Miktoarm star polymers: advances in synthesis, self-assembly, and applications
Kunal
Khanna
a,
Sunil
Varshney
b and
Ashok
Kakkar
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke St West, Montreal, Quebec H3A 2K6, Canada. E-mail: ashok.kakkar@mcgill.ca; Tel: +1 (514) 398-6912
bPolymer Source, 124 Avro Street, Dorval, Quebec H9P 2X8, Canada
First published on 25th May 2010
Abstract
Miktoarm polymers are a relatively new and unique class of macromolecules, and constitute a topical area of research due to their intriguing properties which can be tailored by varying their polymer arms. Much emphasis has been placed in the recent past in developing synthetic methodologies to these star polymers, and examining their self-assembly in solution. This review summarizes the progress made in the area of miktoarm star polymers in terms of their synthesis, behavior in solution, and applications. The different synthetic strategies to construct a variety of miktoarm star polymers are described, and each methodology strikes a balance between ease of synthesis and control over the final architecture. The self-assembly of miktoarm polymers in solution is then elaborated, which is frequently studied as a function of either arm-length (an intrinsic property of the star) or the application of an external stimulus (pH, temperature, etc.). This is followed by an overview of the applications of these stars in areas including drug delivery.
 Kunal Khanna | Kunal Khanna received his BS in Chemistry from Emory University (USA) in 2007, where he conducted research in the group of Professor James T. Kindt. He then came to McGill University and completed his MSc degree in 2010 under the guidance of Professor Ashok Kakkar. His graduate research was concerned with the synthesis and self-assembly of ABC-type miktoarm polymers, and their biological applications. |
 Sunil Varshney | Sunil K. Varshney is the founder and Chairman of Polymer Source Inc. established in 1994 in Montreal, Quebec, Canada. He completed his PhD in Polymer Technology from Indian Institute of Technology, Delhi, India in 1980. He has served as the group leader and consultant at Elf Petrochemicals, France, and was also associated with University of Leige, Belgium from the period 1985 to 1991. He has 29 patents and more than 150 peer-reviewed publications. Polymer Source is a major provider of high valued polymers that are commonly used in pharmaceutical industries, federal laboratories and universities for fundamental and applied research. |
 Ashok Kakkar | Ashok Kakkar is an Associate Professor in the Department of Chemistry at McGill University. He obtained training in Chemistry under the directions of Professor Todd B. Marder (PhD University of Waterloo), Professor The Lord Lewis (NSERC Post-doctoral Fellow, University of Cambridge) and Professor Tobin Marks (NSERC Post-doctoral Fellow, Northwestern University). His research interests include developing methodologies to complex architectures (hyperbranched macromolecules including dendrimers, miktoarm polymers and self-assembled monolayers), and studying their supramolecular chemistry in solution and at interfaces. |
Introduction
Polymers constitute an intricate part of our everyday lives, as well as they continue to be at the cutting edge of research.1 While our understanding of polymers has grown tremendously over the past few decades, much needs to be still done to advance them into the novel materials applications that have yet to be developed. For instance, a greater understanding of their self-assembly in an aqueous medium could yield novel and sophisticated drug delivery vehicles.2 In fact, drug delivery is an extremely important and promising field of research, where various classes of polymers have offered the potential to increase the bioavailability and therapeutic index of drug molecules without leaving toxic byproducts in the body.3 Polymers also offer a facile and cleaner alternative route towards successful gene delivery, and the use of specifically designed macromolecules has generated much promise in gene delivery.4 While linear polymers have traditionally dominated in these areas, recent advances in polymer synthesis have led to the ability to build more complex polymeric architectures, such as gradient polymers,5 polymer brushes,6 graft polymers,7 dendrimers,8 and star polymers.9 Each of these various architectures impart a whole new set of intriguing properties, such as unique morphologies and assemblies in the bulk, that were never thought possible for linear polymers. It is essential to continue to develop each of these specific fields of polymer research, and to elucidate where the greatest promise and potential lies. To this end, the work summarized in this review is oriented towards a specific class of star polymers called miktoarm star polymers.10 Miktoarm star polymers (sometimes called asymmetric star polymers, heteroarm star polymers, or simply miktoarm polymers) are star-shaped polymers where any number of various types of polymer arms emanate from a core. These polymer arms should vary by chemical identity and/or molecular weight (Fig. 1). This specific class of polymers should not be confused with other classes of polymers, such as graft copolymers, H-shaped copolymers, etc., which lack a star-shaped architecture with polymer arms emanating from a focal point (as opposed to a polymeric unit).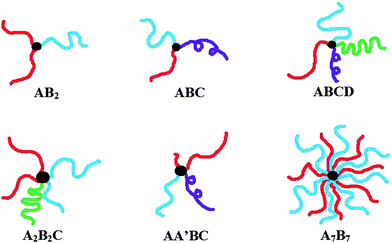 | ||
| Fig. 1 Some different types of miktoarm polymers, whose polymer arms vary by the chemical identity or molecular weight. | ||
Miktoarm star polymers are a synthetically challenging class of polymers. Multiple protection/deprotection strategies, orthogonality, and combination of different polymerization methods are typically necessary for the synthesis of these polymers, regardless of the specific type of desired miktoarm star polymer (A2B, ABC, AB2C2, etc.). Considerable advances in synthetic strategies, self-assembly and applications have recently occurred, making scientific community become increasingly aware of the potential of miktoarm polymers. The scope of this review is to focus on work done within the past decade on miktoarm polymers. An excellent review of work done prior to this decade can be found elsewhere.10 While computational simulations and theoretical chemistry have been combined to elucidate the morphology of miktoarm polymers,11 this review will instead focus on experimental work.
Synthetic strategies
Chlorosilane compounds
One of the most established synthetic strategies for miktoarm stars involves linking chlorosilane compounds which serve as a core, with polymers with reactive chain ends synthesized using living anionic polymerization. Hadjichristidis and coworkers have used tetrachlorosilane to synthesize an A(AB)3-type miktoarm polymer, composed of polystyrene (PS) and polystyrene-b-polyisoprene (Scheme 1).12 In order to ensure that one polystyrene arm was attached to the core, excess tetrachlorosilane was reacted with the anionic chain end of polystyrene, and any unreacted tetrachlorosilane was then easily removed by vacuum. A 20% excess of PS-b-PILi was used to replace the remaining chlorine atoms on the core, thus completing the final miktoarm star. Size exclusion chromatography (SEC) of this star polymer revealed impurities whose resolution was much better resolved by temperature gradient interaction chromatography (TGIC). In fact, TGIC allowed each eluted peak to be characterized as either the product miktoarm star, or some byproduct of the synthesis, such as (PS-b-PI)4 or even (PS-b-PI)5 star copolymers. One notable feature of the chlorosilane method evident from this example is the necessity of carefully reacting the living chain end of a polymer with the Si–Cl bond, for achieving controlled addition of a polymer arm onto the chlorosilane compound. This is often done by adding the chlorosilane compound in excess to ensure monosubstitution. For the creation of miktoarm polymers with more than two types of arms, titration becomes necessary for achieving monosubstitution of a single Si–Cl bond in the chlorosilane core, assuming a polymer is already attached to the core. In this case, titration is done to ensure monosubstitution for two reasons: simpler purifications and for substituting the remaining Si–Cl bonds with a different polymer. This technique has been used to create ABCD miktoarm star polymers as a result of careful introduction of living polymers on the basis of steric hindrance.13 Finally, it is important to note that the chlorosilane methodology involves stringent reaction conditions, such as moisture-free conditions and complicated reactors that typically involve break-seal technology, due to its dependence on living anionic polymerization.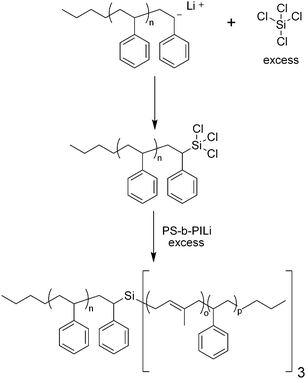 | ||
| Scheme 1 Synthesis of A(AB)3 miktoarm polymer using living anionic polymerization and a chlorosilane linking agent. | ||
Sometimes it is beneficial to combine chlorosilane chemistry with other types of coupling reactions to build the desired miktoarm star. To this end, Hadjichristidis's group created a core composed of two chlorosilane groups and diphenylethylene for the synthesis of an ABCD-type miktoarm star with four mutually incompatible arms: polystyrene, polyisoprene, polydimethylsiloxane (PDMS), and poly(2-vinylpyridine) (P2VP).14 To create the necessary diblock copolymer precursor, titration was used sequentially on the core—first for the monosubstitution of one chlorine with PI and a second time for the monosubstitution of the second chlorine with PDMS (Scheme 2). Because of steric hindrance, the reactivity of the second chlorine is reduced after substitution of the first chlorine with PI. SEC was used to monitor these reactions and determine the endpoint. In a new reactor, a living polystyrene chain (PSLi) was synthesized, and to it was added an equimolar amount of the diblock copolymer. The living polystyrene chain end reacted on the vinyl group of the diphenylethylene moiety on the core, thus creating a triblock copolymer. Finally, the resulting anion was exploited for the living anionic polymerization of 2-vinylpyridine, and the resulting polymer arm was capped with methanol to construct the final ABCD miktoarm star polymer. Purification of such a complex miktoarm polymer from its residual byproducts could only be achieved using a Soxhlet apparatus. Nonetheless, three miktoarm stars were prepared using this strategy, with Mn of approximately 75 kg mol−1 and polydispersities (PDIs) between 1.06 and 1.11. This methodology demonstrates how different synthetic strategies can be combined to create a desired miktoarm star polymer. Furthermore, it demonstrates how the use of a vinylic compound such as diphenylethylene requires a carefully designed synthetic protocol, just as is necessary for chlorosilane protocols. Even though the use of chlorosilane linking agents is one of the oldest strategies for making miktoarm star polymers, it is still an effective strategy and continues to be employed in the synthesis of miktoarm stars.15
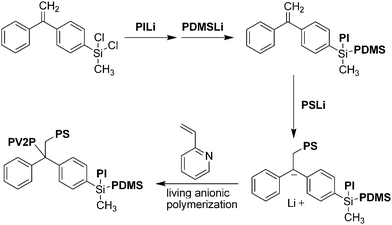 | ||
| Scheme 2 Combination of two synthetic strategies—chlorosilane groups and diphenylethylene (a vinylic compound)—in a single core for the attachment of four different polymers. | ||
Iterative method
The iterative method has been developed by Hirao and coworkers as a versatile synthetic strategy to create a wide variety of complex miktoarm stars through living anionic polymerization. The primary attraction of the iterative method of miktoarm polymer synthesis lies in the ability to make well-defined miktoarm stars without a complex reaction sequence. In an excellent demonstration of this strategy, ABC-, ABCD-, and ABCDE-type miktoarm star polymers were synthesized by living anionic polymerization techniques and 1-(4-(3-bromopropyl)phenyl)-1-phenylethylene which serves as a linking agent.16 The synthesis for these polymers required two steps: (1) linking one living anionic polymer to a second polymer functionalized with the diphenylethylene compound, to create in situ an anion at the branching point, and (2) introduction of another diphenylethylene compound to the growing polymer via the anion. Repetition of these two steps led to the production of miktoarm polymers with excellent polydispersities below 1.03. GPC, 1H NMR, and elemental analysis confirmed that the stars were properly made, with both controlled architecture and desired molecular weight of each polymer arm.The same concept was applied to create an ABCDEFG-type miktoarm star polymer in the exact same manner, except this time a butadiene-derivatized compound was used as the linking agent.17 Other complex miktoarm polymers have been synthesized such as ABCD2, AB2C2, and ABCD2E2.18 The versatility of this iterative methodology was clearly demonstrated in the synthesis of AB2C2D2E4-, AB2C2D4E4-, AB2C4D8E8-, and AB2C4D8E16-type miktoarm stars.19 These stars were made by coupling one of two different diphenylethylene-derivatized linking agents to the in situ generated anion of the growing polymer. These two linking agents contained either one or two diphenylethylene groups, so that either one or two living polymer arms were subsequently coupled to the growing polymer. Many other synthetic examples have been reported with success using this iterative method.20
“Core-first” method
There are many examples throughout the literature where the “core-first” method is used as a synthetic strategy. The first step in this methodology requires the synthesis of a multifunctional initiator, also called a “multifunctional core,” or simply a “core” containing orthogonal initiating sites. From this core, each arm is grown outwards through a combination of different polymerization techniques, such as living anionic polymerization, ring-opening polymerization (ROP),21 or a variety of controlled/“living” radical polymerization (CRP)22 techniques including atom transfer radical polymerization (ATRP),23 nitroxide mediated polymerization (NMP),24 reversible addition–fragmentation chain transfer (RAFT) polymerization,25etc. Because of the combination of different polymerization methods, it is easy to introduce a wide variety of monomers into the final polymeric structure. Also, orthogonality plays a key role in the design of the multifunctional initiator—a successful synthesis of miktoarm stars requires one to polymerize a single type of initiating site on the core while leaving the other sites intact, thus ensuring that each polymerization occurs as intended on the core. Because of this nature of the multifunctional initiator, the number of polymerization initiating sites on the core is the same as the number of polymeric arms on the final miktoarm star. For instance, a multifunctional core with two ATRP initiating sites and one ROP initiating site will yield an A2B miktoarm star polymer.Using the “core-first” method, Tunca and coworkers built an AB2 miktoarm star polymer, in which the first step was the synthesis of a multifunctional initiator designed for sequential ROP and ATRP (Scheme 3).26 After characterization, the core was used for ROP of ε-caprolactone through the alcohol, to generate a polymer functionalized with two bromine end groups. These sites were polymerized via ATRP using either tert-butyl acrylate or methyl methacrylate to create two different types of AB2-type miktoarm star polymers. The polydispersities (PDIs) of these stars were relatively low (<1.23).
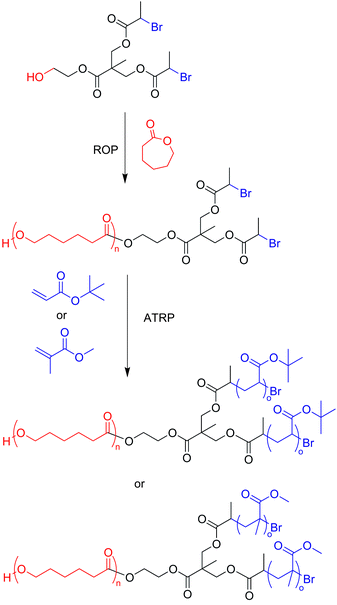
Though the synthesis of ABC miktoarm stars requires a greater deal of sophistication in the synthetic strategy, this can still be easily accomplished using the “core-first” method. Hizal and coworkers synthesized a multifunctional initiator with three different initiating sites for ATRP, ROP, and stable free-radical polymerization (SFRP) to create an ABC miktoarm star polymer where each polymerization step does not require end-group modification for subsequent polymerization reactions (Scheme 4).27 This polymer was composed of polycaprolactone (PCL), polystyrene (PS), and poly(tert-butyl acrylate) (PtBA). Because of the variety of polymerization methods and their accompanying choice of monomers, it is easy to see why the “core-first” technique is a versatile and efficient synthetic strategy, and it has been widely used.28 A simple substitution of one monomer for another can lead to a library of ABC-type miktoarm stars. For instance, in Scheme 3 the ROP of ε-caprolactone could just as easily be replaced with the ROP of glycolic acid or lactide to create two more miktoarm stars.
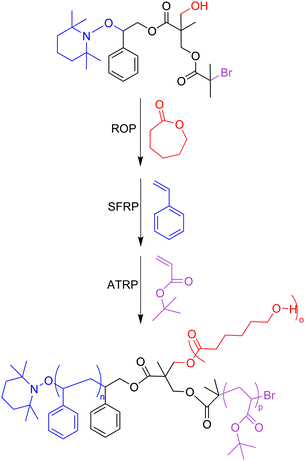 | ||
| Scheme 4 Synthesis of an ABC miktoarm star polymer made with the “core-first” method by three different polymerization methods. | ||
Variations to the “core-first” strategy have become prevalent throughout the literature. For example, it is possible to employ two ROP and one ATRP from a multifunctional initiator to create an ABC miktoarm star provided that protection/deprotection strategies are used.29 In this regard, a core compound composed of one bromide chain-end and two alcohols—one protected with a triphenylmethyl group and the other deprotected—was prepared. ROP of the deprotected alcohol using ε-caprolactone as monomer followed by ATRP with styrene yielded a diblock copolymer. Because the final step involved ROP, it was necessary to functionalize the chain-end of the PCL arm with a protecting group in order to avoid subsequent ROP reactions on both the PCL chain-end as well as the third alcohol. Finally, acidic conditions were employed to deprotect the alcohol at the core so that subsequent ROP with L-lactide (LLA) could occur, yielding an ABC miktoarm star composed of PS, PLLA, and PCL.
Another more commonly seen variation of the “core-first” strategy involves functionalization of the chain-end of a polymer arm, sometimes called the linear macroinitiator. The functional group at the chain end contains multiple types of initiating sites, thus serving as a branching point for successive polymerization reactions, and eventually yielding the final miktoarm star. This variation of “core-first” is an attractive strategy because the synthesis of a core designed to polymerize n arms is replaced by the synthesis of a chain-end functionalized polymer which allows the growth of n − 1 arms.30 Jérôme and coworkers have used this strategy to create a novel, fully biocompatible ABC-type miktoarm star with poly(ethylene glycol), poly(ε-caprolactone), and poly(benzyl β-malolactonate).31 Monohydroxy PEG was capped at its chain-end with one ε-caprolactone unit. Subsequent hydrolysis of this polymer yields a linear macroinitiator with two functionalities (Scheme 5). Anionic ROP of benzyl β-malolactonate was then achieved without any evident ROP on the core's alcohol. The terminal chain end of poly(benzyl β-malolactonate) was capped with a methyl group using trimethylsilyldiazomethane. As in the previous example, this protection step was necessary to avoid unwanted ROPs on the chain end of poly(benzyl β-malolactonate). Finally, ROP on the diblock copolymer's alcohol resulted in the final miktoarm star polymer with a polydispersity of 1.50. This increase in PDI occurred during the ROP of ε-caprolactone, most likely due to slow ROP initiation of the diblock copolymer. Though this method was successful in producing the desired miktoarm polymer, some optimization is necessary—mostly to reduce the PDI—in order to make these polymers more optimal for their intended biological applications.
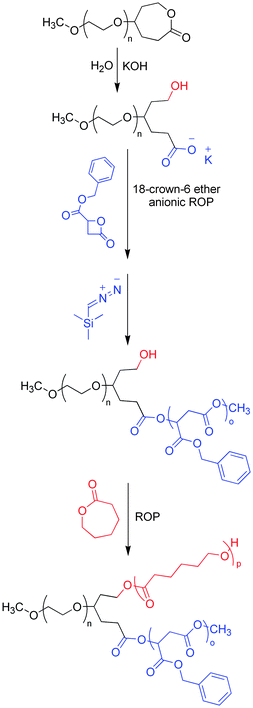 | ||
| Scheme 5 Synthesis of a biocompatible ABC miktoarm star by functionalization of a linear macroinitiator (PEG) followed by successive ROPs. | ||
“Arm first” method
Although the “arm-first” method had typically been used to create homoarm star polymers,32 Gao and Matyjaszewski were the first to use it to demonstrate the synthesis of miktoarm star polymers, where miktoarm stars with many different arms (the number of arms ranged anywhere from 35 to 84) were synthesized using either two or five different types of polymers.33 Generally, in this methodology, the chain ends of many linear macroinitiators, which are formed from a number of CRP methods, are used to polymerize a divinyl compound, typically divinylbenzene (DVB). Many polymers were used to initiate this polymerization in a one-pot fashion to yield miktoarm stars in high yields. The final miktoarm polymer consisted of a crosslinked microgel core composed of DVB, which ties together the many polymer arms that initiated the DVB polymerization, with the polymers emanating outwards (Scheme 6).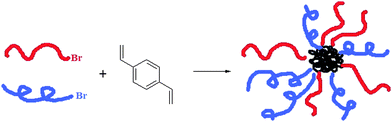 | ||
| Scheme 6 General synthesis of a miktoarm polymer by the “arm-first” method. | ||
According to a study done by Gao and Matyjaszewski, the molar ratio of the various polymers used to initiate polymerization determines the molar ratio of each polymer in the final miktoarm polymer star. For instance, in using a 50/50 mixture of poly(methyl acrylate) (PM) to poly(n-butyl acrylate), a miktoarm star with a 52/48 ratio of these two polymers was achieved, while a starting mixture of 90/10 yielded a miktoarm star with an 87/13 arm ratio. Furthermore, differing chemical identities of the polymeric macroinitiators did not alter the ratios in the final star, which were similar to those of the initial reaction mixture. It was noted that SEC was not sufficient to determine whether the final product was pure, i.e., whether the final product represented a miktoarm polymer or a contaminated mixture containing both miktoarm polymer and two homoarm star polymers. The use of liquid chromatography and gradient polymer elution chromatography (GPEC) helped to confirm the presence of miktoarm polymer and the absence of contaminant homoarm star polymers. These polymers were typically of high molecular weight (over 100 kg mol−1) with PDI generally around 1.40. The “arm-first” method, in general, is one of the less commonly used techniques for miktoarm polymer synthesis.34
“In–out” method
The “in–out” method of miktoarm polymer synthesis can be seen as a cross between the “core-first” and “arm-first” methodologies. Here, a “living” macroinitiator (typically made by CRP methods such as ATRP) initiates the polymerization of a cross-linking agent (such as DVB) to form a homoarm star polymer with polymer arms emanating from the core. Because the macroinitiator's initiating sites are preserved within the core, these initiating sites are used for subsequent polymerization of a second type of monomer to produce a miktoarm star (Scheme 7). Unlike the “core-first” or “arm-first” technique, only AnBm-type miktoarm polymers can be built, i.e., only two types of polymer arms can exist in a miktoarm star polymer synthesized by the “in–out” technique.35 Furthermore, the number of arms of the polymer grown from the core is always less than the number of arms from the macroinitiator (therefore m < n). This can be due to many factors, but the most important reason is the steric hindrance that the initiating sites suffer in the congested core of the homoarm star polymer.36 | ||
| Scheme 7 General synthesis of a miktoarm star by the “in–out” method (the green dots represent bromine atoms). | ||
The “in–out” method has been used successfully to create a miktoarm star polymer composed of PCL and PS linked to a DVB microgel core.37 This was accomplished by first synthesizing PCL which was end-functionalized with a bromine atom. This polymer was subsequently reacted via ATRP with DVB to form a homoarm star polymer macroinitiator. The bromine atoms remaining on the core were subsequently polymerized via ATRP with styrene to form the final miktoarm star polymer. Mn ranged from 70–85 kg mol−1 on the basis of SEC, while the PDI was rather high (1.8–2.7). Further manipulations of this miktoarm star polymer demonstrated the potential applications that exist within this unique class of polymers. To this end, basic hydrolysis of the PCL arm resulted in a homoarm star polymer composed of PS arms. This polymer was subsequently utilized as a nanoenvironment which could facilitate formation of PbS nanoparticles within its microcavities. Electron diffraction studies of the resultant nanoparticles demonstrated the formation of a cubic structure.
Coupling method
Coupling methods have become very widespread for the synthesis of miktoarm polymers in the past decade due to facile synthetic methodologies that have become available, and as these methods help to ensure the overall integrity of the final structure.38 This method is highlighted by coupling of the reactive end of at least one polymer arm to a multifunctional core using highly efficient and orthogonal reactions. The remaining arms on a miktoarm polymer are typically grown using CRP methods, and often by ROP, living anionic polymerization, etc. The popularity of the coupling methods can partially be attributed to the rising popularity of “click” chemistry in macromolecular synthesis.39 “Click” reactions are characterized by many features that lend themselves to macromolecular synthesis, such as simple reaction conditions, orthogonality to a variety of functional groups, simple workups, relatively simple purifications, insensitivity to different solvents, and high yields. Many different “click” reactions have been used to this end, such as thiol–ene40 and Diels–Alder.41 The most popular reaction, the so-called “cream of the crop” is the Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition of an azide to an alkyne to yield a 1,2,3-triazole ring (Fig. 2).42 This specific “click” reaction has found widespread use throughout the synthesis of miktoarm polymers through a coupling strategy.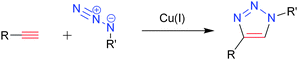 | ||
| Fig. 2 The azide–alkyne Huisgen cycloaddition, a 1,3-dipolar cycloaddition which yields a 1,2,3-triazole ring. | ||
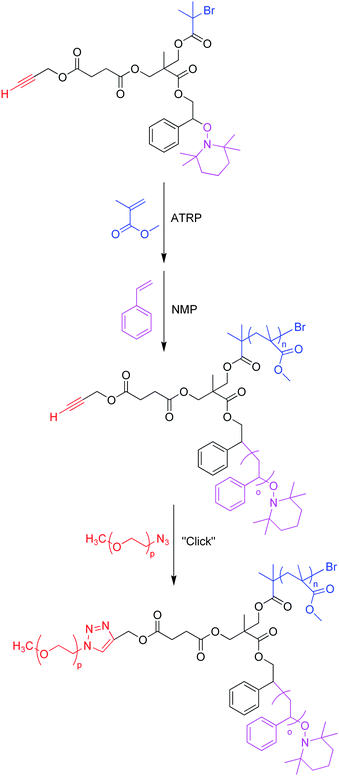
One of the most standard examples of the coupling technique arises from Tunca and coworkers. In their study, ATRP and NMP were used to polymerize PMMA and PS respectively from a trifunctional core, leaving a block copolymer with an alkyne as the only remaining functionality which is located at the junction. Subsequent “click” reaction with either azide-terminated PEG or PtBA yielded an ABC-type miktoarm star polymer (Scheme 8).43 For µ(PMMA-PS-PtBA), the final Mn was 11.2 kg mol−1 with a PDI of 1.15, as determined by GPC equipped with an RI detector. Furthermore, 1H NMR spectroscopy confirmed the incorporation of three distinct polymer arms onto a single scaffold, by appearance of the characteristic peaks of three different polymers, as well as the triazole ring's C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH peak at 7.65 ppm. The synthesis of a second miktoarm polymer, µ(PMMA-PS-PEG), was also confirmed through careful examination of 1H NMR spectra. An Mn of 9.6 kg mol−1 and a PDI of 1.21 were obtained from GPC, which are comparable results to µ(PMMA-PS-PtBA). In considering the fact that these two polymers were made by coupling two different azide-terminated polymers to a block copolymer precursor, these results suggest that a “click” reaction can efficiently couple a variety of azide-terminated polymers to a block copolymer scaffold to create a miktoarm star polymer. Tunca's group has demonstrated that the azide–alkyne “click” reaction can efficiently couple the chain-end of a polymer to the congested core of a block copolymer. Furthermore, the orthogonality of “click” coupling allows tolerance of initiating groups from prior polymerization reactions, the bromide atom for ATRP or the alcohol from ROP for instance, which obviates the need for protection/deprotection strategies for these functionalities. Protection/deprotection strategies can be used, however, to create miktoarm star polymers if the same “click” reaction is to be used in succession. Kakkar and coworkers built an ABC-type miktoarm star polymer using two azide–alkyne Huisgen cycloadditions by selectively deprotecting two alkyl–silyl protected alkynes on the multifunctional core, and then completing the miktoarm star polymer by ROP with ε-caprolactone monomer.44 It should be noted that the “click” coupling method requires a careful control of reaction conditions, as well as a purification strategy for separating a miktoarm star from its polymeric precursors.
CH peak at 7.65 ppm. The synthesis of a second miktoarm polymer, µ(PMMA-PS-PEG), was also confirmed through careful examination of 1H NMR spectra. An Mn of 9.6 kg mol−1 and a PDI of 1.21 were obtained from GPC, which are comparable results to µ(PMMA-PS-PtBA). In considering the fact that these two polymers were made by coupling two different azide-terminated polymers to a block copolymer precursor, these results suggest that a “click” reaction can efficiently couple a variety of azide-terminated polymers to a block copolymer scaffold to create a miktoarm star polymer. Tunca's group has demonstrated that the azide–alkyne “click” reaction can efficiently couple the chain-end of a polymer to the congested core of a block copolymer. Furthermore, the orthogonality of “click” coupling allows tolerance of initiating groups from prior polymerization reactions, the bromide atom for ATRP or the alcohol from ROP for instance, which obviates the need for protection/deprotection strategies for these functionalities. Protection/deprotection strategies can be used, however, to create miktoarm star polymers if the same “click” reaction is to be used in succession. Kakkar and coworkers built an ABC-type miktoarm star polymer using two azide–alkyne Huisgen cycloadditions by selectively deprotecting two alkyl–silyl protected alkynes on the multifunctional core, and then completing the miktoarm star polymer by ROP with ε-caprolactone monomer.44 It should be noted that the “click” coupling method requires a careful control of reaction conditions, as well as a purification strategy for separating a miktoarm star from its polymeric precursors.
 | ||
| Scheme 8 Synthesis of an ABC miktoarm star from a trifunctional core by ATRP, NMP, and “click” chemistry. | ||
Monteiro's group demonstrated in an excellent study that facile synthesis of miktoarm AB2 stars can be easily achieved by optimizing the “click” coupling reaction conditions, resulting in low PDI and high yield in relatively short reaction times.45 For instance, during the growth of polymeric precursors, ATRP was allowed to occur until a conversion of 50% was achieved, at which point the reaction was terminated. This technique reduces the presence of dead polymers in the final product, thus enhancing the purity of chain end-functionalized polymers. Also, to overcome the kinetic barriers associated in “click” reactions with long polymer arms, the core (in this case, tripropargylamine) was added with a syringe pump so as to maximize coupling between the core and the polymer. To this end, monosubstitution of one polymer onto the tripropargylamine core was achieved by adding the core in excess to a solution of the first polymer. Afterwards, a solution of this polymer was added with a syringe pump to a heated solution of the second polymer, affording AB2-type miktoarm stars with a variety of arms (PS and PtBA, PS and PMA, PMA and PtBA, etc.). These conditions yielded a variety of miktoarm stars which all featured high yield in short time (usually above 80% in 5 hours), along with excellent PDI (from 1.03 to 1.07). Further optimization of the “click” coupling reaction was achieved by using the same technique, i.e., feeding tripropargylamine through a syringe pump to a heated polymer solution to create a 3-arm homoarm star polymer. Optimization of both the reaction time and overall yield revealed that addition of the core with a syringe pump yield stars with high yield and much shorter reaction times. Also, the overall yield increased by 10% when unpurified azide-terminated polymer arms from the functionalization of the chain-end of the polymer from a bromide atom to an azide were coupled to the core. These efforts to optimize the “click” coupling reaction to produce a miktoarm star clearly paid off, as some of the best PDI and yields attained from the coupling method were reported in this study.
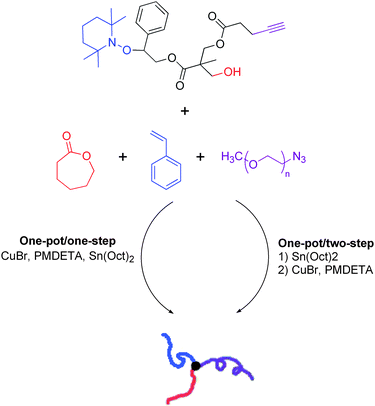
Liu and coworkers combined the “click” reaction between an azide and an alkyne with ROP and ATRP to synthesize an ABC miktoarm polymer composed of PCL, poly((2-dimethylamino)ethyl methacrylate) (PDMA) and either PS or PEG in a one-pot fashion.46 The trifunctional core, ε-caprolactone monomer, 2-(dimethylamino)ethyl acrylate monomer, and an azide-terminated polymer (PS or PEG) were mixed together with catalysts tin(II) 2-ethylhexanoate (Sn(Oct)2), copper(I) bromide (CuBr), and N,N,N′,N′,N″-pentamethyldiethylenetriamine (CuBr/PMDETA) and stirred at 80 °C for 12 hours. Sn(Oct)2 works asa catalyst for ROP while CuBr/PMDETA doubles as a catalyst for both ATRP and the “click” reaction. Regardless of whether PS or PEG was clicked to the core, relatively low PDIs of 1.18 and 1.20, respectively, were attained in a miktoarm star with an approximate Mn of 18 kg mol−1 (both determined by GPC). Similarly, the group of Tunca has created an ABC miktoarm polymer in a one-pot fashion by combining NMP (instead of ATRP), ROP, and the same “click” coupling reaction.47 NMP was used to grow PS, ROP for PCL, and either azide-functionalized PtBA, PMMA, or PEG were clicked to the core to yield the final star (Scheme 9). They varied the technique to make the full miktoarm-star in two ways in an attempt to optimize the synthesis. The NMP and ROP reactions were simultaneously conducted prior to addition of azide-terminated polymer (one-pot/two-step); or the NMP, ROP, and “click” reaction were all allowed to proceed simultaneously (one-pot/one-step). In making miktoarm stars with Mn ranging from 13.5–16.5 kg mol−1, a lower PDI was found from two different miktoarm stars made by the one-pot/two-step technique (1.10 and 1.03) as compared to the one-pot/one-step technique (1.29 and 1.21) (Mn and GPC were obtained from triple detection GPC). Compared to the work of Liu and coworkers, longer reaction time and higher temperatures were used to obtain miktoarm stars. It is evident from these two studies that a variety of conditions and polymers can be used to create a library of miktoarm stars in a one-pot fashion, as long as a well-defined multifunctional core is synthesized. It should be noted that any impurities that were obtained in these reactions, such as an unreacted PS-b-PCL block copolymer, were easily removed by filtration and precipitation. One notable study combined the azide–alkyne “click” coupling reaction with an atom transfer nitroxide radical coupling (ATNRC) reaction to create an ABC miktoarm star polymer in a one-pot synthesis.48 Unlike the previous two examples, this study demonstrated the synthesis of a miktoarm polymer by combining three end-functionalized polymers (rather than a mixture of polymers and monomers) in a one-pot fashion.
 | ||
| Scheme 9 Synthesis of an ABC miktoarm star by one of two methods in a one-pot fashion. | ||
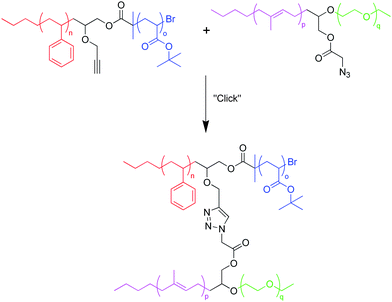
The studies mentioned for the coupling method so far have been used to make relatively simple miktoarm stars—AB2 and ABC—which could also be made using a variety of other techniques, such as the “core-first” method or through the use of chlorosilane compounds. While many synthetic benefits from the coupling method have become evident relative to these other techniques, including a wider variety of monomers, it is important to note that any useful synthetic protocol for the synthesis of miktoarm stars must allow increasingly complex structures to be formed. In one example, ABCD miktoarm star polymers (rarely found in the literature due to stringent reaction conditions) were made by simply coupling two diblock copolymers together through azide–alkyne “click” chemistry (Scheme 10).49 One of the two diblock copolymers was made starting from polystyrene synthesized by living anionic polymerization. Capping the chain end with ethoxyethyl glycidyl ether yielded a polymer with a chain end-functionality that was modified through a series of steps to yield a bifunctional polymer containing an alkyne and 2-bromoisobutyl bromide. Subsequent ATRP with the monomer tert-butyl acrylate yielded the diblock copolymer PS-b-PtBA with an alkyne at the junction. The second diblock copolymer was synthesized starting from the living anionic polymerization of a polymer, in this case PI, and capping the living end with ethoxyethyl glycidyl ether. Ethylene oxide was directly polymerized from this macroinitiator and end-capped using bromoethane to yield a diblock copolymer. Finally, to prepare a diblock copolymer PI-b-PEO with an azide at the junction point, the ethoxyethyl group at the center was converted to an alcohol, whereupon subsequent introduction of a bromide atom and its conversion to an azide yielded the desired diblock copolymer. Finally, these two diblock copolymers were coupled with an azide–alkyne “click” reaction. An excess of one of the diblocks was deliberately used (∼1.5 : 1) to ensure the complete removal of one of the two diblock copolymers in the final product, leaving behind both the 4-arm miktoarm polymer and a single type of unreacted diblock copolymer. The different solubilities of these two types of polymers in water were taken advantage of during the separation of these two polymers, yielding a purified ABCD-type miktoarm star polymer in approximately 70% yield. Seven different polymers with differing molecular weights were obtained with approximate PDI varying from 1.19–1.26 and Mn (determined from 1H NMR) from roughly 12–28 kg mol−1. The same strategy of coupling two block copolymers together through “click chemistry” to create the final miktoarm star was employed to yield a unique H-shaped polymer composed of five different arms (PS, PCL, PtBA, PEG, and PMMA) in moderately high yield.50 This was achieved by conjugating an end-functionalized polymer arm on an ABC-type miktoarm star polymer with a diblock copolymer functionalized at the junction point.
 | ||
| Scheme 10 Synthesis of an ABCD miktoarm star polymer by “click” coupling between two diblock copolymers. | ||
Though most examples of miktoarm polymer synthesis in the literature employ a coupling strategy by utilizing the Cu(I)-catalyzed Huisgen cycloaddition between an azide and an alkyne, there are other reactions that are available to achieve both efficient coupling and a facile synthetic route.51 Other popular variants of the “click” reaction that are widespread in macromolecular synthesis include thiol–ene coupling reactions and Diels–Alder reaction, yet only the Diels–Alder reaction has been utilized in the synthesis of miktoarm polymers. Altintas et al. coupled PCL functionalized with anthracene to PtBA functionalized with maleimide in a Diels–Alder reaction to create a block copolymer, so that subsequent NMP of PS followed by free radical photopolymerization of PMMA from the diblock copolymer junction yielded the final ABCD miktoarm star.52 The Diels–Alder reaction has also been used to make ABC miktoarm polymers.53 Though coupling reactions outside of the classification of “click” reactions are rather sparse, one notable example arose recently where an A2B2 miktoarm polymer was made by coupling two diblock copolymers together at their junction points in an alkyne–alkyne homocoupling reaction.54 Yields and polydispersity were comparable to those already described by the Huisgen “click” reaction.
Self-assembly and applications
Changes in morphology from varying polymer arm length
As discussed above, many methods for the synthesis of miktoarm polymers have emerged in the past decade. The discovery of these reliable synthetic protocols for a variety of miktoarm polymers has undoubtedly simplified our ability to pursue studies of their self-assembly and applications. This interest in miktoarm star polymers stems from the combination of virtually any type and number of polymer arms into a single unique architecture. The widespread research and success in applications of linear block copolymers55 have increased the desire to understand what can happen when the same set of polymer arms are constructed as a star. In observing the general arrangement of core–shell–corona micelles formed from linear triblock copolymers, Timothy Lodge best expressed the source of interest in understanding ABC-type miktoarm polymers: “the mandatory convergence of the three immiscible blocks at one common junction suppresses the formation of concentric structures and leads to an array of new morphologies with compartmentalized micellar cores.”56 With this in mind, the self-assembly and potential applications of any type of miktoarm star (ABC, A2BC, ABCDE, and so on) ought to be worth examining.One of the landmark studies in the self-assembly of miktoarm polymers demonstrated the changing morphologies in aqueous solution which can be attained as the sizes of various arms are adjusted.57 A series of ABC miktoarm polymers were made by a variant of the “core-first” technique using living anionic polymerization methods, whose arms were composed of poly(ethylene oxide) (PEO), polyethylethylene (PEE), and poly(perfluoropropylene oxide) (PFPO) (i.e. one hydrophilic PEO arm and two immiscible hydrophobic PEE and PFPO arms). Many unique morphologies were observed for the first time by cryo-TEM, and a reduction of the size of PEO arms led to transitions from so-called “hamburger” micelles to a mixture of hamburger micelles and worm-like structures in aqueous solution. This reduced size of the hydrophilic PEO arm was believed to have led to the transition from hamburger micelles to worm-like structures, as the short hydrophilic arms of many micelles were believed to group together and create a shared PEO corona so as to best protect the larger hydrophobic inner domains (Fig. 3). Increases in the size of hydrophobic PEE and hydrophobic PFPO further led to a preponderance of worm-like micelles and a marked reduction of individual micelles, an observation which corroborated their overall theory of self-assembly of these systems. They postulated that, within the micellar core of the observed hamburger micelles, PEE segments provided a layer which shielded any interaction between PEO and PFPO blocks.
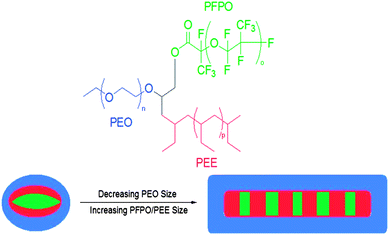 | ||
| Fig. 3 The transition in morphology in aqueous solution of an ABC-type miktoarm star, from “hamburger” micelles (left) to segmented worms (right), as hydrophobicity of the polymer increases. The colors in the picture of the self-assembled structures (bottom) correspond to the polymer's structure (above). | ||
This work was extended to further understand various morphologies that can be attained by simply varying the length of the two hydrophobic arms, revealing a vast variety of morphologies that were attained in aqueous solution.56 A series of miktoarm polymers where the length of PEO was systematically decreased revealed a transition from hamburger micelles to a mixture of worm-like micelles with varying lengths. Further reduction of PEO size past a distinct volume fraction resulted in a mixture of many morphologies, including segmented ribbons, Y-junctions, network micelles, segmented bilayers, and toroids, and finally to a mixture of large bilayer sheets and vesicles. This general transition (vesicles to worms to spheres) had been documented for diblock copolymers.58 A second series of polymers featured variations in the size of PFPO. As the size of this arm increased, a transition in morphology was observed, from segmented worms to a mixture of worms and Y-junctions, and eventually into a wide variety of morphologies including segmented worms, raspberry-like micelles, and multicompartmentalized worms. In these series of miktoarm polymers, the transition from one set of morphologies to another was understood as a result of the changes in polymer arm length in a series of miktoarm polymers. For instance, most of the changes described occurred below a certain volume fraction of PEO, yet above this volume fraction, however, hamburger micelles were consistently observed. Once again, the changes in morphology were understood in terms of the mutual incompatibility of the three arms, as well as the protection of segregated hydrophobic domains by hydrophilic coronas. Studies which seek to understand the relationship between arm size and morphology are important for understanding structure–property relationships for miktoarm star polymers. Increasing research in recent years is being done to this end for various types of ABC miktoarm star polymers.59
pH-induced changes in morphology
While variations in polymer arm size can lead to an array of new morphologies, the use of responsive polymer arms can lead to dynamic changes within a single miktoarm star, and therefore offer enormous potential in applications of these polymers.60 For instance, in a “core-first” fashion with a linear PEG macroinitiator, Armes and coworkers synthesized a variety of AB2 miktoarm stars with various pH-responsive methacrylic monomers by ATRP, such as poly(2-(diethylamino)ethyl methacrylate) (PDEA).61 AB2 stars of PEG(PDEA)2 were fully dissolved in acidic solution, but once the pH rose above 7.3, the PDEA polymers became increasingly deprotonated and hydrophobic, resulting in the formation of micelles whose properties differed from their linear counterparts. Other similar polymers in their library of AB2 stars also demonstrated similar pH-dependent micellization and properties.Jérôme and coworkers built an ABC miktoarm star composed of PEO, PCL, and pH-responsive P2VP (poly(2-vinyl pyridine)).62 Micelles were made in acidic solution, resulting in a micelle with a core composed of PCL and a mixed corona composed of PEO and protonated P2VP. Addition of NaOH to the micelle solution resulted in deprotonation of the P2VP block. Because neutral P2VP could no longer favorably interact with the surrounding solution, the P2VP block in basic conditions was believed to have collapsed from the corona into the core to form a micelle with a corona composed of only PEG (and a core composed of PCL and P2VP) (Fig. 4). This transition from mixed corona micelles to mixed core micelles was accompanied by a marked decrease in micelle size (also referred to as hydrodynamic diameter, or DH) as observed by dynamic light scattering (DLS) studies and transmission electron microscopy (TEM). Two general trends were observed by DLS from a study of a series of these miktoarm polymeric micelles. First, in a micelle with neutral P2VP, the size of the micelle increased with increasing PCL length. Second, in acidic conditions, the size of the micelle increased with increasing P2VP length. Here, just two general types of micelles were observed. The synthesis of a miktoarm star with two pH-responsive polymer arms has been accomplished by Liu and coworkers to demonstrate more complex changes and morphologies that can be achieved with simple pH changes in miktoarm stars. Aqueous micelles from ABC miktoarm polymer composed of PEG, PMMA, and poly(2-(diethylamino)ethyl methacrylate) (PDEA) (which was obtained from PtBA by hydrolysis with trifluoroacetic acid) were prepared.63 At pH 10, PMMA was fully ionized while PDEA was fully neutral, so that a micelle was formed with a mixed corona of PEG and PMMA and a core of PDEA. At pH 6, PEG formed the corona and PMMA and PDEA formed a mixed hydrophobic core due to charge compensation between two partially charged polymer arms. At pH 2, PMMA was fully neutral while PDEA was fully protonated so as to yield a core composed of PMMA and a mixed corona of PEG and PDEA.
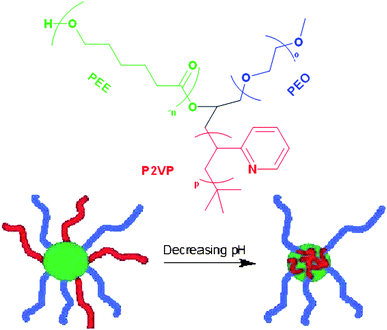 | ||
| Fig. 4 Depiction of the change in morphology in aqueous solution of an ABC-type miktoarm polymer (above), where the P2VP arm (red) shifts from the core to the corona with decreasing pH in aqueous media. | ||
With intended applications in biological systems, an AB2-type miktoarm polymer composed of one poly(ε-benzyloxycarbonyl-L-lysine) (PZLL) arm and two poly(γ-benzyl-L-glutamate) (PBLG) arms was synthesized with Mn of 26.7 kg mol−1 and PDI of 1.13.64 Once this biologically relevant polymer was synthesized via a combination of ROP and “click” coupling, hydrolysis with trifluoroacetic acid resulted in deprotection of the peptide monomers, yielding a miktoarm star with one PLL (poly(L-lysine)) and two PLGA (poly(L-glutamate)) arms. The interest in this polymer lies in the water solubility of its component arms. PLL is hydrophilic in acidic conditions while PLGA is hydrophilic in basic conditions. The solubility of the miktoarm polymer in aqueous solution at varying pH's reflected the characteristic hydrophilicity of each arms outside the pH range of 4.6–6.2. Within this range, however, macroscopic phase separation occurred due to the insolubility of both arms in their surrounding environment. These solubility results were confirmed by 1H NMR, where the hydrophilic polymer is not in a restricted environment and thus exhibits its characteristic spectral peak. A very slight increase in DH was observed in going from a PLL corona micelle to a PLGA corona micelle. Previous studies of the linear counterparts of these polymers indicated the formation of vesicles, yet these miktoarm stars yielded spherical micelles, thereby demonstrating the unique properties that can be attained from miktoarm stars as a result of a simple change in the chain architecture.
Electrolysis-induced micellization
There are many other ways to induce changes in the morphology of a self-assembled solution of miktoarm stars besides pH changes. A fascinating study by Plamper et al. allowed the manipulation of micelles composed of miktoarm polymers with one longer PEG arm and 5 shorter arms of poly[{2-(methacryloyloxy)ethyl}trimethylammonium chloride] (PMOTAC) (Fig. 5). PMOTAC is a polyelectrolyte polymer that reacts in the presence of certain counterions.65 This is the only example of such manipulation of miktoarm polymer micelles in the literature. At high enough concentration of the counterion in aqueous micellar solution, PMOTAC was expected to become hydrophobic, thus driving a phase transition from unimers to micelles. Indeed, micellization of this miktoarm polymer was induced in an aqueous solution of this polymer with a specific molar ratio of hexacyanoferrate(III)/hexacyanoferrate(II) counterions at a specific concentration. It is important to note that inversing the molar ratio of the counterion at the same concentration that induced micellization could lead to the prevalence of unimers in solution. Electrolysis was used to reversibly oxidize (complete in 35 min) and reduce (complete in 20 min) the counterions in solution which in turn resulted in micelles and unimers in solution, respectively. Cryo-TEM studies of this polymer indicated that even the morphology in aqueous solution could be tuned by electrochemical stimuli due to the different morphological transition states that occur as the miktoarm polymer transitions from unimer to the final state where vesicles were observed.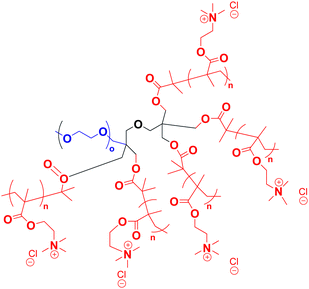 | ||
| Fig. 5 Structure of the miktoarm polymer, with PEG in blue and PMOTAC in red. | ||
Thermoresponsive micelles
Thermosensitive polymers are currently widespread in polymer science because of their suggested industrial applications. To incorporate thermosensitivity in miktoarm polymers, Liu's group synthesized an ABC miktoarm polymer composed of PS, PCL, and poly(N-isopropylacrylamide) (PNIPAM) (Fig. 6).66 PNIPAM polymers were expected to remain soluble in water below 32 °C (the lower critical solution temperature, or LCST) and insoluble above this temperature. These miktoarm polymers formed aqueous micelles with hydrophilic PNIPAM coronas and mixed hydrophobic PCL and PS cores at room temperature. An ∼10 nm size decrease of these micelles was observed by DLS upon heating of the aqueous micelle solution to 45 °C, whereupon the PNIPAM coronas collapse upon themselves due to their decreasing solubility with the surrounding solvent. This work was further expanded by combining a different hydrophobic polymer poly(tert-butyl methacrylate) (PtBMA) with thermoresponsive PNIPAM and hydrophilic PEG to form an ABC miktoarm star.67 Compared to the previous study, similar qualitative results were obtained here as well. At room temperature micelles formed in aqueous solution with PtBMA cores and mixed PNIPAM and PEG coronas with an intensity–average hydrodynamic radius 〈Rh〉 of 30 nm (from DLS). As the temperature of the aqueous micellar solution was gradually increased, a phase transition was observed between 40 and 47 °C where 〈Rh〉 increased to the final steady-state value of 110 nm. This change in size is likely accompanied with the formation of mixed PtBMA/PNIPAM cores and PEG coronas, as a result of PNIPAM's temperature-induced insolubility in water as the system rises above the LCST. One very interesting observation was found from this study: the LCST transition for PNIPAM was higher than that of free PNIPAM linear chains in solution. This transition was thought to occur as a result of the interaction between the two polymers composing of the PEG/PNIPAM corona during the phase transition, where PEG is hydrophilic and PNIPAM is gradually becoming insoluble in the surrounding solvent.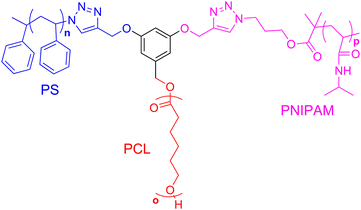 | ||
| Fig. 6 Structure of ABC miktoarm polymer with PCL (red), PS (blue), and PNIPAM (pink). | ||
Light-responsive micelles
Relatively few studies of light-responsive miktoarm polymers have been conducted. Tunca and coworkers made an A2B2-type miktoarm star composed of PMMA and PS (made by CRP methods) and a core functionalized with an azobenzene moiety.68 With the polymer dissolved in chloroform, UV light with λ < 350 nm was found to incur a photochemical isomerization from trans-azobenzene to cis-azobenzene, though this transition was slow (∼7 h). This isomerization was accompanied by a reduction in hydrodynamic volume of the dissolved polymer. Complete back isomerization of the polymer from cis-azobenzene to trans-azobenzene took approximately 2 days to occur in the dark.The only other reported example of light-responsive micelles arose from Plamper et al., where a miktoarm star composed of two PEO arms and 3 or 4 poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) arms (A2B3 and A2B4) resulted in so-called “confused micelles” in aqueous solution.69 PDMAEMA is an extremely unique polymer which has both an LCST and an upper critical solution temperature (UCST), which imparts unique hydrophilic properties in response to temperature and light (pH-dependent properties were not studied). As a result, their miktoarm stars formed self-assembled structures both above and below the LCST and UCST respectively, while unimers were found in aqueous solution between these two critical temperatures (multivalent counterions were necessary in order to access the UCST). Regardless of temperature, the micellization of this polymer was characterized by a PEG corona and a PDMAEMA core. Furthermore, morphology was found to be dependent on the number of PDMAEMA arms—spherical micelles were formed from A2B4 polymers while vesicles were formed from A2B3 polymers. Finally, because of the unique interaction between PDMAEMA and hexacyanocobaltate(III) below the UCST temperature, UV light induced the irreversible disassembly of micelles into unimers in solution. These two examples clearly illustrate the unique self-assembly behavior that can be attained by incorporation of light-responsive moieties or polymers within the overall structure of the miktoarm star.
Solvent-induced changes in micellization
One of the only studies in the literature of a micelle transition induced by solvent mixtures for miktoarm stars was reported by Lodge et al. for an ABC-type miktoarm star polymer with PEO, PEE, and PFPO arms (structure shown in Fig. 3).70 As the hydrophilic PEO arm length decreased in size, they had previously observed a general transition from micelles to segmented worms to vesicles, and it was thought that a gradual change in composition of a solvent mixture THF/H2O could induce this transition within a single aqueous micellar solution. Tetrahydrofuran (THF) is a solvent selective for PEE and PEO blocks, while water is only selective for PEO. The different miktoarm polymers they examined in pure water adopted morphologies resembling multicompartment disks. In increasing THF/H2O mixtures, a transition to smaller disks, then a mixture of worms and spherical micelles, and finally an oblate ellipsoid-shaped micelle was observed. This oblate micelle was also observed by cryo-TEM of miktoarm polymers in pure THF. To assist their studies of solvent mixture-inducing transitions in morphology, they examined self-assembled linear diblock copolymers of PEE and PEO in aqueous media as they added increasing amounts of THF, finding two distinct transitions in morphology as a transition from spherical micelles (in pure aq. solution) to worms/vesicles and eventually to unimers in solution was observed. The specific solvent mixture that led to unimers in block copolymer solution (60 wt% THF) also led to a similar transition with miktoarm polymers, where the PEE arm seemed to shift completely from the core to the corona based on 1H NMR spectroscopy, as this arm became fully solvated by the surrounding solvent mixture, thus forming oblate ellipsoid-shaped micelles. The correlation between linear diblock copolymer and miktoarm star polymer phase transition is fascinating, as the solvent mixture which induces a micellar phase transition in miktoarm polymer solutions can be predicted from its linear diblock counterpart. This correlation of phase transition between two polymers of different architectures suggests how self-assembly behavior of copolymer solutions is primarily dictated by the identity of composite polymer arms, rather than the architecture. At the same time, however, two completely different morphologies were observed in the linear and miktoarm star solutions past the phase transition point, simply because of the polymer chain architecture.Schizophrenic micelles
So-called “schizophrenic” micelles were characterized by a complete inversion of the polymers composing the core and corona in response to external stimuli including temperature, pH, ionic strength, etc. The effect of stimuli on schizophrenic micelles is different from the aforementioned examples thus far, where a miktoarm star was composed of one polymer arm that displays responsive hydrophilicity in response to external stimuli and a second hydrophilic polymer arm which was unresponsive. Schizophrenic micelles, on the other hand, undergo complete inversion in the composition of the micellar corona and core in response to stimuli, so that no polymer arm remains hydrophilic throughout micellization. Examples of these polymers have become widely investigated in linear block copolymers.71 A few publications have emerged in recent years which report schizophrenic micellization in miktoarm polymers. The first example of schizophrenic micelles in self-assembled miktoarm polymers was found by Armes and coworkers.72 They prepared an AB2 miktoarm polymer composed of two rarely studied arms: one Jeffamine arm (a statistical copolymer of ethylene oxide and propylene oxide) and two poly(sulfobetaine methacrylate) (PSBMA) arms. PSBMA homopolymer is known to be hydrophobic at temperatures below the UCST. Consequently, at low temperatures of aqueous solution of this AB2 miktoarm star, micelles were formed with PSBMA core and Jeffamine coronas (Fig. 7). Upon increasing the temperature above the UCST of PSBMA, complete dissolution of these polymer arms resulted in unimers in solution as both arms are dissolved. As the temperature was further increased, the critical micelle temperature was reached at which point micelles were again formed, but this time with Jeffamine cores and PSBMA coronas.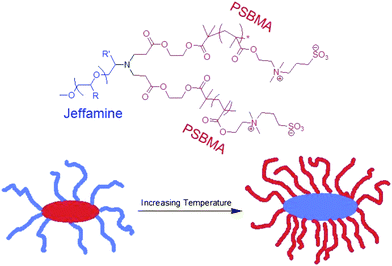 | ||
| Fig. 7 Schizophrenic micelle where the core and corona are inverted upon an increase in temperature in aqueous solution. The color of the polymers in the molecular structure (above) corresponds to the location of the polymer within the micelle (below); R = H or CH3. | ||
Another example of schizophrenic micelles arose from the synthesis of zwitterionic AB2 miktoarm polymers, composed of one PDEA arm and two poly(succinyloxyethyl methacrylate) (PSEMA) arms.73 Schizophrenic micelles were formed by the response of these two polymers to acidic and basic solutions. At low/high pH, PDEA is charged/hydrophobic, while PSEMA is hydrophobic/charged. Therefore, at low pH, micelles were formed with PSEMA cores and PDEA coronas, and vice versa for high pH. These results were confirmed by 1H NMR in various deuterated solvents. In between the pH at which these polymers become soluble, precipitation occurred. Subsequent adjustment of solution pH to either acidic or basic conditions induced formation of self-assembled structures as one of the two polymer arms became soluble in the surrounding solvent. DLS did not indicate the formation of micelles at acidic pH, but instead suggested the formation of large undefined aggregates. Meanwhile, DLS from basic pH indicated the formation of well-defined micelles. In this study, pH was the only variable that needed to be controlled in order to regulate the formation of micelles. The combination of polymers that respond to two different stimuli can also lead to the formation of schizophrenic micelles. Indeed, such micelles were achieved with AB4-type miktoarm star polymers with one PNIPAM arm and four PDEA arms.74 The choice of these two polymer arms with response to different stimuli (temperature and pH) required specific control of two different variables in order to achieve the formation of schizophrenic micelles instead of unimers or precipitates. The micelles formed with PDEA cores were almost twice as large as those formed with PNIPAM cores, as measured by DLS.
Small molecule storage and release
In one of the best examples of miktoarm polymer applications, Lodge and coworkers exploited the unique architecture and resultant self-assembly of miktoarm polymers, as they demonstrated the potential biological applications of this unique class of polymers. Using the ABC-type star composed of PEE, PFPO, and PEO, they achieved the simultaneous and segregated storage of two immiscible types of dyes within the multicompartment core of aqueous micelles (Fig. 8).75 Because the two hydrophobic dye molecules, pyrene and 1-naphthyl perfluoroheptanyl ketone (or NFH), were found to selectively associate with PEE and PFPO respectively in a block copolymer mixture of PEE/PEO and PFPO/PEO, it was believed that an aqueous micelle would similarly sequester these two dyes into separate compartments. UV-Vis absorption spectrophotometry helped to confirm this theory. Thus, each of the two linear counterparts of the miktoarm star helped to successfully predict the uptake of the two small molecules in aqueous miktoarm polymeric micelles.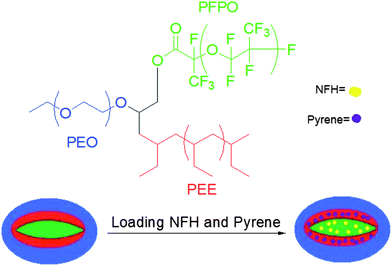 | ||
| Fig. 8 Loading of two immiscible dyes into separate domains in a multicompartment micelle in aqueous solution. | ||
To study the encapsulation and release of paclitaxel from AB2-type miktoarm stars, Nederberg et al. constructed stars composed of one PEG arm and either two poly(D-lactide) (PDLA) arms, two poly(L-lactide) (PLLA) arms, or a mixture of both.76 Both PEG–PLLA–PDLA and a blend of PEG–PLLA–PLLA and PEG–PDLA–PDLA were found to exhibit a lower critical micelle concentration than PEG–PLLA–PLLA and PEG–PDLA–PDLA. Loading these micelles with paclitaxel induced dramatic change in size of the micelles, according to DLS, of at least 100 nm. However, the loaded micelles maintained their narrow polydispersity. The miktoarm polymer blend allowed paclitaxel loading of 11.6 wt%, while PEG–PDLA–PLLA showed loading of approximately 10 wt%. Furthermore, these miktoarm polymers demonstrated a sustained release of paclitaxel without any initial burst under simulated biological conditions, thus proving that miktoarm polymers have great potential as drug delivery vehicles. PEG–PLLA–PLLA miktoarm star polymers were loaded with doxorubicin hydrochloride in a separate study by a different group.77 With smaller overall Mn, excellent encapsulation of the small molecule (within polymerosome structures) was also achieved. Release of the drug was accomplished in similar conditions as the other study, where an initial burst for 2 hours released 30% of the drug, followed by steady and prolonged release of the drug. The correlation between these two studies is encouraging as the success of loading and release of drug molecules is reproducible. However, these results demonstrate how more work needs to be done with small molecule loading and release in new types of miktoarm star polymers in order to establish their benefits and shortcomings relative to their linear counterparts. Although there have been few published studies which focus on miktoarm polymers in terms of drug delivery,78 yet these results demonstrate the promise that lies ahead.
Conclusions
The field of miktoarm polymers has grown considerably in the past decade and the future looks very promising for these star-shaped macromolecules. Many methods have emerged to synthesize miktoarm polymers, each with its own set of benefits and disadvantages. For instance, the “in–out” and “arm-first” techniques lack the ability to synthesize a structure with a defined number of arms, but it is a relatively quick and easy way to synthesize miktoarm star polymers. Living anionic polymerization techniques are still used to combine living polymers with chlorosilane linking agents or divinyl compounds to synthesize stars. However, the benefits of controlled radical polymerization have become unavoidably evident, such as the wide variety of monomers that can be synthesized under relatively benign reaction conditions, thereby yielding polymers which easily lend themselves to postpolymerization functionalization. Not only has CRP found increasingly widespread use in miktoarm polymer synthesis in recent years, but also has “click” chemistry due to its efficiency, orthogonality to other functional moieties present, simple workup, and compatibility with CRP techniques. Of course, numerous elements of different synthetic strategies can be combined to create one miktoarm star polymer. The choice of a specific synthetic strategy depends heavily on the type of miktoarm polymer desired. Nonetheless, careful design of synthetic protocols has been highly beneficial, as increasingly complicated miktoarm polymers are easier to build now than they were ten or fifteen years ago. For instance, there are numerous examples available of ABCD-type miktoarm polymers being made in an efficient manner.A growing understanding of the behavior of miktoarm polymers in aqueous solution and the relation between polymer arm length and overall morphology is becoming increasingly important. Lodge's group has done excellent studies to lead the way in developing an understanding of this relationship, but it needs to be established for more types of miktoarm stars—ABC stars with chemically different arms, or AB2 and ABCD stars, for instance. Many polymer arms have been used for miktoarm polymer synthesis. It is important now to understand how they influence the morphology of the assembled structure in solution. A study of the response of morphology to external stimuli has just begun, and studies related to pH-response, temperature, light, redox chemistry, and solvent have corroborated with predicted behavior of miktoarm stars in solution, and have shown fascinating behavior. However, thorough studies of these stimuli have not yet been conducted. Part of this can be attributed to the fact that much of the work with stimuli-responsive behavior has come about in the past five years as the synthesis of miktoarm polymers has become easier and better understood. Indeed, interesting results have been achieved which most certainly precludes further studies and applications.
One of the most interesting areas of research in macromolecules is in small molecule encapsulation and delivery. Multicompartment micelles offer the potential to uniquely sequester small molecules. Furthermore, the unique assembly of unimers into micellar aggregates offers different properties as compared to their linear counterparts (such as lower critical micelle concentration in water or well-defined structures), which renders miktoarm polymers as attractive candidates for further studies in drug delivery and biological applications. Since preliminary work in this field has been done with AB2 and ABC systems, extension of these studies is important to pursue and further understand these promising systems, and also perhaps include other types of miktoarm stars such as an ABCDE-type star as drug delivery vehicles.
Acknowledgements
We thank NSERC (Discovery and Collaborative Research and Development Grants) of Canada, Polymer Source, and Center for Self-Assembled Chemical Structures (FQRNT, Quebec Canada) for financial assistance.Notes and references
- C. K. Ober, S. Z. D. Cheng, P. T. Hammond, M. Muthukumar, E. Reichmanis, K. L. Wooley and T. P. Lodge, Macromolecules, 2009, 42, 465 CrossRef CAS.
- (a) R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198 CrossRef CAS; (b) E. M. Martín del Valle, M. A. Galán and R. G. Carbonell, Ind. Eng. Chem. Res., 2009, 48, 2475 CrossRef.
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581 CrossRef CAS.
- D. Greszta and K. Matyjaszewski, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1996, 37, 569 CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437 CrossRef CAS.
- C. J. Hawker, Angew. Chem., Int. Ed. Engl., 1995, 34, 1456 CAS.
- R. Hourani and A. K. Kakkar, Macromol. Rapid Commun., 2010 DOI:10.1002/marc.200900712.
- K. Matyjaszewski, P. J. Miller, J. Pyun, G. Kickelbick and S. Diamanti, Macromolecules, 1999, 32, 6526 CrossRef CAS.
- (a) N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857 CrossRef CAS; (b) N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas and A. Avgeropoulos, Prog. Polym. Sci., 2005, 30, 725 CrossRef CAS; (c) N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS.
- (a) Y. Zhu, R. K. Yu Li and W. Jiang, Chem. Phys., 2006, 327, 137 CrossRef CAS; (b) W. Kong, B. Li, Q. Jin, D. Ding and A.-C. Shi, J. Am. Chem. Soc., 2009, 131, 8503 CrossRef CAS; (c) X. He, L. Huang, H. Liang and C. Pan, J. Chem. Phys., 2003, 118, 9861 CrossRef CAS; (d) C.-I. Huang and H.-T. Yu, Polymer, 2007, 48, 4537 CrossRef CAS; (e) D. Kou, Y. Jiang and H. Liang, J. Phys. Chem. B, 2006, 110, 23557 CrossRef CAS; (f) T. Lu, X. He and H. Liang, J. Chem. Phys., 2004, 121, 9702 CrossRef CAS; (g) E. B. Zhulina and O. V. Borisov, Macromolecules, 2008, 41, 5934 CrossRef CAS; (h) R. Wang and T. Xu, Polymer, 2007, 48, 4601 CrossRef CAS.
- D. Cho, S. Park, T. Chang, A. Avgeropoulos and N. Hadjichristidis, Eur. Polym. J., 2003, 39, 2155 CrossRef CAS.
- H. Iatrou and N. Hadjichristidis, Macromolecules, 1993, 26, 2479 CrossRef CAS.
- A. Mavroudis and N. Hadjichristidis, Macromolecules, 2006, 39, 535 CrossRef CAS.
- (a) P. Fragouli, H. Iatrou, N. Hadjichristidis, T. Sakurai, Y. Matsunaga and A. Hirao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6587 CrossRef CAS; (b) P. Fragouli, H. Iatrou, N. Hadjichristidis, T. Sakurai and A. Hirao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 614 CrossRef CAS.
- Y. Zhao, T. Higashihara, K. Sugiyama and A. Hirao, J. Am. Chem. Soc., 2005, 127, 14158 CrossRef CAS.
- A. Hirao, T. Higashihara and K. Inoue, Macromolecules, 2008, 41, 3579 CrossRef CAS.
- Y. Zhao, T. Higashihara, K. Sugiyama and A. Hirao, Macromolecules, 2007, 40, 228 CrossRef CAS.
- T. Higashihara, T. Sakurai and A. Hirao, Macromolecules, 2009, 42, 6006 CrossRef CAS.
- (a) A. Hirao, T. Higashihara, M. Nagura and T. Sakurai, Macromolecules, 2006, 39, 6081 CrossRef CAS; (b) T. Higashihara, M. Nagura, K. Inoue, N. Haraguchi and A. Hirao, Macromolecules, 2005, 38, 4577 CrossRef CAS; (c) A. Hirao, M. Hayashi and T. Higashihara, Macromol. Chem. Phys., 2001, 202, 3165 CrossRef CAS; (d) A. Hirao and T. Higashihara, Macromolecules, 2002, 35, 7238 CrossRef CAS; (e) A. Hirao, K. Kawasaki and T. Higashihara, Macromolecules, 2004, 37, 5179 CrossRef CAS; (f) A. Hirao and Y. Tokuda, Macromolecules, 2003, 36, 6081 CrossRef CAS; (g) T. Higashihara, M. Kitamura, N. Haraguchi, K. Sugiyama, A. Hirao, J.-H. Ahn and J.-S. Lee, Macromolecules, 2003, 36, 6730 CrossRef CAS; (h) K. Sugiyama, K. Inoue, T. Higashihara, H. Hayashi and A. Hirao, React. Funct. Polym., 2009, 69, 480 CrossRef CAS.
- H. R. Krischeldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- E. Rizzardo, J. Chiefari, R. Mayadunne, G. Moad and S. Thang, Macromol. Symp., 2001, 174, 209 CrossRef CAS.
- T. Erdogan, Z. Ozyurek, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2313 CrossRef CAS.
- U. Tunca, Z. Ozyurek, T. Erdogan and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4228 CrossRef CAS.
- (a) P.-J. Shi, Y.-G. Li and C.-Y. Pan, Eur. Polym. J., 2004, 40, 1283 CrossRef CAS; (b) W. Zhang, W. Zhang, J. Zhu, Z. Zhang and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6908 CrossRef CAS; (c) S. Kakwere and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6396 CrossRef; (d) D. Priftis, M. Pitsikalis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5164 CrossRef CAS; (e) L. K. Breland and R. F. Storey, Polymer, 2008, 49, 1154 CrossRef CAS; (f) A. Heise, M. Trollsas, T. Magbitang, J. L. Hedrick, C. W. Frank and R. D. Miller, Macromolecules, 2001, 34, 2798 CrossRef CAS; (g) Y. Miura, K. Yamaoka and A. Mannan, Polymer, 2006, 47, 510 CrossRef CAS; (h) G. Wang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1136 CrossRef CAS; (i) H. Durmaz, S. Aras, G. Hizal and U. Tunca, Des. Monomers Polym., 2005, 8, 203 Search PubMed; (j) Y. Miura, Y. Sakai and K. Yamaoka, Macromol. Chem. Phys., 2005, 206, 504 CrossRef CAS; (k) Y. Shi, Z. Fu, B. Li, L. Zhang, X. Cai and D. Zhang, Eur. Polym. J., 2007, 43, 2612 CrossRef CAS; (l) T. He, D. Li, X. Sheng and B. Zhao, Macromolecules, 2004, 37, 3128 CrossRef CAS; (m) C. Celik, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2542 CrossRef CAS; (n) Y. Yamazaki, N. Ajioka, A. Yokoyama and T. Yokozawa, Macromolecules, 2009, 42, 606 CrossRef CAS; (o) C. Liu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 325 CrossRef CAS.
- C. Gordin, C. Delaite, H. Medlej, D. Josien-Lefebvre, K. Hariri and M. Rusu, Polym. Bull., 2009, 63, 789–801 CrossRef CAS.
- (a) Z. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2004, 37, 8933 CrossRef CAS; (b) Y.-M. Guo, J. Xu and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 437 CrossRef CAS; (c) R. Matmour and Y. Gnanou, J. Am. Chem. Soc., 2008, 130, 1350 CrossRef CAS; (d) G. Wang and J. Huang, Macromol. Rapid Commun., 2007, 28, 298 CrossRef CAS; (e) X.-S. Feng and C.-Y. Pan, Macromolecules, 2002, 35, 4888 CrossRef CAS; (f) X. S. Wang, M. A. Winnik and I. Manners, Macromol. Rapid Commun., 2003, 24, 403 CrossRef CAS; (g) R. Francis, B. Lepoittevin, D. Taton and Y. Gnanou, Macromolecules, 2002, 35, 9001 CrossRef CAS; (h) J. Babin, C. Leroy, S. Lecommandoux, R. Borsali, Y. Gnanou and D. Taton, Chem. Commun., 2005, 1993 RSC.
- J. Rieger, O. Coulembier, P. Dubois, K. V. Bernaerts, F. E. Du Prez, R. Jérôme and C. Jérôme, Macromolecules, 2005, 38, 10650 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 3154 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 11828 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2008, 41, 4250 CrossRef CAS.
- (a) T. Glauser, C. M. Stancik, M. Moller, S. Voytek, A. P. Gast and J. L. Hedrick, Macromolecules, 2002, 35, 5774 CrossRef CAS; (b) J. Du and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2263 CrossRef CAS; (c) J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 9018 CrossRef CAS; (d) J. T. Wiltshire and G. G. Qiao, Macromolecules, 2008, 41, 623 CrossRef CAS; (e) H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 7216 CrossRef CAS.
- J. Du and Y. Chen, Macromolecules, 2004, 37, 3588 CrossRef CAS.
- (a) A. Vora, K. Singh and D. C. Webster, Polymer, 2009, 50, 2768 CrossRef CAS; (b) O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1218 CrossRef CAS; (c) C. N. Urbani, C. A. Bell, M. R. Whittaker and M. J. Monteiro, Macromolecules, 2008, 41, 1057 CrossRef CAS; (d) O. Altintas, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5916 CrossRef CAS; (e) J. Zhu, X. Zhu, E. T. Kang and K. G. Neoh, Polymer, 2007, 48, 6992 CrossRef CAS; (f) G. Deng, D. Ma and Z. Xu, Eur. Polym. J., 2007, 43, 1179 CrossRef CAS; (g) W. Zhang, W. Zhang, Z. Zhang, J. Zhu, Q. Pan and X. Zhu, Polym. Bull., 2009, 63, 467 CrossRef CAS; (h) L. Yang, H. Zhou, G. Shi, Y. Wang and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6641 CrossRef CAS; (i) Y. Y. Yuan, Y.-C. Wang, J.-Z. Du and J. Wang, Macromolecules, 2008, 41, 8620 CrossRef CAS; (j) M. S. Rahman, M. Changez, J.-W. Yoo, C. H. Lee, S. Samal and J.-S. Lee, Macromolecules, 2008, 41, 7029 CrossRef CAS; (k) O. Altintas, G. Hizal and U. Tunca, Des. Monomers Polym., 2009, 12, 83 Search PubMed.
- (a) R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS; (b) C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200 CrossRef CAS; (c) B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef CAS; (d) G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536 RSC.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) G. Franc and A. K. Kakkar, Chem. Commun., 2008, 5267 RSC; (c) V. Ladmiral, T. M. Legge, Y. Zhao and S. Perrier, Macromolecules, 2008, 41, 6728 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699 CrossRef CAS.
- K. Khanna, S. Varshney and A. K. Kakkar, Macromolecules, 2010 Search PubMed , submitted.
- M. R. Whittaker, C. N. Urbani and M. J. Monteiro, J. Am. Chem. Soc., 2006, 128, 11360 CrossRef CAS.
- Y. Zhang, C. Li and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3066 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3588 CrossRef CAS.
- Q. Fu, G. Wang, W. Lin and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 986 CrossRef CAS.
- G. Wang, X. Luo, C. Liu and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2154 CrossRef CAS.
- E. Gungor, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4459 CrossRef CAS.
- C. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, Des. Monomers Polym., 2009, 12, 83 Search PubMed.
- H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 499 CrossRef CAS.
- E. Gungor, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6703 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Langmuir, 2006, 22, 9409 CrossRef CAS.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef CAS.
- (a) J. Bang, S. Jain, Z. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Macromolecules, 2006, 39, 1199 CrossRef CAS; (b) S. Jain and F. S. Bates, Science, 2003, 300, 460 CrossRef CAS.
- (a) Z. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2006, 39, 765 CrossRef CAS; (b) Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245 CrossRef CAS; (c) N. Saito, C. Liu, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2008, 41, 8815 CrossRef CAS; (d) H. Hückstädt, A. Göpfert and V. Abetz, Macromol. Chem. Phys., 2000, 201, 296 CrossRef CAS.
- (a) C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2009, 25, 13718 CrossRef CAS; (b) Y. Zhang, H. Liu, J. Hu, C. Li and S. Liu, Macromol. Rapid Commun., 2009, 30, 941 CrossRef CAS; (c) X. Wang, J. He and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4818 CrossRef CAS.
- Y. Cai, C. Burguiere and S. P. Armes, Chem. Commun., 2004, 802 RSC.
- K. Van Butsele, C. A. Fustin, J. F. Gohy, R. Jérôme and C. Jérôme, Langmuir, 2009, 25, 107 CrossRef CAS.
- H. Liu, C. Li, H. Liu and S. Liu, Langmuir, 2009, 25, 4724 CrossRef CAS.
- J. Rao, Y. Zhang, J. Zhang and S. Liu, Biomacromolecules, 2008, 9, 2586 CrossRef CAS.
- F. A. Plamper, L. Murtomäki, A. Walther, K. Kontturi and H. Tenhu, Macromolecules, 2009, 42, 7254 CrossRef CAS.
- Y. Zhang, H. Liu, H. Dong, C. Li and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1636 CrossRef CAS.
- C. Li, Z. Ge, H. Liu and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4001 CrossRef CAS.
- T. Erdogan, E. Gungor, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1396 CrossRef CAS.
- F. A. Plamper, J. R. McKee, A. Laukkanen, A. Nykänen, A. Walther, J. Ruokolainen, V. Aseyev and H. Tenhu, Soft Matter, 2009, 5, 1812 RSC.
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2008, 24, 12001 CrossRef CAS.
- V. Bütün, S. Liu, J. V. M. Weaver, X. Bories-Azeau, Y. Cai and S. P. Armes, React. Funct. Polym., 2006, 66, 157 CrossRef.
- Y. Cai, Y. Tang and S. P. Armes, Macromolecules, 2004, 37, 9728 CrossRef CAS.
- Y. Cai and S. P. Armes, Macromolecules, 2005, 38, 271 CrossRef CAS.
- Z. Ge, Y. Cai, J. Yin, Z. Zhu, J. Rao and S. Liu, Langmuir, 2007, 23, 1114 CrossRef CAS.
- T. P. Lodge, A. Rasdal, Z. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608 CrossRef CAS.
- F. Nederberg, E. Appel, J. P. K. Tan, S. H. Kim, K. Fukushima, J. Sly, R. D. Miller, R. M. Waymouth, Y. Y. Yang and J. L. Hedrick, Biomacromolecules, 2009, 10, 1460 CrossRef CAS.
- H. Yin, S.-W. Kang and Y. H. Bae, Macromolecules, 2009, 42, 7456 CrossRef CAS.
- (a) T. K. Georgiou, L. Phylactou and C. S. Patrickios, Biomacromolecules, 2006, 7, 3505 CrossRef CAS; (b) T. K. Georgiou, M. Vamvakaki, M. Phylactou, L. Phylactou and C. S. Patrickios, Biomacromolecules, 2005, 6, 2990 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |