DOI:
10.1039/C0PY00022A
(Paper)
Polym. Chem., 2010,
1, 678-684
A polythiophene derivative with octyl diphenylamine-vinylene side chains: synthesis and its applications in field-effect transistors and solar cells
Received
20th January 2010
, Accepted 2nd February 2010
First published on
9th March 2010
Abstract
A new polythiophene derivative with octyl diphenylamine-vinylene conjugated side chains, DPAV-PT, was synthesized by the Stille coupling reaction, and characterized by 1H NMR, GPC, TGA, UV-Vis absorption spectroscopy, and cyclic voltammetry. The copolymer is readily soluble in the common organic solvents and exhibits good thermal stability with 5% weight loss temperature of 267 °C. DPAV-PT possesses a broad absorption band at 300–650 nm (with an optical bandgap of 1.85 eV). Cyclic voltammetry displays a HOMO energy level of −5.01 eV. The weight average molecular weight (Mw) of DPAV-PT is 3.1 × 104 with the polydispersity index of 1.3. A polymer solar cell with the configuration of ITO/PEDOT:PSS/DPAV-PT:PCBM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 w/w)/Ca/Al has a power conversion efficiency of 0.7% under the illumination of AM1.5, 100 mW cm−2. The field effect hole mobility of the polymer reached 6.1 × 10−4 cm2 V−1 s−1 with an on/off ratio of 103 and a threshold voltage of −7 V after 180 °C annealing.
:1 w/w)/Ca/Al has a power conversion efficiency of 0.7% under the illumination of AM1.5, 100 mW cm−2. The field effect hole mobility of the polymer reached 6.1 × 10−4 cm2 V−1 s−1 with an on/off ratio of 103 and a threshold voltage of −7 V after 180 °C annealing.
1. Introduction
Conjugated polymers have been the subject of much research in recent years due to their promising applications in electroluminescence (EL),1 organic field-effect transistors (OFETs),2 electrochromic devices (ECDs),3 solar cells4 and sensors.5 In particular, polymer solar cells (PSCs) have attracted considerable attention due to their unique attractive properties such as being lightweight, flexible and low cost. For the conjugated polymers in PSCs as electron donor, broad absorption and higher hole mobility play important role for efficient photovoltaic materials.6 To this end, Li and co-workers do much original work on side chain conjugated polythiophenes. For example, their group synthesized a series of polythiophene and poly(thienylene vinylene) derivatives with conjugated phenylene-vinylene,7 thienylene-vinylene,8 terthiophene-vinylene9 or phenothiazine vinylene side chains.10 The polymers with the conjugated side chains showed broad absorption in the visible region and higher hole mobility,7–10 therefore leading to PCE of up to 3.2%.8
OFETs attracted extensive interest due to their promising potentials in sensors, low-cost large area memories, smart cards, and driving circuits for large-area displays.11 Easy processability and good compatibility with flexible plastic substrates offer excellent opportunities for polymers in fabricating low-cost OFETs. Although much effort has been devoted to this, to date the excellent semiconductors are still limited. Therefore, design and synthesis of new conjugated polymers for FETs are of great interest.
Diphenylamine (DPA) is a well-known hole transporting unit. To the best of our knowledge, the photovoltaic and field effect properties of DPA-based polymers have been scarcely explored. Based on this information, in order to shed light on the effect of DPA structure on the properties of the conjugated side chain polythiophene derivatives, we synthesized a new polythiophene derivative with octyl diphenylamine units as conjugated side chains, DPAV-PT (see Scheme 1), via the Stille coupling reaction. A polymer solar cell with the configuration of ITO/PEDOT:PSS/DPAV-PT:PCBM (1:1 w/w)/Ca/Al was fabricated, and the power conversion efficiency (PCE) of 0.7% was obtained under the illumination of AM1.5, 100 mW cm−2, which is higher than that of triphenylamine vinylene conjugated polythiophene derivative, OTPAV-PT (PCE: 0.2%).12 The reason may be from its broader absorption and relatively higher hole mobility due to its better planarity. The field effect hole mobility of the polymer reached 6.1 × 10−4 cm2 V−1 s−1 with an on/off ratio of 103.
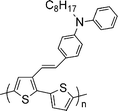 |
| | Scheme 1 Chemical structure of DPAV-PT. | |
2. Experimental
2.1. Materials
3-Methyl thiophene was purchased from Aldrich Chemical Co, Pd (PPh3)4, (C4H9)3SnCl and n-BuLi were obtained from Alfa Asia Chemical Co, and they were used as received. Toluene was dried over molecular sieves and freshly distilled prior to use. (2,5-Dibromo-thiophen-3-ylmethyl)-phosphonic acid diethyl ester and 2,5-bis(tributylstannyl)thiophene was synthesized according to the literature.8 The other chemical reagents were common commercial products and used as received without further purification.
2.2. Characterization
1H NMR spectra were recorded using a Bruker AM-400 spectrometer, with tetramethylsilane (TMS) as the internal reference, chemical shifts were recorded in ppm. Molecular weight and polydispersity of the polymer were determined by size exclusion chromatography (SEC) analysis with polystyrene as the standard (Waters 515 HPLC pump, a Waters 2414 differential refractometer, and three Waters Styragel columns (HT2, HT3, and HT4)) using THF (HPLC grade) as eluent at a flow rate of 1.0 mL min−1 at 35 °C. Thermogravimetric analysis (TGA) was conducted on a Shimadzu DTG-60 thermogravimetric analyzer with a heating rate of 10 K min−1 under a nitrogen atmosphere. Differential scanning calorimetry (DSC) was recorded with a Thermal Analysis (TA) DSC-2010 in nitrogen. The UV-Vis absorption spectra were recorded on a JASCO V-570 spectrophotometer. For solid state measurements, polymer solution in chloroform was drop cast on quartz plates. Optical bandgap was calculated from the onset of the absorption band. The cyclic voltammogram was recorded with a computer controlled Zahner IM6e electrochemical workstation (Germany) using polymer film on platinum disk as the working electrode, platinum wire as the counter electrode and Ag/Ag+ (0.1 M) as the reference electrode in an anhydrous and argon-saturated solution of 0.1 M of tetrabutylammonium hexafluorophosphate (Bu4NPF6) in acetonitrile. Electrochemical onsets were determined at the position where the current starts to differ from the baseline.
2.3. Fabrication and characterization of polymer solar cell
The polymer solar cells were fabricated in the configuration of the traditional sandwich structure with ITO positive electrode and metal negative electrode. The ITO glass was cleaned by sequential ultrasonic treatment in detergent, deionized water, acetone, and isopropanol, and then treated in an ultraviolet-ozone chamber (Ultraviolet Ozone Cleaner, Jelight Company, USA) for 20 min. A thin layer of poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) (Baytron, PVP 4083, Germany) was spin-coated on the ITO glass and dried in vacuum oven at 150 °C for 15 min. The thickness of the PEDOT: PSS layer was ca. 40 nm. Subsequently, the active layer was prepared by spin coating the o-dichlorobenzene solution of polymer DPAV-PT: PCBM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, w/w) with the polymer concentration of 10 mg mL−1 on the top of the PEDOT:PSS layer, giving a thickness of ca. 77 nm determined by a surface profilometer (XP-2, USA). The devices were completed by evaporating Ca/Al metal electrodes defined by masks. The Ca electrode (20 nm) capped with Al (100 nm) was thermally deposited on the active layer at a pressure of 3 × 10−5 Pa, The active area of a device was 4 mm2. The current–voltage (I–V) measurement of the PSCs was conducted on a computer-controlled Keithley 236 source measure unit. A Xenon lamp with AM1.5 filter was used as a white-light source and the optical power was 100 mW cm−2. All the measurements were automatically controlled by a computer system, and performed under ambient atmosphere at room temperature.
:1, w/w) with the polymer concentration of 10 mg mL−1 on the top of the PEDOT:PSS layer, giving a thickness of ca. 77 nm determined by a surface profilometer (XP-2, USA). The devices were completed by evaporating Ca/Al metal electrodes defined by masks. The Ca electrode (20 nm) capped with Al (100 nm) was thermally deposited on the active layer at a pressure of 3 × 10−5 Pa, The active area of a device was 4 mm2. The current–voltage (I–V) measurement of the PSCs was conducted on a computer-controlled Keithley 236 source measure unit. A Xenon lamp with AM1.5 filter was used as a white-light source and the optical power was 100 mW cm−2. All the measurements were automatically controlled by a computer system, and performed under ambient atmosphere at room temperature.
2.4. Fabrication of OFET devices
Thin-film OFETs were fabricated with top-contact configuration. An n-doped Si wafer with a thermally grown silicon dioxide layer (thickness of 450 nm) was used as the substrate. The substrates were cleaned in water, alcohol, acetone, and rinsed in deionized water. Thin polymer films were prepared by spin coating of a 0.3 wt% solution of DPAV-PT in chloroform onto the bare SiO2/Si substrates at a speed of 3000 rpm (revolutions per minute) for 40 s at room temperature. After dried at 80 °C and annealed at 180 °C under N2 for half an hour, gold film (50 nm) was deposited on the organic layer to form the drain and source electrodes, for a typical device, the drain-source channel length (L) and width (W) are 50 μm and 3000 μm, respectively. OFET measurements were performed at room temperature using a Keithley 4200SC semiconductor parameter analyzer under ambient conditions. AFM images were obtained using a Veeco's Dimension V atomic force microscopic (AFM) in the tapping mode.
2.5. Synthesis of monomers and polymer
The synthetic routes to the monomers and polymer are shown in Scheme 2. The detailed synthetic procedures are as follows.
 |
| | Scheme 2 The synthetic routes to the monomers and DPAV-PT. | |
2.5.1.
N-Octyldiphenylamine 1.
Diphenylamine (8.5 g, 50 mmol), sodium hydroxide (20.0 g, 500 mmol), and dimethyl sulfoxide (100 mL) were placed in a 250 mL three-necked flask, the mixture was stirred for half an hour, octyl bromide (7.7 mL, 55 mmol) was added dropwise to the reaction mixture in 20 min, and then this mixture was stirred for 24 h at room temperature. The reaction mixture was poured into water, extracted with methylene dichloride, and then dried with MgSO4. The resulting liquid was purified by column chromatography using petroleum ether as eluent, colorless oil was obtained (9.8 g, 70%). GC-MS: m/z = 281. 1H NMR (400 MHz, CDCl3): 7.49–6.91 (m, 10H), 3.68 (t, 2H), 1.77–1.36 (m, 12H), 0.91 (t, 3H).
2.5.2. 4-Formyl-N-octyldiphenylamine 2.
A 100 mL three-necked flask containing 4.4 mL (52.8 mmol) of anhydrous DMF was cooled in an ice bath. To this solution, 1.4 mL (14.4 mmol) of phosphorus oxychloride was added dropwise over 30 min. Compound 1 (1.35 g, 4.8 mmol) in 30 mL of 1,2-dichloroethane was added to the above solution and heated to ca. 90 °C for 48 h. This solution was cooled to room temperature, poured into ice water, and neutralized to pH 6–7 by dropwise addition of saturated aqueous sodium hydroxide solution. The mixture was extracted with chloroform. The combined organic layer was dried with anhydrous MgSO4 and then concentrated under reduced pressure. The titled product was obtained (1.07 g, 72%) using petroleum ether and ethyl acetate (10:1) as eluent by column chromatography under reduced pressure. GC-MS: m/z = 309. 1H NMR (400 MHz, CDCl3): 9.86 (s, H), 7.3 (d, 2H), 7.2 (d, 2H), 7.1 (d, 2H), 6.77 (d, 3H), 3.68 (t, 2H), 1.75–1.25 (m, 12H), 0.88 (t, 3H).
2.5.3. 2,5-Dibromo-3-(n-octyldiphenylamine-vinylene)thiophene 3.
Under an ice-water bath, (2,5-dibromothiophen-3-ylmethyl)phosphonic acid diethyl ester (3.35 g, 8.3 mmol) was dissolved in 10 mL of DMF, and CH3ONa (0.6 g, 11 mmol) was added. After 5 min, compound 2 (2.56 g, 8.3 mmol) was added dropwise to the solution. After 2 h, the solution was poured into methanol and filtered, and a yellow liquid of 3 was obtained (2.36 g, 52%) using petroleum ether as eluent. GC-MS: m/z = 547. 1H NMR (400 MHz, CDCl3): 7.68 (d, 2H), 7.3 (d, 2H), 7.2 (d, 2H), 6.95 (q, 2H), 6.77 (d, 1H), 6.6 (s, 1H), 6.5 (d, 2H), 3.68 (t, 2H), 1.75–1.25 (m, 12H), 0.88 (t, 3H).
2.5.4. Synthesis of Polymer DPAV-PT.
Pd(PPh3)4 (30 mg, 0.026 mmol), monomer 3 (0.547 g, 1 mmol), and 2,5-bis(tributylstannyl)thiophene (0.667 g, 1 mmol) were put into a three-necked flask. The mixture was flushed with N2 for 10 min, and then 18 mL of toluene was added. Under the protection of N2, the reactant was heated to reflux for 12 h. The mixture was cooled to room temperature and poured into 30 mL of methanol and then filtered into a Soxhlet thimble. Soxhlet extractions were performed with methanol, hexane, and CHCl3. The polymer was recovered from the CHCl3 fraction by rotary evaporation. The solid was dried under vacuum overnight. The dark-red polymer of DPAV-PT was obtained for 440 mg (yield: 94%). 1H NMR (400 MHz, CDCl3): 7.3–6.8 (br, 14H), 3.68 (t, 2H), 1.75–1.25 (m, 12H), 0.88 (t, 3H).
3. Results and discussion
3.1 Synthesis and characterization of DPAV-PT
The synthesis of the monomers and the corresponding polymer are outlined in Scheme 2. Diphenylamine was used as a starting material for preparation of compound 1, 1 was in turn converted to 2 by Vilsmeier reaction, monomer 3 was obtained using 2via Wittig reaction. The polymer was synthesized with 2,5-bis(tributylstannyl)thiophene in the presence of dibromide 3 using Stille type cross-coupling condensation polymerization. The DPAV-PT was purified by continuous extractions with methanol, hexane, and chloroform using Soxhlet apparatus, and the chloroform part was recovered. The chemical structure of the polymer was verified by 1H NMR as shown in Fig. 1. The characteristic signals at 7.3–6.8 ppm can be assigned to the resonance of protons on phenyl ring, thiophene ring and vinylene group. –CH2– protons linking to nitrogen are at 3.68 ppm, the signals at 1.75–0.88 ppm correspond to the protons of the long alkyl chain. SEC result (using polystyrene standards and THF as eluent) has shown weight-average molecular weight (Mw) value is 3.1 × 104 with the polydispersity index (PDI) of 1.3.
3.2 Thermal stability
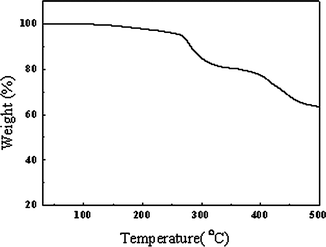
Thermal stability of the polymer is important for device fabrication. Fig. 2 displays the TGA thermogram of DPAV-PT. The TGA analysis reveals that, under the protection of an inert atmosphere, the onset points of the weight loss (5%) of DPAV-PT is ca. 267 °C. Tg was not observed in the DSC thermogram. Good thermal stability of the resulting copolymer prevents the deformation of the copolymer morphology and the degradation of the polymeric active layer under applied electric fields.
3.3 Optical properties
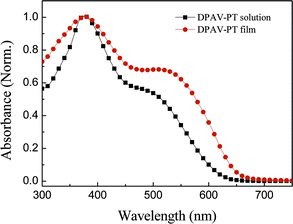
The photophysical characteristic of the polymer DPAV-PT was investigated by UV-Vis absorption spectra in dilute chloroform solution and in solid film spinning-coated on a quartz substrate. Fig. 3 shows the UV-Vis absorption spectra of the polymer solution and film. In solution, DPAV-PT shows an absorption peak at ca. 378 nm and a shoulder at ca. 502 nm. The peak of 378 nm belongs to the absorption of the diphenylamine-vinylene conjugated side chains, and the 502 nm shoulder peak could be ascribed to the π–π* transition absorption of the conjugated polythiophene main chains. The main chain absorption of the polymer film in the visible region got broadened, red-shifted in comparison with that of the polymer solution, because of the aggregation and stronger interchain interactions between the conjugated main chains of the conjugated polymer. The absorption edge of the polymer film is at ca. 670 nm, corresponding to an optical bandgap (Eoptg) of 1.85 eV.13
3.4. Electrochemical properties
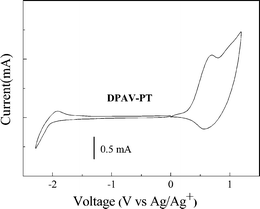
The onset oxidation and reduction potentials obtained from the cyclic voltammograms correspond to the HOMO and LUMO energy levels, respectively.14,15Fig. 4 shows cyclic voltammograms of a DPAV-PT film on a Pt electrode with 0.1 mol L−1 tetrabutylammonium hexafluorophosphate (Bu4NPF6)/CH3CN as the electrolyte at a scan rate of 50 mV s−1. It can be seen that DPAV-PT exhibits quasi-reversible or reversible p-doping/dedoping (oxidation/re-reduction) processes over a positive potential range and irreversible n-doping/dedoping (reduction/re-oxidation) processes over a negative potential range. In a positive potential region, the onset oxidation potential (Eoxon) is 0.3 V vs. Ag/Ag+ for DPAV-PT. In the negative potential region, the onset reduction potential (Eredon) located at −1.75 V vs. Ag/Ag+ for DPAV-PT.
From the onset oxidation potential (Eoxon) and the onset reduction potential (Eredon) of the polymer DPAV-PT, we calculated the HOMO and LUMO energy levels of the polymer according to the equations.16
| HOMO= −e(Eoxon + 4.71) (eV) |
| LUMO= −e(Eredon + 4.71) (eV) |
The ELUMO and EHOMO values of DPAV-PT are −2.96 eV and −5.01 eV, respectively, and the corresponding electrochemical bandgap (EECg) was 2.05 eV. The electrochemical bandgap is slightly larger than the optical bandgap due probably to the interface barrier for the charge injection.
3.5. Theoretical calculations
The optimal geometries and electronic state wavefunction distribution of HOMO and LUMO of the model compound (monomer) were obtained at the DFT B3LYP/6-31G* level using the Gaussian 03 program suit (Fig. 5).17 To simplify the calculations, the alkyl chain was replaced by a –CH3 group. DFT/B3LYP/6-31G* has been found to be an accurate method for calculating the optimal geometry and electronic structures of many molecular systems. Ab intitio calculations on the model compound for DPAV-PT show that the electrons are delocalized within the entire molecule due to the pi-conjugation. The electronic wavefunction of the HOMO was distributed entirely over conjugated molecules, which is beneficial for obtaining higher hole mobility. From the DFT B3LYP/6-31G* level calculations,18 HOMO and LUMO energy levels of DPAV-PT are −5.3 and −3.24 eV and Eg is 2.06 eV, which are in good agreement with the experimental values for the energy gap and the HOMO and LUMO energy levels. Therefore, the DFT calculations performed here on the repetitive units can provide good estimations of the HOMO, LUMO, and bandgap energy trends, thus allowing a rapid screening of the most promising polymeric structures.
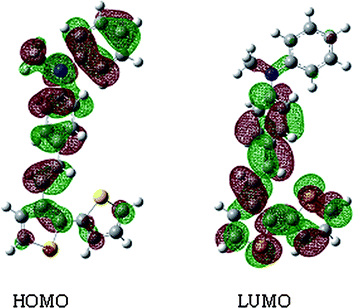 |
| | Fig. 5 Molecular orbital iso-surfaces of HOMO and LUMO of the model compound for DPAV-PT, calculated at the DFT B3LYP/6-31G* level. | |
3.6 Photovoltaic properties
In order to investigate the potential applications of the copolymer in solar cells, bulk heterojunction PSCs were fabricated with a device structure of ITO/PEDOT:PSS/DPAV-PT:PCBM(1:1 w/w)/Ca/Al. The PSC device was tested under simulated 100 mW cm−2 AM 1.5G illumination. The active area was 4 mm2 for the solar cell device discussed in this work, and the thickness of the active layer was 77 nm. Fig. 6 shows the current density versus voltage (J–V) curve of the device and the corresponding open-circuit voltage (Voc), short circuit current (Jsc), fill factor (FF). It can be seen that the Voc, Jsc and FF values of the device with DPAV-PT as electron donor are 0.68 V, 2.78 mA cm−2, 0.35, respectively. PCE of the preliminary device based on DPAV-PT is 0.7%. Compared with the photovoltaic device based on OTPAV-PT, 12 the device based on DPAV-PT possessed better photovoltaic performance, which may be contributed to by the higher open circuit voltage, relatively broader and red-shifted absorption, relative higher hole mobility in the blend of DPAV-PT and PCBM.
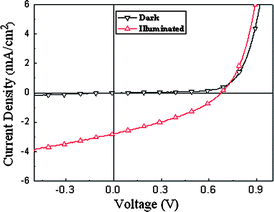 |
| | Fig. 6
J–V curves of the polymer solar cell based on DPAV-PT in the dark and under the illumination of AM 1.5 G, 100 mW cm−2. | |
3.7 Organic field-effect transistors
OFETs based on DPAV-PT were fabricated using solution processing. Top-contact OFETs were fabricated on a highly n-doped silicon wafer with a 450 nm thick thermally grown SiO2 dielectric layer. The surface of the substrate was modified with octadecyltrichlorosilane (OTS). The semiconducting layer (thickness about 40 nm) was spin-cast onto the substrate at 3000 rpm from 0.3 wt% chloroform solution. Gold source and drain electrodes were deposited through a shadow mask on top of active layer. The OFETs of the DPAV-PT exhibited typical p-channel OFET characteristics.
In the OFETs, drain current (IDS) can be described with the following equation:
| IDS = μ(W/2L)Ci(VG − VT)2 |
Where Ci is the capacitance per unit area of the gate dielectric layer, VG is the gate voltage, VT is the threshold voltage, μ is the field-effect mobility, and W and L are the channel width and length dimensions, respectively. The mobility was calculated from the slope of the curve of IDS1/2vs. VG. The threshold voltage of the device was determined from the linear fit of the square root of IDS at the saturated regime vs. gate voltage VG, and VT was determined by extrapolating the measured data to IDS = 0.
Thin films of DPAV-PT exhibit relative low hole mobility without thermal annealing. The mobility of DPAV-PT films deposited on bare and OTS treated substrates were about 7.91 × 10−5 cm2 V−1 s−1 and 1.10 × 10−4 cm2 V−1 s−1, respectively. OTS surface modification reduced the threshold voltage from −17–−21 V to −2–−6 V, and slightly improved the on/off ratio as shown in Fig. 7. Due to low HOMO of DPAV-PT (−5.0 eV), the current at zero gate voltage of the OFET devices was very low, affording ideal switch on characteristics and superior stability against doping, which is dramatically better than the polymer P3HT.
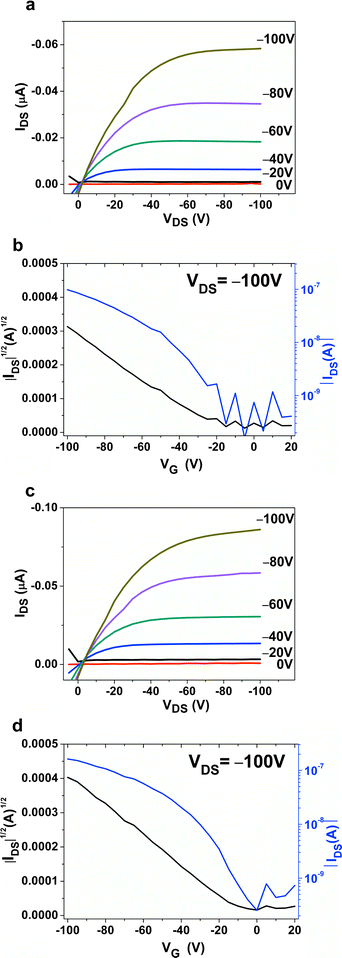 |
| | Fig. 7 Output characteristics and transfer characteristics of DPAV-PT thin films deposited on (a) and (b), bare SiO2; (c) and (d), OTS modified SiO2. | |
Fig. 8a–d show the output curves and the transfer characteristics of DPAV-PT, without and with OTS modifications after being annealed at 180 °C. After annealing, OFETs of DPAV-PT fabricated on OTS treated substrate afforded a hole mobility of 6.1 × 10−4 cm2 V−1 s−1, an on/off ratio of 103 and a threshold voltage of −7 V, respectively. This device performance showed apparent improvement compared to that of the devices without annealing. In addition, further improvement of the mobility of DPAV-PT could be achieved by using more favorable device fabrication conditions (e.g. different solvents, surface treatment, thermal treatment, etc).
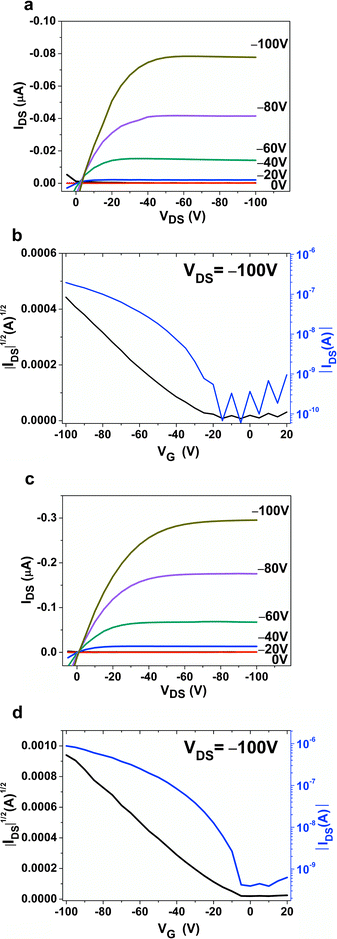 |
| | Fig. 8 Output characteristics and transfer characteristics of annealed (180 °C) DPAV-PT thin films deposited on (a) and (b), bare SiO2; (c) and (d), OTS modified SiO2. | |
Fig. 9a and b show the AFM images of films spin-cast with DPAV-PT on bare and OTS modified substrate, Fig. 9c and d show the AFM images of the annealed films. Both the presence of the OTS monolayer at the polymer/dielectric interface and the thermal annealing increase surface smoothness of DPAV-PT thin films. As-spun films of DPAV-PT are always more rough than those of annealed thin films, independent of substrate chemistry. The morphology of the films is in agreement with their trend of charge transport performances. The hole mobility was increased after device annealed and it was apparently correlated with the surface morphology, indicating a strong relationship between morphology and device performances.
 |
| | Fig. 9 AFM imagines of DPAV-PT thin films deposited on (a) and (c), bare SiO2; (b) and (d), OTS modified SiO2, (c) and (d) are annealed (180 °C). | |
4. Conclusion
In summary, we have synthesized a new side chain conjugated polythiophene derivative, DPAV-PT, by the Stille coupling reaction. The polymer possesses good solubility in common organic solvents. DPAV-PT film exhibited broad absorption in the visible region from 300 nm to 650 nm, a relatively low HOMO level, which is beneficial for the air-stability and the increase of Voc of the PSC. A DPAV-PT based polymer solar cell afforded a PCE of 0.7% under the illumination of AM 1.5, 100 mW cm−2, a FET device demonstrated an average hole mobility of 6.1 × 10−4 cm2 V−1 s−1 after 180 °C annealing. The preliminary results indicate that DPAV-PT could be a good candidate for applications in polymer solar cell and organic field effect transistors.
Acknowledgements
The authors acknowledge Xuesen Mo for the synthesis of the intermediates and appreciate great help from Prof. Yongfang Li from Institute of Chemistry and Dr Dequan Xiao from Yale University. This work was supported by National Natural Science Foundation of China, the Opening Fund of State Key Laboratory of Powder Metallurgy and Start-up fund of Central South University.
References
-
(a) J. Liu, Z. Xie, Y. Cheng, Y. Geng, L. Wang, X. Jing and F. Wang, Adv. Mater., 2007, 19, 531 CrossRef CAS;
(b) H. Wu, Y. Lei, W. Yang and Y. Cao, Chem. Soc. Rev., 2009, 38, 3391 RSC.
-
(a) Y. He, W. Wu, G. Zhao, Y. Liu and Y. Li, Macromolecules, 2008, 41, 9760 CrossRef CAS;
(b) X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246 CrossRef CAS;
(c) B. S. Ong, Y. Wu, P. Liu and S. Gardner, Adv. Mater., 2005, 17, 1141 CrossRef CAS;
(d) K. Lu, C. Di, H. Xi, Y. Liu, G. Yu, W. Qiu, H. Zhang, X. Gao, Y. Liu, T. Qi, C. Du and D. Zhu, J. Mater. Chem., 2008, 18, 3426 RSC.
-
(a) C. B. Nielsen, A. Angerhofer, K. A. Abboud and J. R. Reynolds, J. Am. Chem. Soc., 2008, 130, 9734 CrossRef CAS;
(b) Y. Zou, G. Sang, M. Wan, S. Tan and Y. Li, Macromol. Chem. Phys., 2008, 209, 1454 CrossRef CAS.
-
(a) N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295 CrossRef CAS;
(b) Y. Zou, D. Gendron, R. Badrou-Aïch, A. Najari, Y. Tao and M. Leclerc, Macromolecules, 2009, 42, 2891 CrossRef CAS.
-
(a) I. B. Kim, B. Erdogan, J. N. Wilson and U. F. H. Bunz, Chem.–Eur. J., 2004, 10, 6247 CrossRef CAS;
(b) Y. Zou, M. Wan, G. Sang, M. Ye and Y. Li, Adv. Funct. Mater., 2008, 18, 2724 CrossRef CAS.
- Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952 CrossRef CAS.
- J. Hou, L. Huo, C. He, C. Yang and Y. Li, Macromolecules, 2006, 39, 594 CrossRef CAS.
- J. Hou, Z. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911 CrossRef CAS.
- E. J. Zhou, Z. A. Tan, L. J. Huo, Y. J. He, C. H. Yang and Y. F. Li, J. Phys. Chem. B, 2006, 110, 26062 CrossRef CAS.
- Y. Zou, W. Wu, G. Sang, Y. Yang, Y. Liu and Y. Li, Macromolecules, 2007, 40, 7231 CrossRef CAS.
- G. B. Blanchet, Y. Loo, J. A. Rogers, F. Gao and C. R. Fincher, Appl. Phys. Lett., 2003, 82, 463 CrossRef CAS.
- Y. Zou, G. Sang, W. Wu, Y. Liu and Y. Li, Synth. Met., 2009, 159, 182 CrossRef CAS.
- Y. Zou, D. Gendron, R. Neagu-Plesu and M. Leclerc, Macromolecules, 2009, 42, 6361 CrossRef CAS.
- Y. F. Li, Y. Cao, J. Gao, D. L. Wang, G. Yu and A. J. Heeger, Synth. Met., 1999, 99, 243 CrossRef CAS.
- Y. Zou, J. Hou, C. Yang and Y. Li, Macromolecules, 2006, 39, 8889 CrossRef CAS.
- Q. Sun, H. Wang, C. Yang and Y. Li, J. Mater. Chem., 2003, 13, 800 RSC.
- B. Liu, A. Najari, C. Pan, M. Leclerc, D. Xiao and Y. Zou, Macromol. Rapid Commun., 2010, 31, 391 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. N. Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.