investigated by MALDI-(CID)-MS/MS coupled to LC. The fragmentation of suitable parent mass ions resulted in typical fragment ion patterns and provided a clear differentiation between cyclic and linear oligomers. End-groups of isomeric linear oligomers were easily differentiated. In addition, this technique could be used for a detailed examination of longer copolymer sequences.45
Recently, the coupling of ultra performance liquid chromatography (UPLC) with ESI-TOF-MS was reported for the characterization of complex synthetic copolymers (see Fig. 3).46 For the first time, the determination of the conditions for LACCC of PEO and poly(propylene oxides) by means of this coupling method was demonstrated. These conditions were applied for an investigation of various PEOs and their copolymers.
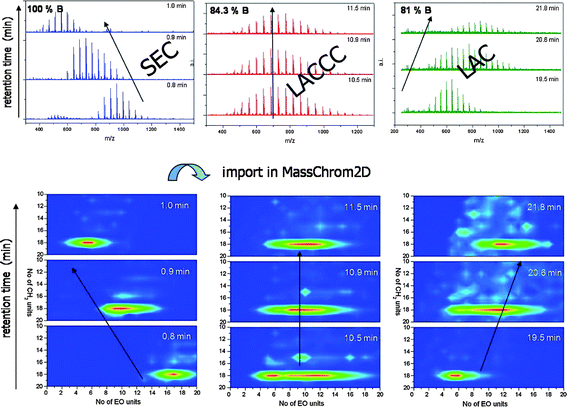 | ||
| Fig. 3 Determination of critical conditions of adsorption for PEO with UPLC/ESI-TOF-MS.46 In contrast to established, mostly laborious routines to find suitable chromatographic separation conditions, the chosen coupling method enables a very fast adjustment of parameters. Similar to LC/MALDI-MS a two-dimensional analysis of homo- and copolymers regarding chemical structure (functionality-type distribution), copolymer composition and molar mass can be performed simultaneously. Furthermore, there is no need to use specific polymer standards of different molar masses for a determination of critical conditions and SEC calibration. The figure is taken from ref. 46 with kind permission of the American Chemical Society. | ||
Desmazieres et al. described the advantages and limits of an APCI Interface for HPLC- and SEC-MS analysis of synthetic polymers.47 Industrial polymers with molecular masses of up to 5 kg mol−1 such as polyethers, polysiloxanes and their copolymers were investigated. The results were discussed with regard to what is obtained by classical techniques, namely SEC and MALDI-MS. In-source decomposition, which was observed above m/z = 2000–3000 Th was the main drawback of the APCI interface. This can induce an underestimation of average molar masses. However, APCI allows the detection within a wide range of sample/solvent polarity and appears to be complementary to ESI.
Barman et al. determined polyethylene oxide (PEO) impurities in two monofunctional poly(ethylene oxides) (PEO methyl ether [M-PEO] and PEO vinyl ether [V-PEO]) quantitatively by HPLC coupled to APCI-MS.48 A quantification, which is a well-known but unsolved problem using ELS or MS detectors, was achieved by comparing LC-MS mass spectra of different ratios of monofunctional PEO and PEO, and matching peak retention times with those of available PEO standards for all M-PEO and V-PEO sample types. This information is helpful in selecting the appropriate PEO standard to determine PEO content in each sample type. ELSD response factors for various PEO standards were also compared. Schneider et al. investigated polydimethylsiloxanes for medical applications, comparing various analytical techniques: Py/GC-MS, ESI-MS, MALDI-TOF-MS, LC-MS.49 Among these techniques, LC/APCI-MS coupling allowed the fastest and most effective analysis. In addition, the complexity of the mass spectra deduced from these LC/MS experiments was simplified compared to the mass spectra obtained by MALDI-TOF. The authors demonstrated how the LC/APCI-MS coupling permits the complete characterization of end-groups being present in very small quantities for this class of polymers.
A further contribution concerning on-line coupling of LACCC with ESI tandem MS addresses the characterization of PEO/PS block copolymers at critical separation conditions of PEO, using a mobile phase containing the cationizing agent to avoid dilution effects. Samples were investigated in both MS and MS/MS mode,50 Oligomers were successfully separated according to the PS block size and additional information on the structure was obtained from simplified MS spectra. The microstructure of this copolymer, synthesized by nitroxide-mediated polymerization, was unambiguously identified in LCCC/ESI-MS/MS experiments, since the PS block size could be determined by its chromatographic behavior and MS/MS pattern.
Tandem mass spectrometry
Much attention has been paid to the application of tandem MS techniques in polymer analysis over the recent years. Tandem mass spectrometry (MS/MS or MSn) employs multiple stages of mass analysis in order to examine selectively the fragmentation of particular mass-selected ions. In this technique, often two mass analyzers are separated by a collision cell in which an inert gas (e.g. nitrogen or noble gas) is allowed to collide with a mass-selected sample precursor from the first MS unit. This is known as collision induced dissociation (CID). In the second MS unit, product (or fragment) ions are separated and detected. The most common combinations are quadrupole-TOF (q-TOF) and TOF/TOF instruments which allow the products of a single fragmentation process (MS2) to be detected with great mass precision. Quadrupole ion traps and ion cyclotron resonance (ICR) mass spectrometers allow storage, selection, fragmentation and analysis of the precursor and product ions in the same cell, thereby allowing multiple fragmentations (MSn with n > 10) to be carried out.While fragmentation analysis was always a current practice for the mass spectral characterization of biopolymers (e.g. peptide sequencing), it was not until the past few years that the number of applications of MS/MS in the analysis of synthetic polymers started to increase considerably.
Especially in MALDI-MS, some technological advances are notable: recent years have brought about an increasing number of commercially available MALDI instruments with the capability to perform collision induced dissociation (CID) of mass selected precursor ions. In contrast to earlier reflectron time-of-flight (TOF) instruments, featuring post source decay (PSD) as the only fragmentation mechanism, these new instruments provide high spectral resolving power and controlled fragmentation.
Hercules pointed out the advantages of performing MALDI-MS/MS for polymer structural characterization.51 An unequivocal assignment of both individual end-groups is obtainable from tandem MS, as compared to only the combined end-group mass from single stage MS. Benefits also lie in the structural analysis of polymers of isomeric monomers such as 4-alkyl-substituted polystyrenes and poly(methyl styrene) isomers. Hercules and co-workers analyzed these polymers by CID as well as poly(phenyl sulfone) and poly(sulfone)s in further detail.32,40,52 Fragmentation patterns in MALDI-TOF/TOF could be assigned and, based on information gained from additional pyrolysis-GC-MS experiments, preferential cleavage sites of the polymer were identified.
MALDI and ESI-MS/MS fragmentation analysis was applied for the structural investigation of isomeric dendrimers53 and for the elucidation of the gas-phase fragmentation of half- and first-generation polyamidoamine (PAMAM) dendrimers.54 Furthermore, electron capture dissociation (ECD) and CID of complexes of these dendrimers with metal ions were determined by CID followed by Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS).55 The mechanism of fragmentation of Fréchet dendrons was investigated by Baytekin et al.56
Baumgaertel et al. used MALDI-CID-TOF/TOF for the structural investigation of OH-functional PMMA57 as well as for methacrylate functional and non-functionalized, OH-terminal 2-ethyl-2-oxazoline oligomers.58 The authors found a significant influence of the end-group on fragmentation: the polyoxazoline macromonomer underwent initial cleavage of the methacrylate followed by (charge-induced) unzipping of the polymer backbone. In contrast, the OH-terminal polymer fragmented via multiple bond cleavages in the backbone, explained by McLafferty type reactions, as well as elimination of ethane or hydrogen in a mechanism similar to that observed for the structurally similar PEOs. The authors furthermore suggested that future studies into the CID of polyoxazolines carrying end-groups of different leaving ability will pave the way towards the construction of MS/MS product ion libraries. These may eventually aid in the automated assignment of CID-spectra of homo and copolymers. Giordanengo et al. investigated the tandem mass spectrometry of poly(methacrylic acid) (PMAA) ionized by negative mode59 and positive mode ESI.60 Two major fragmentation pathways were found to be operative: dehydration and cleavage of H2O between two adjacent acrylic acid units to form the anhydride (charge remote) and decarboxilation were most likely to occur in a charge-induced mechanism. Both reactions could potentially be used to identify the MAA content in MAA-containing copolymers. Other examples include the structural investigation of (R,S)-3-hydroxybutyrate/(R,S)-3-hydroxy-4-ethoxybutyrate copolyesters by ESI-MS/MS, as well as isocyanate oligomers61,62 and poly(ester amide)s from sebacic acid and 4-amino-1-butanol by MALDI-CID-MS.63
The mechanism of LACCC separation of copolyesters was confirmed by MALDI-MS/MS and the structure of different end-groups as well as short copolymer sequences was determined by Weidner et al.45 The ability of MALDI- and ESI-MS/MS for a structural assignment of individual end-groups of PMMA and PPO was shown by Jackson et al.64 The nature of the end-groups of novel azo-functional oligoesters synthesized through bulk ring opening of ε-caprolactone and D,L-lactide (LA) was shown in an ESI-MS/MS fragmentation experiment by Peptu et al.65
Polyesters and polyacrylates were investigated by Simonsick et al. by means of laser desorption (LD) FT-ICR-MS.66 The authors showed the advantages of using ultrahigh resolution FT-ICR-MS for the structural analysis and elucidation of the isomeric composition.
The degradation of the backbone of silver-cationized polystyrenes and backbone substituent effects were studied by Polce et al. using ESI-MS/MS and MALDI-MS/MS.67 They demonstrated that the spectral abundance depends on the collision energy and noted a significant influence of the flight times on the spectra, using different qTOF, q-trap instrumentation. It was also shown that CID exhibits some similarities to thermal degradation, and that changes in PS backbone structure can have dramatic effects on the dissociation pathway. Similar studies were performed for linear and branched polyacrylates revealing that MS/MS can aid in determining the branching architecture of hyperbranched oligomers.68 The fragmentation chemistry of gas-phase adducts of poly(dimethylsiloxane) oligomers was compared using both, metal and organic adduct ions. The results indicated that fragmentation of the oligomer ions highly depends on the adducted charged species.69 In a series of publications, Charles and co-workers focussed on the analysis of a styrene/ethylene oxide block-copolymer (PEO-b-PS) synthesized by nitroxide mediated polymerization of styrene, employing an oligo(ethylene oxide)-functional controlling agent (see species 7,4 in Scheme 1 for structural details).70–72 In a first study, the authors characterized the copolymer by ESI-CID-MS/MS.70 For both, the lithium as well as the silver adducted macroion, dissociation proceeded via an initial radical cleavage of the nitroxide group. This was followed by a radical unzipping of the PS block (see Scheme 1) from which the size of the PS block could be determined.
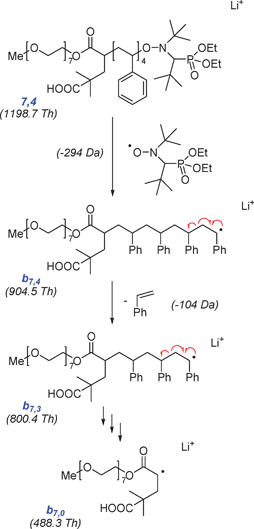 | ||
| Scheme 1 During collision induced dissociation (CID) MS/MS, nitroxide terminal poly(ethylene oxide-b-styrene) 7,4 undergoes an initial homolytic cleavage of the nitroxyl radical. The radical polymer fragment b7,4 subsequently undergoes a sequence of depolymerisation steps, until only the cationized poly(ethylene oxide)-functional leaving group radical is left.‡ The scheme has been adapted from ref. 70. | ||
The observed mechanism differed significantly from the fragmentation mechanism observed for PS carrying stable end-groups (alkyl, OH), as only b-type fragments were observed,67 highlighting the great influence labile functional end-groups can play in determining polymer fragmentation pattern. Structural information on the cleaved nitroxide radical was gained from minor product ions in the fragmentation spectra. The same group was also involved with tandem MS of doubly charged PEO oligomers.73
Mazarin et al. later used quantum chemical calculations in addition to NMR-measurements to rationalize the observed fragmentation reactions of nitroxide functional PEOs with different adducted cations.74 This interesting study showed that an advanced theoretical understanding of the complex ionization and dissociation processes of synthetic polymers can help in tuning and interpreting the structural information obtained in (co)polymer analysis by MS/MS. The prediction of polymer fragmentation patterns based (partially) upon quantum chemical calculations and computational modelling studies may be of increasing value in the future.
Copolymer characterization
Due to the convolution of the compositional variation and the molecular weight distribution, copolymer spectra are usually quite complex. The different ionization abilities of the constituting monomers may inhibit quantitative compositional analysis. Nevertheless, the past decade has brought about a number of successful approaches towards qualitative and semi-quantitative copolymer characterization by MALDI-TOF MS and to a lesser extent ESI-MS.75 The application of tandem mass spectrometry to investigate the co-monomer sequence from the fragmentation pattern of selected oligomer ions adds yet another dimension to the analysis of copolymers by MS.5Information on the constitution of co-oligomers can be represented in an easily interpretable form by the use of a 2D compositional plot, also called copolymer ‘fingerprint’, shown in Fig. 4. These fingerprints were recently used by Willemse and van Herk to derive accurate sets of free radical copolymerization parameters from a MALDI-MS analysis of the produced copolymer (see radical polymerization).76 A good example of this type of diagram can also be found in a study by Kasperczyk et al. (discussed in polymer degradation) who monitored the changes in the composition of a glycolide/ε-caprolactone copolymer upon degradation in an aqueous medium.77
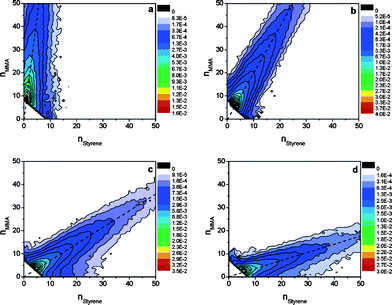 | ||
| Fig. 4 Copolymer fingerprint plots obtained from the pulsed laser initiated radical copolymerization of methyl methacrylate (MMA) and styrene at a molar feed ratio (xSt) of: xSt = 0.053 (a), xSt = 0.249 (b), xSt = 0.600 (c), and xSt = 0.792 (d). The dashed lines in the copolymer fingerprint plots are indicative of the average chemical composition of the copolymer. The figure is taken from ref. 76 with kind permission of the American Chemical Society. | ||
Polyurethanes (PU) are industrial polymers that find a range of applications as foams, fibres, coatings and elastomers. The final polyurethane usually consists of a network formed by crosslinking isocyanate co-oligomers, whose monomer composition greatly influences the properties of the final material. Characterization of PUs by spectroscopic methods such as NMR or FT-IR as well as by chromatography delivers only average monomer compositional information. In order to determine the co-monomer sequence of a sample made of crosslinked methylenebiphenyl diisocyanate and toluene diisocyanate, Pasch and co-workers therefore reverted to MALDI-TOF-MS.62 After hydrolysis of the initial network material, the authors showed that MALDI not only identified the co-oligomer composition of the formed individual tri- and tetramers; using collision induced dissociation (CID) experiments, mass peaks were identified, which were specific for block-like and random monomer segments.
Adamus et al. analyzed multi- and triblock copolymers based on 1,5-dioxepan-2-one and ε-caprolactone (DXO/CL), used in biomedical applications such as tissue implants and artificial cartilages.78 The copolymer composition, average block lengths and molecular weights were determined by 1H, 13C-NMR and SEC. Due to the small mass difference of the two co-monomers of only 2 g mol−1, the mass spectra of the formed copolymer remained quite interpretable, so that the end-groups and structures (linear/cyclic) of the DXO/CL copolymer could be determined by the authors. Due to mass discrimination effects DXO-enriched hydrophilic polymer ions dominated the mass spectrum. It was concluded that despite of this, MALDI clearly revealed useful information on the microstructure of these block copolymers. 3-Hydroxybutyrate/3-hydroxy-4-ethoxybutyrate copolyesters formed by anionic ring-opening polymerization were another type of copolymer analyzed by Adamus.61 ESI-CID-MS/MS was employed to study in detail the copolymer composition and end-groups. The author established that in CID chain fragmentation mainly proceeded by β-hydrogen type rearrangements. Significant differences were found in the tandem mass spectra of diblock- and random copolyesters. MS/MS could therefore be employed to identify the microstructure (block or random) of these copolymers similar to the study of Pasch and co-workers.62
In the case of a PS/PEO block copolymer investigated by Mazarin and co-workers, the two blocks were linked together by a weak ester bond (see 7,4 in Scheme 1). It was therefore possible to analyze both blocks individually after a saponification reaction.71 This greatly reduced spectral complexity and discrimination effects due to the amphiphilic nature of the block copolymer. Molecular weights of the individual blocks determined by MALDI-MS were compared to those derived from NMR and SEC. The authors noted that using MALDI with a DCTB/silver trifluoroacetate matrix, the nitroxide functional PS could not be detected, ESI-MS with silver nitrate yielded the intact nitroxide capped PS macroion.
The prospect of applying mass spectrometry to copolymer characterization is alluring, as no other technique provides structural information itemized for each co-monomer repeat unit, thereby allowing detailed sequence information to be derived from the mass spectra. At the moment, some expertise is required in the interpretation of the obtained data, and one is far from being able to extract quantitative compositional and sequence information from tandem MS spectra. The great challenge lies in transforming tandem mass spectral data of copolymers into chemically useful information with a minimum of required user input, ideally by quantitative and automated interpretation of the complex copolymer fragmentation patterns.
Quantitative mass spectrometry
The determination of accurate molecular weight distributions of synthetic polymers as well as the quantification of the detected species in mass spectrometry remains an exciting and challenging task. The potential applications of accurate mass and concentration data from mass spectrometry include fields such as synthetic end-group transformations, polymer degradation and the kinetic analysis of polymerization processes. The kinetic parameters of polymerization processes are often determined from the molecular weight distribution obtained from a controlled polymerization experiment. In these experiments, any improvement in the accuracy of molecular weight data will benefit the evaluation of the kinetic rate coefficients. The availability of a technique that allows the accurate determination of molecular weights regardless of the analyzed polymer class is therefore highly desirable. The application of mass spectrometry is an interesting option, firstly because – in contrast to SEC – molecular weights determined by MS are far more accurate and the accuracy does not depend on the type of polymer analyzed. Additionally, the potential of gaining structural and compositional information on functional polymers and copolymers in conjunction with molecular weight is a great advantage of mass spectrometry. These data contain valuable additional information for the polymer kineticist. The possibility of putting an accurate concentration value on each species in a mixture of polymers or in a copolymer has potential to greatly benefit the yield of mechanistic and kinetic data from polymerization experiments. However, everyday synthetic polymer chemistry may also benefit from mass spectrometry, as a tool to quantitatively monitor molecular weight of produced polymers, end-group conversion of functional polymers or the formation of side products during the polymerization. Mass spectrometry delivers this information at a high speed and relatively hassle-free.However, quantitative mass spectrometry remains a challenging field. The past decade of extensive research into the ability of ESI- and MALDI-MS at reproducing molecular mass distributions has brought about the realization of the limitations of this technique. The dependence of the ionization process (especially in ESI), ion transmission and ion detection on the molecular weight and composition of the polymer poses limits to the ability to achieve a quantitative polymer analysis without additional means of calibration. Instrumental mass bias will skew the apparent MWD of polymers to lower (or higher) masses therefore leading to a systematic error in the obtained molecular weight averages.
In MALDI as well as in ESI, there have been notable developments towards solving long standing limitations imposed by mass bias. Aksenov and Bier applied a new type ion detector – a superconducting tunnel junction (STJ) cryodetector – in conjunction with MALDI-TOF mass spectrometry towards the analysis of PS into the mega Dalton mass range.79 A great advantage of the STJ cryodetector over traditional electron multipliers is that in theory this detector shows no loss in sensitivity at high masses, aiding to decrease mass bias in polymer MS. The detector is energy dispersive, which allows for the discrimination of the detected species based on charge state, which may help to deconvolute the overlapping charge envelopes frequently observed in ESI-MS.43
The ionization process itself is arguably the greatest source of mass bias in polymer mass spectrometry. Schlosser et al. investigated the influence of the matrix-to-analyte ratio in MALDI-TOF MS of poly(L-lysine).80 In a detailed investigation of an equimolar mixture of three monodisperse lysine oligomers (10-, 15-, 25-mer) the relative intensities varied by over an order of magnitude as a result of simply changing the analyte-to-matrix ratio. At low relative polymer concentrations, it was argued that instrumental discrimination favoured low molecular mass oligomers, leading to an overrepresentation of these species in the mass spectrum. At high polymer concentrations higher mass oligomers were overrepresented. This was explained by saturation effects or ion-molecule reactions in the MALDI-plume, favouring ionization of the heavier oligomers. As a consequence it was concluded that significant attention needs to be paid to whether analyte concentration effects alter the observed concentration distributions whenever attempting quantitative MALDI analysis.
Fourier transform ion cyclotron mass spectrometry (FT-ICR-MS) is today the highest resolving mass spectrometric method available. In contrast to time-of-flight mass spectrometry, which features theoretically no upper mass limit, analysis by FT-MS is usually limited to a narrow mass-to-charge range. Miladinovic et al. investigated the application of novel modes, using ramped trapping potentials to allow storage of sodiated PEO over a wide mass range of up to 1 × 104 Th‡ in the cyclotron cell for subsequent analysis.81 The method was compared with an earlier ‘integral’ trapping technique as well as with linear and reflectron mode MALDI-MS. It was shown that high resolution, high mass analysis is possible by FT-MS, but limitations are reached if the individual masses are to be quantified by the new method, whereas the integral trapping technique was shown to be superior in the investigated m/z range. Tandem mass spectrometry using sustained off-resonance irradiation collision-induced dissociation (SORI-CID) was furthermore shown to be ineffective for m/z in excess of 6 × 103 Th.
Schnöll-Bitai et al. advised that the quantitative evaluation of MALDI-TOF spectra to derive molecular weight distributions requires a number of data transformations to be made prior to analysis.82 These include a proper baseline estimation, coordinate transformations from the time-of-flight to the mass axis and – most importantly – corrections for instrumental mass bias to be made for broad distributions. Determination of truly accurate molecular weight distributions can still be challenging and requires significant analytical expertise, even for polymer samples that fulfil the criterion of narrow polydispersity. Guttman et al. recently presented the result of a years-long journey towards providing an absolute mass distribution standard (NIST SRM 2881), of which the MWD was determined by MALDI mass spectrometry alone.83 An internal calibration employing end-group functional PS as internal standard allowed these authors to account for the inherent mass bias of MALDI-MS. The meticulous approach also comprised the creation of novel methods in reproducible MALDI sample preparation,84 extensive testing of the robustness and repeatability of MALDI by inter-laboratory comparison,85 numerical optimization of instrumental parameters86 and novel approaches at the unsupervised analysis of mass spectra.87 This enabled the authors not only to provide molecular mass and concentration data from mass spectrometric methods alone, certification also involved an estimation of the random and systematic errors in molecular weight distributions acquired by the MALDI method. The authors noted that MALDI-sample preparation was the greatest source of uncertainty in the data (also refer to MALDI sample preparation).
Pullulans are polysaccharides made of a sequence of maltotriose subunits. These polymers are of interest as calibration standards for aqueous SEC. Pullulans easily undergo fragmentation in MALDI which has so far precluded them from MWD analysis by mass spectrometry. Schnöll-Bitai and co-workers tested a series of matrices for the characterization of these fragile polymers.88 It was found that the ionic liquid matrix (ILM) 2,5-dihydroxybenzoic acid butylamine (DHBB) was superior to the crystalline matrices 2,5-dihydroxybenzoic acid (DHB) and 2,4,6-trihydroxyacetophenone (THAP). With this matrix, better reproducibility was achieved due to the more homogeneous sample distribution and less chemical background noise was observed, which is a general advantage of ILMs. Model simulations of the fragmentation events showed that during ionization with DHBB significantly less fragmentation occurred and that the obtained molecular weight data were of high reliability.
MALDI-MS coupled offline to size exclusion chromatography can be used as an accurate molecular weight sensitive detector in SEC. Chikh et al. used this approach together with traditional calibration by PS standards to investigate the molecular mass and branching structure of hyperbranched polyesters based on 2,2-bis(hydroxymethyl)propanoic acid.89 Surprisingly, the ‘apparent’ molecular weight calibration obtained with PS standards was in close agreement with the ‘true’ MS-derived calibration. This was explained by two antagonist effects: the loss in hydrodynamic volume due to chain branching and the greater hydrodynamic volume of linear polyesters as compared to PS of the same molecular weights (caused by increased chain-stiffness).
In contrast to MALDI, polymer molecular weight analysis by ESI is somewhat limited due to the fact that ESI produces multiply charged ions and may exhibit stronger mass bias. As a great advantage, electrospray ionization can be coupled online to chromatographic separations, allowing fast sample throughput. This is not possible for MALDI-MS which – as a solid state technique – has to be interfaced offline to SEC. Under many circumstances, ESI may also provide a softer ionization process than MALDI, which is of great importance in the analysis of functional polymers. Online ESI-MS as a means of molecular weight calibration in SEC, pioneered by Simonsick and co-workers in 1993 has been established for some time now.90 However, there is a plethora of additional information in the mass spectra derived from online coupling SEC to mass spectrometry, which remains unused in a simple calibration approach.
Gruendling et al. have recently applied a sophisticated data processing algorithm based on the maximum entropy (MaxEnt) principle to data obtained from polymer analysis by SEC/ESI-MS.91 The retention time of oligomers as a function of molecular weight is only one parameter that can be obtained by online MS. The full chromatographic profile of the oligomers eluting from the size exclusion column is intrinsically recorded by online MS (see Fig. 5, left). This data contains information on the band broadening effects which usually deteriorate molecular weight data from SEC. Maximum entropy processing allowed the individual chromatographic elution profiles recorded by MS to be used to restore the original molecular weight distributions corrected for chromatographic band broadening from the original RI-chromatogram. Samples with molecular weights of up to 15 kg mol−1 could be analyzed by this procedure using simple quadrupole ion trap instrumentation. The manufacturer-indicated molecular weight averages of narrow PMMA standards, determined by light scattering were shown to be higher than the SEC/MS-derived values by 5–14%. Still, more information is inherent to the online mass spectrometric data, when samples are analyzed that contain mixtures of polymers differing in their chemical end-groups. The unique capability of online mass spectrometry to identify these functional polymers as well as to quantify them with respect to each other was used by the same authors to restore individual molecular weight distributions of binary and ternary mixtures of functional PMMA.92
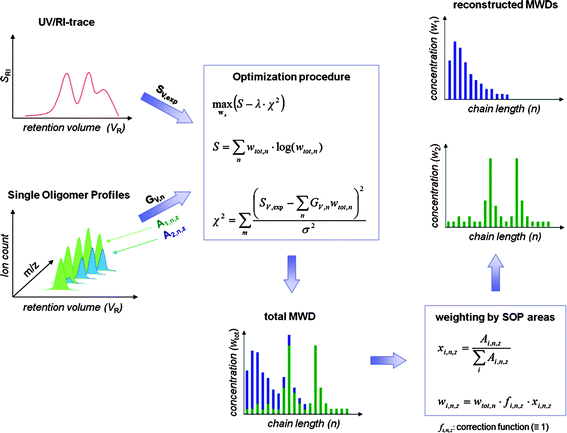 | ||
| Fig. 5 A flow diagram of the principal approach used to arrive at the individual molecular weight distributions of the components in a mixture of two end-group-functional polymers. The instrumental calibration and band-broadening data derived by MS, as well as the RI detector trace are processed to arrive at the deconvoluted total MWD. Weighting by the areas of the SOPs of the functional oligomers yields the reconstructed individual molecular weight distributions. The figure is taken from ref. 92 with kind permission of the American Chemical Society. | ||
Restoration of the individual MWDs is possible by a simple weighting procedure of the total molecular weight distribution derived by the MaxEnt algorithm with the relative peak areas recorded by mass spectrometry (see Fig. 5, lower right).
Radical polymerization mechanisms
The elucidation of reaction mechanisms is paramount for the determination of optimum reaction conditions as well as for the design of agents that can control polymerizations. Soft ionization mass spectrometry offers a unique opportunity to investigate the mechanism – and to some extent also the kinetics – of polymerization processes as a detailed image of the end-groups is obtained. In addition, modern mass spectrometry hyphenated with on-line (or off-line) chromatography techniques allows for an absolute determination of the molecular weight distribution (see above), which can also be applied to elucidate the kinetics and mechanism of polymerization reactions. In the following section, we will explore the latest developments in the application of mass spectrometry towards the study of reaction mechanism and kinetics for variable radical polymerization systems. Recent investigations have also focussed on non-radical chain- and step-growth polymerizations. However, due to personal expertise, we limit our discussion to the trends that have emerged in radical polymerization within the past 5 years, i.e. since we last reviewed the field.1Propagation
The study of the propagation reaction in free radical polymerization is a domain that is in the majority of cases addressed by the IUPAC recommended pulsed laser polymerization–size exclusion chromatography (PLP/SEC) technique.93 Mass spectrometry was initially thought to be an ideal tool to substitute for the statistical error-prone SEC analysis, yet it became increasingly clear that the shape of the molecular weight distribution could only be satisfactorily determined in selected cases,82,94 as chain length dependent ionization and instrumental biases often lead to a distorted representation of the molecular weight distribution. Recently, however, significant progress has been made in the determination of absolute molecular weight distributions via coupled SEC/ESI-MS as noted above,90,91 opening an opportunity to revisit the substitution of SEC for the analysis of PLP samples. Barner-Kowollik and colleagues have demonstrated the determination of virtually error free molecular weight distributions for the homologous series of polymethyl-, ethyl- and butyl methacrylate prepared via PLP.95 Not surprisingly, the obtained propagation rate coefficients and associated Arrhenius parameters (activation energy, EA, and pre-exponential factor, A) are beset with a very small random error (in the range of less than 0.2 kJ mol−1 for EA). While the molecular weight accuracy of the approach is extremely high, it is limited by the accessible molecular weight range. When monomers are employed that lead to well-ionisable polymers (such as acrylates or methacrylates), molecular weights of around 15 kg mol−1 are accessible with conventional quadrupole ion trap mass analyzers via the exploitation of higher charge states during the ionization process. The limitation in the molecular weight range requires the use of a fast pulsing laser system (up to 500 Hz) to limit the number of propagation steps that can occur between two consecutive laser pulses. In addition, the molecular weight limitation currently disallows the use of high temperatures for rapidly propagating monomers.In an alternative approach, Willemse and van Herk applied MALDI-TOF-MS to the pulsed laser initiated copolymerization of methyl methacrylate and styrene.76 These authors derived so-called fingerprint plots (see Fig. 4) – 2D contour plots of the probability distribution of the individual co-monomer chain-lengths – from the mass spectra. A fit of the implicit penultimate unit model, resulted in accurate point estimates of the monomer and radical reactivity ratios, r and s (rSt = 0.517, rMMA = 0.420, sSt = 0.296, sMMA = 0.262). Although it was noted that compositional data from MALDI-TOF-MS always has to be interpreted with caution, no significant ionization bias seemed to be present in the current case, which is likely due to the generally low compositional dispersity of chain-growth copolymers. The accuracy of the parameters was confirmed by a comparison with Monte Carlo simulations. A further aspect that has received continued recent attention in both SEC/ESI-MS studies and MALDI-TOF investigations of PLP prepared samples is the question of the chain length dependence of the propagation rate coefficient. The recent PLP/SEC/ESI-MS investigations support – similar to earlier results obtained via PLP/MALDI-MS96,97 – the notion that the propagation rate coefficient is indeed chain length dependent and this dependency can be adequately described by an empirical model recently introduced by Russell and colleagues.96
The coupling of SEC to ESI-MS methods for the analysis of PLP generated distributions has substantial potential to access virtually (statistical) error free propagation rate coefficients. It would thus be highly desirable if ESI-MS could be expanded to even higher mass ranges, as SEC/ESI-MS is the in principal most suitable tool for the analysis of PLP distributions. This may be possible by the application of high resolution instrumentation.
Initiation
The initiation process in radical polymerization is of paramount importance as it determines – at least in non-controlled radical processes – the polymer end-groups. Thus, the study of initiation is a core domain of polymer mass spectrometry. From the multitude of initiation processes that are available to induce macromolecular growth, many have been assessed via mass spectrometric techniques. Most recently, the study of the photo-initiation process, γ-radiation-induced radical generation as well as thermally induced initiation via the use of peroxides has been in the focus of mass spectrometric polymer analysis.Initiation via peroxides is one of the most frequently employed methods to generate radical species. Buback, Vana and colleagues have applied ESI coupled to both quadrupole ion traps as well as FT-ICR mass analyzers to map the reaction pathways that occur during peroxide initiation as well as the initiator efficiency, f, of the employed peroxides. Most recently, these authors presented an ESI-MS based approach to determine f in both bis-3,5,5-trimethylhexanoyl and dibenzoyl peroxide initiated polymerizations of methyl methacrylate with high accuracy.98 The method makes use of a comparative evaluation of mass spectrometric peak intensities of macromolecules that have been initiated by a cocktail of initiators, of which one serves as an internal reference. Currently, the above study is the only example where firm values for f have been obtained via ESI-MS. The employed methodology is limited to polymers that are readily ionisable and have a molecular weight within the analytical ESI-MS range. Nevertheless, the approach represents an attractive option for estimating f. In previous studies, the same authors have investigated the fragmentation pathways of peroxypivalates,99 diacyl peroxides,100 peroxyacetates,101 and peroxydicarbonates102 in variable polymerizations. In addition, Wang and Hutchinson studied the high temperature (138 °C) tert-butyl peroxyacetate initiated polymerization of butyl methacrylate via MALDI-TOF analysis, which demonstrated the occurrence of methacrylate back-biting, a reaction usually associated with acrylate systems.103
Photo-initiation is an important method for generating primary radicals, which is being employed in both fundamental kinetic investigations (e.g. PLP/SEC(/ESI-MS), as well as single pulsed laser polymerization coupled to both time resolved IR and electron spin resonance spectroscopy104,105) as well as industrial photo-curing applications. Most recently, ESI-MS coupled to quadrupole mass analyzers has also been used in our group to both qualitatively and quantitatively study the fragmentation pathways of a multitude of photo-initiators.106,107,108 These investigations are aimed at providing a reactivity map of the initiating radicals towards different monomer species. Interestingly, the literature abounds with investigations into the fragmentation pathways of photo-initiators, yet the more significant question – at least from a polymer chemist's point of view – is which radical fragments are identified as chain ends in what proportions. ESI-MS investigations on photo-initiated polymerization of methyl and butyl acrylate, methyl and butyl methacrylate as well itaconates with variable substitution patterns employing both symmetrical as well as unsymmetrical photo-initiators have demonstrated that the propensity with which primary radicals add to monomers varies greatly.106 For example, Barner-Kowollik and colleagues have recently shown that mesitoyl radical fragments contribute a factor of eight times less than benzoyl fragments to the initiation process of methyl methacrylate.107 These authors employed initiator cocktails (in the above case a mixture of mesitil and benzoin) to systematically vary the proportion of the individual radical fragments in the polymerizing mixture. The photolytic cleavage was induced by a pulsed laser beam (100 Hz) at 351 nm. The pulsing action of the laser ensures – similar to the above discussed PLP experiments – that the generated molecular weights are within the analytical window of the ESI-MS. Quantification was achieved via the integration of the signals associated with radical termination by disproportionation, as these peaks represent species that can be clearly assigned to one initiating radical. Methyl methacrylate is a particularly well-suited monomer for such investigations as its associated propagating radicals mainly terminate via disproportionation. In all ESI-MS studies that seek to quantify specific amounts of end-groups, it is highly important to ensure that no (chain length dependent) ionization bias is affecting the results. Typical approaches to establish whether such biases are affecting the system include an evaluation within each repeat unit pattern and a subsequent plot of the molar fractions of each end-group species in the polymer as a function of the chain length, as well as a recently introduced method, which allows for the deduction of end-group bias free integration results by comparing neighbouring repeat unit intervals.107 Additional evidence for the disparate initiation abilities of initiator derived radicals comes from studies of Wyzgoski et al. who demonstrated via off-line SEC/MALDI-MS that styrene and methyl methacrylate photo-polymerizations initiated with (2,4,6-trimethylbenzoyl)diphenylphosphine oxide mainly carry the highly reactive diphenylphosphinyl group as initiating chain end.108
Among the more recent mass spectroscopic studies directed at unravelling initiation mechanisms is the relatively unusual initiation by γ-radiation. Similarly to photo-irradiation, it can be readily switched off and operated conveniently at ambient temperatures. Its main advantages lie in the very low radical fluxes that can be generated and the absence of any initiator species, as the monomer (or additionally present solvent molecules) are transformed into radicals via the ionizing radiation. An ESI-MS analysis of polymer generated via γ-radiation induced polymerization requires the presence of a species which is able to keep the molecular weights within the mass spectrometric analytical window. Two recent studies into this topic from our laboratories employ reversible addition fragmentation chain transfer (RAFT) agents to control the molecular weight. These studies investigate the bulk RAFT mediated polymerization of methyl and butyl acrylate109 as well as the RAFT mediated polymerization of acrylic acid and N-isopropylacrylamide.110 These studies demonstrate that the initiation can proceed via H radicals derived from the monomer, H or OH radicals derived from water as well as via the homolytic radiolysis of the controlling RAFT agent. In addition, the investigations demonstrate that polymers which contain acidic protons, such as poly(acrylic acid), poly(N-isopropylacrylamide), and even the CH-acidic poly(dialkyl vinylphosphonate)s can lead to mass spectra where these labile protons have been substituted with sodium and/or potassium ions, leading to additional peak series.111 Another rather exotic way to initiate free radical polymerizations is by electron beam irradiation. Pusch and van Herk elucidated the initiating end-groups in the pulsed electron beam initiated emulsion polymerization of methyl-, ethyl- and butyl methacrylate using MALDI-TOF MS.112 Hydrogen and hydroxyl radicals were identified as the main initiating species, as in the case of initiation by γ-radiation. Additionally, an increasing degree of initiation by monomer radicals was observed with decreasing water solubility of the polymerized monomer. The major species formed by radiation induced polymer decomposition were also identified.
The above recent examples highlight that the use of (SEC/)ESI-MS and – to a slightly smaller extent – MALDI-TOF MS has matured to a level where quantitative data on initiation probabilities and reactivities can be accurately derived. The emerging trend of analyzing initiation processes via soft ionization mass spectrometry in a quantitative fashion will undoubtedly continue and provide – for the first time – reliable data on the efficacy of individual radical initiation pathways.
Termination and transfer
The study of termination and transfer events in radical polymerization is traditionally the domain of kinetic investigations such as SP-PLP-ESR, SP-PLP-NIR, RAFT-CLD-T or SP-PLP-RAFT-NIR.105 However, recently progress can be seen in studying aspects of these processes via quantitative mass spectrometry. In the realm of bimolecular radical termination, the vexed question of determining the ratio of combination to disproportionation has recently been convincingly tackled via quantitative ESI-MS on samples of PMMA prepared via peroxide initiated thermal polymerization.113 It was established that the ratio of polymer generated via disproportionation compared to combination is a function of chain length and of fundamental kinetic parameters such as the rate of initiation and the rate coefficients for propagation and termination. Most interestingly, this study revealed that the extent of disproportionation in methyl methacrylate polymerization is not as high as commonly assumed (λ is close to 0.63). Based on this initial study, it seems now a matter of priority to quantitatively assess other monomer systems in a similar fashion, as the current experimental situation with regard to firm numbers on the ratio of combination to disproportionation – especially as a function of temperature – is unsatisfactory.114Transfer reactions lend themselves to an assessment via mass spectrometry, as the transfer activity is (quantitatively) visible in the obtained end-groups. However, only a few recent studies make use of mass spectrometry to study transfer reactions. One has to differentiate between transfer reactions to a deliberately added transfer agent and transfer reaction that involve the monomer, the generated polymer or both. A recent example on elucidating a reaction mechanism that is dominated by transfer to monomer is provided by Wegner, Barner-Kowollik and colleagues. These authors investigated the product mixture resulting from the free radical polymerization of dimethyl vinyl phosphonate, diethyl vinyl phosphonate, and diisopropyl vinyl phosphonate via ESI-MS.115 The resulting mass spectra showed no evidence of polymer chains capped with initiator derived fragments as end-groups. Instead, the identified species indicated that transfer reactions generate activated monomer species (i.e., a monomeric radical), which then act as initiators in the polymerizations. More complex transfer reaction scenarios are addressed in the section below.
Branching
Recently, our understanding of acrylate free radical polymerization has greatly increased. Reliable estimates for the rate coefficients governing the complex reactions occurring in acrylate free radical polymerizations are currently becoming available.116 These include intra- and intermolecular transfer to polymer reactions (including intramolecular random transfer and so-called backbiting) as well as β-scission, termination and propagation of the generated tertiary mid-chain radicals. Mass spectrometry has recently also made (quantitative) contributions to unravelling the prevalence of certain reactions, as characteristic end-group patterns are formed during intra- and intermolecular transfer and its follow-on reactions. Within the time frame under consideration in here, one of the earliest contributions to elucidating acrylate free radical polymerizations via ESI-MS coupled with an FT-ICR mass analyzer came from Simonsick and colleagues.117 These early studies on the high temperature (T > 140 °C) polymerization of ethyl- and butyl acrylate in xylene solution underpinned the previously advanced notion that β-scission of the generated tertiary mid-chain radicals is an important reaction pathway. In addition, these authors found no evidence that acrylate polymerizations can be initiated without the addition of a dedicated initiator. Contrary to these results, Barner-Kowollik, Junkers and colleagues provided ESI-MS evidence that initiator-free high temperature acrylate polymerizations can lead to the formation of highly uniform macromolecules featuring a vinylic function as the chain terminus.118 Such self-initiated and self-organizing acrylate reaction sequences are postulated to proceed via a reversible chain transfer equilibrium, not unlike the one occurring during the RAFT process. In a subsequent study,119 the same authors demonstrated that although the addition of initiator is not required for the synthesis of acrylate based macromonomers, it improves the reaction rates considerably. The exact nature of the acrylate auto-initiation mechanism, however, remains speculative, as the macromonomers are entirely composed of monomer repeat units without any additional end-groups.The study of acrylate polymerisations, specifically those of butyl acrylate, at intermediate reaction temperatures (T close to 60 °C) was recently investigated by our group. In two subsequent studies, it could be demonstrated in a quantitative fashion that the addition of a low molecular weight transfer agent (such as a thiol) can successfully prevent the onward reactions of mid-chain radicals and transform these into non-reactive linear chains.120 As a result, a more uniform and less branched poly(acrylate) is produced. As the suppression of β-scission is readily observable via ESI-MS, it was possible to quantitatively map the individual products as a function of the transfer agent concentration.121 The reduction of the β-scission products as a function of transfer agent concentration is a good indicator for the desired effect of removing (i.e. patching with a proton) the tertiary mid-chain radicals from the polymerization mixture. However, mass spectrometry cannot directly provide information about the degree of branching of polyacrylates, as the resulting branched polymer peaks are isobaric with those of linear polyacrylates (some qualitative information may, however, be gained by employing MS/MS68). In the above studies, the reduced branching level was thus assessed via quantitative 13C-NMR spectroscopy on the quaternary carbon concentration.120 The free radical polymerization of butyl vinyl ether (BVE) leads exclusively to an oligomeric product. When Kumagai et al. analyzed the produced oligomer by MALDI-TOF mass spectrometry, a peak series was identified that could be assigned to aldehyde end-group-functional PBVE.122 This was in accordance with an addition fragmentation mechanism proposed by the authors, in which the expulsion of a butyl radical from the macroradical chain-end by β-scission led to chain transfer and formation of the aldehyde end-group. However, 1H-NMR revealed the major species carried an intra-chain ketone instead of an aldehyde end-group. This information could not be derived from mass spectrometry alone, serving as a good example as of why one should always be aware that mass spectrometry cannot distinguish between isomeric species. The authors reasoned that in the investigated polymerization, backbiting and subsequent mid-chain radical β-scission must play an important role.
From recent studies of branched aramid polymers it has become clear that ion mobility spectrometry is a method that is highly sensitive to the structure and branching of (isomeric) polymers.40 Determination of the extent of branching of acrylate and vinyl ether polymers formed via free radical polymerization may therefore be just another very exciting application for this powerful gas-phase separation technique.
Living/controlled radical polymerization
Living/controlled radical polymerisation has been a constant topic of mass spectrometric investigations, especially the ATRP and RAFT process. Both have been extensively subjected to MALDI-TOF as well as (SEC/)ESI-MS analysis to assess end-group fidelity as well as to clarify complex mechanistic questions. Since our last assessment of the state-of-the-art on the application of soft ionization MS towards polymers prepared via living/controlled radical processes,1 research in this area has been steady. It is notable that the investigations have advanced to more complex systems (such as stars, see below). Most importantly, two systematic studies were carried out by Barner-Kowollik, Du Prez and colleagues as well as Ladavière and co-workers, who assessed the differences in ionization protocols (MALDI vs. ESI) for polymers prepared via nitroxide mediated polymerization (NMP), atom transfer radical polymerization (ATRP) as well as RAFT.18,123Recently, both MALDI-TOF and a novel matrix-free ionization method, desorption ionization on silicon (DIOS) were employed to study PMMA chains tethered to porous silicon and anodic aluminium oxide surfaces via surface initiated ATRP by Gorman et al.14 The study effectively demonstrated that ATRP surface tethered chains can be efficiently analyzed via mass spectrometry techniques. Matrix-free DIOS-MS was effective for the direct analysis of the polymers up to a molecular weight of close to 6 kg mol−1. Beyond this molecular weight threshold, the signal to noise ratio rapidly decreased. Based on the MS analysis, the study concluded that under the same polymerization conditions, PMMA grown on both substrates had a significantly lower molecular weight and a broader molecular weight distribution than the polymer formed in solution. In a study investigating Co(II) mediated living radical polymerization Langlotz et al.124 employed liquid injection field desorption/ionization mass spectrometry (LIFDI-MS) to monitor the reaction intermediates on-line during the polymerization process. The LIFDI methodology is a very soft ionization method and especially suited for systems where moisture and air must be rigorously excluded from the reaction system.
Nitroxide mediated polymerization systems (NMP) were also recently investigated via MALDI-TOF techniques, for example by Dire et al.,125 who subjected PMMA prepared via SG1 (N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) nitroxide) to MS analysis and found that all polymer chains were terminated by an alkene function in the presence of a large excess of free SG1, implying that β-hydrogen transfer from propagating radicals to the nitroxide is the predominant chain-stoppage event. At low SG1 concentrations, however, these authors could show that two termination events existed. Studer and co-workers investigated the 2,2,6,6-tetraethylpiperidin-4-on-N-oxyl mediated polymerization of N-isopropylacrylamide via MALDI-TOF and provided evidence that chain end degradation occurs during the MALDI process.126 Yet they also noted that the degree of chain end degradation varies with the applied laser intensity.
While the above studies concentrated on the elucidation of the structure of linear chains, the formation of star polymers via living radical polymerization has also been studied via ESI-MS methods. Barner-Kowollik and colleagues have in two studies investigated the formation of star polymers via the RAFT process, in so-called R-group approach RAFT polymerizations, where the core itself carries one or multiple radical functionalities during macromolecular growth. Both acrylate as well as styrene systems were investigated,134 proving that the coupling of the core with propagating chains as well as star-star coupling events readily occur. In the case of acrylate systems, additional species originating from the generation of mid-chain radicals and their follow-on reactions (i.e. termination and β-scission) were identified with high certainty.
Polymer degradation
Soft ionization MS is ideally suited as a tool to observe the loss of polymer integrity upon chemical degradation. As most degradation processes will inevitably lead to a change in the end-group structure, the polymer backbone, and chemical composition in the case of copolymers valuable conclusions about the degradation chemistry can be derived from the changes in the mass spectral peak pattern observed. The propensity of mass spectrometry to (semi-)quantitatively follow the molecular weight distribution during degradation allows chain-breakage events to be directly recognized from the spectrum.Poly(1,5-dioxepan-2-one) (PDXO) and poly(ε-caprolactone) (PCL) are two aliphatic polyesters often used in biomedical applications. Hakkarainen et al. monitored the hydrolysis of these polymers by ESI-MS, and compared the degradation product pattern to mass loss, changes in pH, molecular weight and copolymer composition.138 They found that hydrolysis of the DXO chains was greatly accelerated when compared to caprolactone, due to the significantly higher solubility of the product oligomer units. In a subsequent study, DXO/CL networks crosslinked with 2,2′-bis(ε-caprolactone-4-yl) propane (BCP) were subjected to degradation.139 The water-soluble degradation products were analyzed by ESI-MS. The variation in the intensities of ions corresponding to DXO and CL/DXO oligomers with or without attached crosslinking agent BCP correlated with the extent of hydrolytic degradation in the polyester matrix. With proceeding degradation time an increasing number of chains with attached BCP were identified using MS/MS, indicating that disruption of the network structure had occurred. In accordance with these earlier studies, co-monomer content played a crucial role in determining the hydrolytic stability of the network, with the more hydrophobic CL-rich networks exhibiting greatly increased stability. Kasperczyk et al. used ‘copolymer fingerprints’ obtained from ESI-MS together with 1H-NMR to monitor the changes in the composition of a glycolide/ε-caprolactone copolymer upon degradation in an aqueous medium.77 NMR allowed changes of the average sequence distribution and composition of the components to be followed quantitatively, while ESI-MS allowed to intimately follow the changes in copolymer structural composition and formation of degradation products with proceeding degradation. Pulkkinen et al. analyzed the degradation of a novel oxazoline-linked poly-ε-caprolactone (PCL-O), a polymer with possible drug delivery applications.140 Oligomeric products formed upon enzymatic degradation were unequivocally identified by gradient HPLC coupled online to ESI-MS/MS. Mass spectrometric results provided important information of the enzymatic degradation process and the structure of the polymer, which is otherwise difficult to ascertain by conventional methods.
Bernhard et al. used ESI-MS and MALDI-TOF-MS to assess the changes in molecular weight and end-group structure during the biodegradation of PEO in municipal wastewater and in marine water.141
Fluorosilicones are chemically inert and insoluble in hydrocarbon fuel or mineral oils making them an important starting material for sealants and gaskets in fuel tank applications. Kählig et al. studied the behaviour of poly[(3,3,3-trifluoropropyl)methylsiloxane] (PTFPMS) under treatment with typical solvents, acetone, ethyl acetate and the non-solvent methanol. 1H-, 19F- and 29Si-NMR spectroscopy, GC-MS and negative mode ESI-MS were used for characterization.142 Complete degradation of PTFPMS occurred in acetone and methanol at longer incubation times and elevated temperatures. Using ESI-MS, the main degradation product was identified as cyclic tetrasiloxane. Cyclic tri-, penta- and hexasiloxane were formed in lower concentrations.
Weathering, e.g. the UV radiation-, heat- and moisture-induced degradation of polymers is of great industrial importance. Polymers with applications in coating formulations are at the focal point of studies into the chemical mechanisms behind the weathering process. These polymers have to provide excellent optical properties as well as optimum corrosion protection under harsh environmental conditions. Malanowski et al. studied in detail the photooxidative degradation of poly(neopentyl isophthalate) – a typical polymer in coating formulations – focussing on the molecular mechanisms leading to fragmentation of the polymer.143 Attenuated total reflection (ATR) FT-IR spectroscopy and MALDI-MS were employed in the study. Based upon the spectroscopic evidence, the degradation process was divided into two stages: an earlier stage which was dominated by Norrish I photocleavage, with subsequent hydrogen abstraction and formation of mainly isobutyl and phthalic acid end-groups, visible by MALDI. Reactions with O2 then lead to alcohol and acid group formation in the later stages and a number of further reactions eventually causing oxidation induced chain breakage were postulated. Carroccio et al. studied poly(1,4-butylene terephthalate) (PBT) commonly used as base plastic in automotive electrical parts.144 Films of PBT were treated at high temperature and under UV-exposure. MALDI-MS was then used to establish the photo- and thermal oxidation processes. The formation of radicals followed by α-hydrogen abstraction, hydroperoxide formation and thermal cleavage as well as the formation of biphenyl bridges between PBT chains were discussed.
Bennet et al. studied the degradation of (terminally saturated and unsaturated) PMMA – a common component in protective surface coatings – at harsh environmental temperatures (95 °C) and high UV radiation.145 It was found that terminal unsaturation is necessary to enable degradation at these comparably low temperatures, in which case a cyclic non-radical mechanism is operative during thermal degradation. These studies therefore complement earlier pyrolysis studies at much increased temperatures. The mechanism was further confirmed by synthesis and degradation of epoxy-functional PMMA, an intermediate product in the cycle and the accelerating effects of the UV radiation on degradation were discussed.
To better understand the mechanistic pathways during the ultrasonic degradation of vinyl polymers, Takeda et al. used uniform PMMA as a model polymer.146 These authors observed much more rapid degradation of PMMA carrying a tert-butyl end-group than of hydrogen terminal PMMA. Interestingly, this characteristic is similar to the behaviour observed during tandem mass spectrometry of vinyl polymers, where the end-group plays a significant role in determining the fragmentation pathway.67 No tert-butyl functional degradation products were observed in the degraded solutions. This suggested a quick release of the end-group, but the authors excluded an unzipping process due to the lack of MMA monomer in the degraded solutions, identifiable by GC-MS. Further studies will have to reveal the exact causes of the observed behaviour.
The reversible addition fragmentation chain transfer (RAFT) process offers powerful control over polymer composition and architecture and is compatible with a vast range of monomer and solvent systems. Barner-Kowollik, Stenzel and co-workers have recently introduced a novel orthogonal and highly efficient conjugation reaction of RAFT-functional vinyl polymers to achieve fast access to block-copolymer architectures. This process uses the functional dithioester end-group of a RAFT polymer as the dienophile in a hetero-Diels–Alder (HDA) cycloaddition with a second diene-functionalized polymer to produce the block copolymer in high yields. In the product copolymer, a 3,6-dihydro-2H-thiopyran ring links both blocks together via a dithioacetal C–S–C bond. The degradation stability of this moiety was the primary concern of a recent study.147 Using ESI-MS, the stability of the dithioacetal linkage in presence of strong acid and base as well as temperatures up to 100 °C was confirmed. At temperatures in excess of 120 °C, the authors discovered cleavage reactions including a retro HDA reaction, which leads to a fully reversible conjugation for some of the employed model compounds. Gruendling et al. focused on the in-solution stability of dithioester-functional PMMA and PS in (peroxide containing) tetrahydrofuran as well as in an inert solvent, dichloromethane.148 Mass spectrometry indicated the formation of an unexpected degradation product in the ether solution; hydroperoxide functional PMMA was the exclusive degradation product. A radical degradation mechanism was postulated based on the mass spectrometric evidence. Degradation in inert solvent was significantly less pronounced and mainly produced vinyl terminated polymer due to cleavage of the dithioester as well as some sulfine from reaction with dissolved singlet oxygen. Recently, the same authors exploited this reaction as a synthetic tool towards the functional end-group modification of RAFT polymers, to produce hydroxyl functional polymer.149 Online SEC/ESI-MS was used in this study to quantitatively follow the synthetic conversion of the RAFT end-group.150
Acknowledgements
C.B.-K. acknowledges on-going funding from the Karlsruhe Institute of Technology (KIT) in the context of the Excellence Initiative for leading German universities as well as the German Research Council (DFG) supporting our mass spectrometry research.References
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Polymer, 2004, 45, 7791–7805 CrossRef CAS
.
-
L. Li, MALDI Mass Spectrometry for Synthetic Polymer Analysis (Chemical Analysis: A Series of Monographs on Analytical Chemistry and Its Applications), Wiley, 2010 Search PubMed
- S. M. Weidner and S. Trimpin, Anal. Chem., 2008, 80, 4349–4361 CrossRef CAS
- S. M. A. B. Batoy, E. Akhmetova, S. Miladinovic, J. Smeal and C. L. Wilkins, Appl. Spectrosc. Rev., 2008, 43, 485–550 CrossRef CAS
- A. C. Crecelius, A. Baumgaertel and U. S. Schubert, J. Mass Spectrom., 2009, 44, 1277–1286 CrossRef CAS
- A. V. Walker, Anal. Chem., 2008, 80, 8865–8870 CrossRef CAS
- C. M. Mahoney, Mass Spectrom. Rev., 2009 DOI:10.1002/mas.20233
- R. Rial-Otero, M. Galesio, J.-L. Capelo and J. Simal-Gándara, Chromatographia, 2009, 70, 339–348 CrossRef CAS
- D. Robb, T. Covey and Bruins, Anal. Chem., 2000, 72, 3653–3659 CrossRef CAS
- S. Kéki, J. Török, L. Nagy and M. Zsuga, J. Am. Soc. Mass Spectrom., 2008, 19, 656–665 CrossRef CAS
- L. Nagy, V. Pálfi, M. Narmandakh, Á. Kuki, A. Nyíri, B. Iván, M. Zsuga and S. Kéki, J. Am. Soc. Mass Spec., 2009 DOI:10.1016/j.jasms.2009.1008.1025
- S. Kéki, L. Nagy, Á. Kuki and M. Zsuga, Macromolecules, 2008, 41, 3772–3774 CrossRef CAS
- T. Watanabe, H. Kawasaki, T. Yonezawa and R. Arakawa, J. Mass Spectrom., 2008, 43, 1063–1071 CrossRef CAS
- C. B. Gorman, R. J. Petrie and J. Genzer, Macromolecules, 2008, 41, 4856–4865 CrossRef CAS
- D. Asakawa, L. C. Chen and K. Hiraoka, J. Mass Spectrom., 2009, 44, 945–951 CrossRef CAS
- T. Gruendling, G. Hart-Smith, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41, 1966–1971 CrossRef CAS
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 589–597 CrossRef CAS
- C. Ladavière, P. Lacroix-Desmazes and F. Delolme, Macromolecules, 2009, 42, 70–84 CrossRef CAS
- R. P. Quirk, H. Kim, M. J. Polce and C. Wesdemiotis, Macromolecules, 2005, 38, 7895–7906 CrossRef CAS
- W. Dempwolf, S. Flakus and G. Schmidt-Naake, Macromol. Symp., 2009, 275–276, 166–172 CrossRef CAS
- G. A. Harris, L. Nyadong and F. M. Fernandez, Analyst, 2008, 133, 1297–1301 RSC
- J. P. Williams and J. H. Scrivens, Rapid Commun. Mass Spectrom., 2008, 22, 187–196 CrossRef CAS
- T. Rothenbacher and W. Schwack, Rapid Commun. Mass Spectrom., 2009, 23, 2829–2835 CrossRef CAS
- S. E. Whitson, G. Erdodi, J. P. Kennedy, R. P. Lattimer and C. Wesdemiotis, Anal. Chem., 2008, 80, 7778–7785 CrossRef CAS
- M. C. Jecklin, G. Gamez and R. Zenobi, Analyst, 2009, 134, 1629–1636 RSC
- S. D. Hanton, T. M. McEvoy and J. R. Stets, J. Am. Soc. Mass Spectrom., 2008, 19, 874–881 CrossRef CAS
- S. M. Weidner and J. Falkenhagen, Rapid Commun. Mass Spectrom., 2009, 23, 653–660 CrossRef CAS
- R. D. Deegan, O. Bakajin, T. F. Dupont, G. Huber, S. R. Nagel and T. A. Witten, Nature, 1997, 389, 827–829 CrossRef CAS
- A. Hortal, P. Hurtado, B. Martínez-Haya, A. Arregui and L. Bañares, Appl. Phys. A: Mater. Sci. Process., 2008, 92, 859–863 CrossRef CAS
- M. Mazarin, T. N. T. Phan and L. Charles, Rapid Commun. Mass Spectrom., 2008, 22, 3776–3782 CrossRef CAS
- S. D. Hanton and J. R. Stets, J. Am. Soc. Mass Spectrom., 2009, 20, 1115–1118 CrossRef CAS
- S. T. Ellison, A. P. Gies, D. M. Hercules and S. L. Morgan, Macromolecules, 2009, 42, 5526–5533 CrossRef CAS
- W. J. Erb and K. G. Owens, Rapid Commun. Mass Spectrom., 2008, 22, 1168–1174 CrossRef CAS
- T. Tu, A. D. Sauter Jr., A. D. Sauter 3rd and M. L. Gross, J. Am. Soc. Mass Spectrom., 2008, 19, 1086–1090 CAS
- T. Tu and M. L. Gross, TrAC, Trends Anal. Chem., 2009, 28, 833–841 CrossRef CAS
- A. Lee, H. J. Yang, Y. Kim and J. Kim, Bull. Kor. Chem. Soc., 2009, 30, 1127–1130 CAS
- F. Wurm, D. Wilms, J. Klos, H. Löwe and H. Frey, Macromol. Chem. Phys., 2008, 209, 1106–1114 CrossRef CAS
- A. Berthod, J. A. Crank, K. L. Rundlett and D. W. Armstrong, Rapid Commun. Mass Spectrom., 2009, 23, 3409–3422 CrossRef CAS
- L. Fernandes, R. Rial-Otero, M. Temtem, C. Veiga de Macedo, A. Aguiar-Ricardo and J. L. Capelo, Talanta, 2008, 77, 882–888 CrossRef CAS
- A. P. Gies, M. Kliman, J. A. McLean and D. M. Hercules, Macromolecules, 2008, 41, 8299–8301 CrossRef CAS
- G. R. Hilton, A. T. Jackson, K. Thalassinos and J. H. Scrivens, Anal. Chem., 2008, 80, 9720–9725 CrossRef CAS
- D. Bagal, H. Zhang and P. D. Schnier, Anal. Chem., 2008, 80, 2408–2418 CrossRef CAS
- S. Trimpin and D. E. Clemmer, Anal. Chem., 2008, 80, 9073–9083 CrossRef CAS
- S. Julka, H. Cortes, R. Harfmann, B. Bell, A. Schweizer-Theobaldt, M. Pursch, L. Mondello, S. Maynard and D. West, Anal. Chem., 2009, 81, 4271–4279 CrossRef CAS
- S. M. Weidner, J. Falkenhagen, K. Knop and A. Thünemann, Rapid Commun. Mass Spectrom., 2009, 23, 2768–2774 CrossRef CAS
- J. Falkenhagen and S. Weidner, Anal. Chem., 2009, 81, 282–287 CrossRef CAS
- B. Desmazieres, W. Buchmann, P. Terrier and J. Tortajada, Anal. Chem., 2008, 80, 783–792 CrossRef CAS
- B. N. Barman, D. H. Champion and S. L. Sjoberg, J. Chromatogr., A, 2009, 1216, 6816–6823 CrossRef CAS
- C. Schneider, M. Sablier and B. Desmazières, Rapid Commun. Mass Spectrom., 2008, 22, 3353–3361 CrossRef CAS
- M. Girod, T. N. T. Phan and L. Charles, Rapid Commun. Mass Spectrom., 2008, 22, 3767–3775 CrossRef CAS
- D. Hercules, Anal. Bioanal. Chem., 2008, 392, 571–573 CrossRef CAS
- A. Gies, S. Ellison, M. Vergne, R. Orndorff and D. Hercules, Anal. Bioanal. Chem., 2008, 392, 627–642 CrossRef CAS
- D. Schubert, M. Corda, O. Lukin, B. Brusilowskij, E. Fiscaronkin and C. A. Schalley, Eur. J. Org. Chem., 2008, 2008, 4148–4156 CrossRef
- T. J.-C. Vincent, R. Dolé and C. M. Lange, Rapid Commun. Mass Spectrom., 2008, 22, 363–372 CrossRef CAS
- M. A. Kaczorowska and H. J. Cooper, J. Am. Soc. Mass Spectrom., 2008, 19, 1312–1319 CrossRef CAS
- B. Baytekin, H. T. Baytekin, U. Hahn, W. Reckien, B. Kirchner and C. A. Schalley, Chem.–Eur. J., 2009, 15, 7139–7149 CrossRef CAS
- A. Baumgaertel, C. R. Becer, M. Gottschaldt and U. S. Schubert, Macromol. Rapid Commun., 2008, 29, 1309–1315 CrossRef CAS
- A. Baumgaertel, C. Weber, K. Knop, A. Crecelius and U. S. Schubert, Rapid Commun. Mass Spectrom., 2009, 23, 756–762 CrossRef CAS
- R. Giordanengo, S. Viel, B. Allard-Breton, A. Thévand and L. Charles, J. Am. Soc. Mass Spectrom., 2009, 20, 25–33 CrossRef CAS
- R. Giordanengo, S. Viel, B. Allard-Breton, A. Thévand and L. Charles, Rapid Commun. Mass Spectrom., 2009, 23, 1557–1562 CrossRef CAS
- G. Adamus, Macromolecules, 2009, 42, 4547–4557 CrossRef CAS
- V. Mass, W. Schrepp, B. v. Vacano and H. Pasch, Macromol. Chem. Phys., 2009, 210, 1957–1965 CrossRef CAS
- P. Rizzarelli and C. Puglisi, Rapid Commun. Mass Spectrom., 2008, 22, 739–754 CrossRef CAS
- A. Jackson, S. Slade, K. Thalassinos and J. Scrivens, Anal. Bioanal. Chem., 2008, 392, 643–650 CrossRef CAS
- C. Peptu, V. Harabagiu, B. C. Simionescu, G. Adamus, M. Kowalczuk and J.-M. Nunzi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 534–547 CrossRef CAS
- W. Simonsick and V. Petkovska, Anal. Bioanal. Chem., 2008, 392, 575–583 CrossRef CAS
- M. J. Polce, M. Ocampo, R. P. Quirk, A. M. Leigh and C. Wesdemiotis, Anal. Chem., 2008, 80, 355–362 CrossRef CAS
- K. Chaicharoen, M. Polce, A. Singh, C. Pugh and C. Wesdemiotis, Anal. Bioanal. Chem., 2008, 392, 595–607 CrossRef CAS
- J. Renaud, A. M. Alhazmi and P. M. Mayer, Can. J. Chem., 2009, 87, 453–459 CrossRef CAS
- M. Girod, T. N. T. Phan and L. Charles, J. Am. Soc. Mass Spectrom., 2008, 19, 1163–1175 CrossRef CAS
- M. Girod, M. Mazarin, T. N. T. Phan, D. Gigmes and L. Charles, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3380–3390 CrossRef CAS
- M. Girod, T. N. T. Phan and L. Charles, Rapid Commun. Mass Spectrom., 2009, 23, 1476–1482 CrossRef CAS
- M. Girod, Y. Carissan, S. Humbel and L. Charles, Int. J. Mass Spectrom., 2008, 272, 1–11 Search PubMed
- M. Mazarin, M. Girod, S. Viel, T. N. T. Phan, S. R. A. Marque, S. Humbel and L. Charles, Macromolecules, 2009, 42, 1849–1859 CrossRef CAS
- M. S. Montaudo, Mass Spectrom. Rev., 2002, 21, 108–144 CrossRef CAS
- R. X. E. Willemse and A. M. van Herk, J. Am. Chem. Soc., 2006, 128, 4471–4480 CrossRef CAS
- J. Kasperczyk, S. Li, J. Jaworska, P. Dobrzynski and M. Vert, Polym. Degrad. Stab., 2008, 93, 990–999 CrossRef CAS
- G. Adamus, M. Hakkarainen, A. Höglund, M. Kowalczuk and A.-C. Albertsson, Biomacromolecules, 2009, 10, 1540–1546 CrossRef CAS
- A. A. Aksenov and M. E. Bier, J. Am. Soc. Mass Spectrom., 2008, 19, 219–230 CrossRef CAS
- G. Schlosser, A. Jakab, G. Pocsfalvi, K. Vékey, F. Hudecz and G. Mezö, Rapid Commun. Mass Spectrom., 2009, 23, 1249–1254 CrossRef CAS
- S. Miladinovic, S. Robotham and C. Wilkins, Anal. Bioanal. Chem., 2008, 392, 585–594 CrossRef CAS
- I. Schnöll-Bitai, T. Hrebicek and A. Rizzi, Macromol. Chem. Phys., 2007, 208, 485–495 CrossRef
- C. M. Guttman, K. M. Flynn, W. E. Wallace and A. J. Kearsley, Macromolecules, 2009, 42, 1695–1702 CrossRef CAS
- S. D. Hanton, I. Z. Hyder, J. R. Stets, K. G. Owens, W. R. Blair, C. M. Guttman and A. A. Giuseppetti, J. Am. Soc. Mass Spectrom., 2004, 15, 168–179 CrossRef CAS
- C. M. Guttman, S. J. Wetzel, W. R. Blair, B. M. Fanconi, J. E. Girard, R. J. Goldschmidt, W. E. Wallace and D. L. Van der Hart, Anal. Chem., 2001, 73, 1252–1262 CrossRef CAS
- W. Wallace, C. M. Guttman, K. M. Flynn and A. J. Kearsley, Anal. Chim. Acta, 2007, 604, 62–68 CrossRef CAS
- A. J. Kearsley, W. E. Wallace, J. Bernal and C. M. Guttman, Appl. Math. Lett., 2005, 18, 1412–1417 CrossRef
- I. Schnöll-Bitai, R. Ullmer, T. Hrebicek, A. Rizzi and I. Lacik, Rapid Commun. Mass Spectrom., 2008, 22, 2961–2970 CrossRef
- L. Chikh, M. Tessier and A. Fradet, Macromolecules, 2008, 41, 9044–9050 CrossRef CAS
- L. Prokai and W. J. Simonsick, Rapid Commun. Mass Spectrom., 1993, 7, 853–856 CAS
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Anal. Chem., 2008, 80, 6915–6927 CrossRef CAS
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Macromolecules, 2009, 42, 6366–6374 CrossRef CAS
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191–254 CrossRef CAS
- H. C. M. Byrd and C. N. McEwen, Anal. Chem., 2000, 72, 4568–4576 CrossRef CAS
- T. Gruendling, D. Voll, M. Guilhaus and C. Barner-Kowollik, Macromol. Chem. Phys., 2010, 211, 80–90 CrossRef CAS
- J. P. A. Heuts, G. T. Russell, G. B. Smith and A. M. v. Herk, Macromol. Symp., 2007, 248, 12–22 CrossRef CAS
- R. X. E. Willemse, B. B. P. Staal, A. M. van Herk, S. C. J. Pierik and B. Klumperman, Macromolecules, 2003, 36, 9797–9803 CrossRef CAS
- M. Buback, H. Frauendorf, F. Günzler, F. Huff and P. Vana, Macromol. Chem. Phys., 2009, 210, 1591–1599 CrossRef CAS
- M. Buback, H. Frauendorf and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4266–4275 CrossRef CAS
- M. Buback, H. Frauendorf, F. Günzler and P. Vana, Polymer, 2007, 48, 5590–5598 CrossRef CAS
- M. Buback, H. Frauendorf, F. Günzler and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2453–2467 CrossRef CAS
- M. Buback, H. Frauendorf, O. Janssen and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6071–6081 CrossRef CAS
- W. Wang and R. A. Hutchinson, Macromolecules, 2009, 42, 4910–4913 CrossRef CAS
- M. Buback, M. Egorov, T. Junkers and E. Panchenko, Macromol. Rapid Commun., 2004, 25, 1004–1009 CrossRef CAS
- C. Barner-Kowollik, M. Buback, M. Egorov, T. Fukuda, A. Goto, O. F. Olaj, G. T. Russell, P. Vana, B. Yamada and P. B. Zetterlund, Prog. Polym. Sci., 2005, 30, 605–643 CrossRef CAS
- Z. Szablan, T. Junkers, S. P. S. Koo, T. M. Lovestead, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2007, 40, 6820–6833 CrossRef CAS
- F. Günzler, E. H. H. Wong, S. P. S. Koo, T. Junkers and C. Barner-Kowollik, Macromolecules, 2009, 42, 1488–1493 CrossRef CAS
- F. J. Wyzgoski, M. J. Polce, C. Wesdemiotis and M. A. Arnould, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2161–2171 CrossRef CAS
- T. M. Lovestead, G. Hart-Smith, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2007, 40, 4142–4153 CrossRef CAS
- G. Hart-Smith, T. M. Lovestead, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Biomacromolecules, 2007, 8, 2404–2415 CrossRef CAS
- G. Hart-Smith and C. Barner-Kowollik, Polymer, 2009, 50, 5175–5180 CrossRef CAS
- J. Pusch and A. M. van Herk, Macromolecules, 2005, 38, 8694–8700 CrossRef CAS
- M. Buback, F. Günzler, G. T. Russell and P. Vana, Macromolecules, 2009, 42, 652–662 CrossRef CAS
-
C. Barner-Kowollik, P. Vana and T. P. Davis, in Handbook of Radical Polymerization, ed. T. P. Davis and K. Matyjaszewski, Wiley, New York, 1st edn, 2002, pp. 187–ff Search PubMed
- B. Bingol, G. Hart-Smith, C. Barner-Kowollik and G. Wegner, Macromolecules, 2008, 41, 1634–1639 CrossRef
- T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7585–7605 CrossRef CAS
- C. Quan, M. Soroush, M. C. Grady, J. E. Hansen and W. J. Simonsick, Macromolecules, 2005, 38, 7619–7628 CrossRef CAS
- T. Junkers, F. Bennet, S. P. S. Koo and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3433–3437 CrossRef CAS
- A.-M. Zorn, T. Junkers and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 2028–2035 CrossRef CAS
- T. Junkers, S. P. S. Koo, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2007, 40, 8906–8912 CrossRef CAS
- S. P. S. Koo, T. Junkers and C. Barner-Kowollik, Macromolecules, 2009, 42, 62–69 CrossRef CAS
- T. Kumagai, C. Kagawa, H. Aota, Y. Takeda, H. Kawasaki, R. Arakawa and A. Matsumoto, Macromolecules, 2008, 41, 7347–7351 CrossRef CAS
- G. Hart-Smith, M. Lammens, F. E. Du Prez, M. Guilhaus and C. Barner-Kowollik, Polymer, 2009, 50, 1986–2000 CrossRef CAS
- B. K. Langlotz, J. L. Fillol, J. H. Gross, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2008, 14, 10267–10279 CrossRef CAS
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333–6345 CrossRef CAS
- T. Schulte, K. O. Siegenthaler, H. Luftmann, M. Letzel and A. Studer, Macromolecules, 2005, 38, 6833–6840 CrossRef CAS
- C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. Mcleary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5809–5831 CrossRef CAS
- A. Feldermann, A. Ah Toy, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Polymer, 2005, 46, 8448–8457 CrossRef CAS
- A. Ah Toy, P. Vana, T. P. Davis and C. Barner-Kowollik, Macromolecules, 2004, 37, 744–751 CrossRef
- P. Geelen and B. Klumperman, Macromolecules, 2007, 40, 3914–3920 CrossRef CAS
- G. Zhou and I. I. Harruna, Anal. Chem., 2007, 79, 2722–2727 CrossRef CAS
- M. Bathfield, F. D'Agosto, R. Spitz, C. Ladavière, M.-T. Charreyre and T. Delair, Macromol. Rapid Commun., 2007, 28, 856–862 CrossRef CAS
- D. Konkolewicz, B. S. Hawkett, A. Gray-Weale and S. Perrier, Macromolecules, 2008, 41, 6400–6412 CrossRef CAS
- H. Chaffey-Millar, G. Hart-Smith and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1873–1892 CrossRef CAS
- A. Ah Toy, H. Chaffey-Millar, T. P. Davis, M. H. Stenzel, E. I. Izgorodina, M. L. Coote and C. Barner-Kowollik, Chem. Commun., 2006, 835–837 RSC
- T. Junkers, M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 746–753 CrossRef CAS
- F. Günzler, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1864–1876
- M. Hakkarainen, G. Adamus, A. Höglund, M. Kowalczuk and A.-C. Albertsson, Macromolecules, 2008, 41, 3547–3554 CrossRef CAS
- A. Höglund, M. Hakkarainen, M. Kowalczuk, G. Adamus and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4617–4629 CrossRef
- M. Pulkkinen, J. J. Palmgrén, S. Auriola, M. Malin, J. Seppälä and K. Järvinen, Rapid Commun. Mass Spectrom., 2008, 22, 121–129 CrossRef CAS
- M. Bernhard, J. P. Eubeler, S. Zok and T. P. Knepper, Water Res., 2008, 42, 4791–4801 CrossRef CAS
- H. Kählig, P. Zöllner and B. X. Mayer-Helm, Polym. Degrad. Stab., 2009, 94, 1254–1260 CrossRef
- P. Malanowski, S. Huijser, F. Scaltro, R. A. T. M. van Benthem, L. G. J. van der Ven, J. Laven and G. de With, Polymer, 2009, 50, 1358–1368 CrossRef CAS
- S. Carroccio, P. Rizzarelli, G. Scaltro and C. Puglisi, Polymer, 2008, 49, 3371–3381 CrossRef CAS
- F. Bennet, T. Gruendling, T. P. Davis, P. J. Barker and C. Barner-Kowollik, Macromol. Phys. Chem., 2009 DOI:10.1002/macp.200900625
- Y. Takeda, H. Kawasaki, T. Watanabe, K. Ute and R. Arakawa, Polym. J., 2008, 40, 682–683 CrossRef CAS
- S. Sinnwell, C. V. Synatschke, T. Junkers, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41, 7904–7912 CrossRef CAS
- T. Gruendling, R. Pickford, M. Guilhaus and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7447–7461 CrossRef CAS
- T. Gruendling, M. Dietrich and C. Barner-Kowollik, Aust. J. Chem., 2009, 62, 806 CrossRef CAS
- M. Dietrich, M. Glassner, T. Gruendling, C. Schmid, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010 10.1039/b9py00273a
Footnotes |
| † Dedicated to the memory of Prof. Michael Guilhaus (1954–2009). |
| ‡ The unit Thomson (1 Th ≡ 1 u/e ≡ 1 Da/e) is recommended as an appropriate unit for the mass-to-charge ratio (m/z) in mass spectrometry. |
| This journal is © The Royal Society of Chemistry 2010 |