Investigation into thiol-(meth)acrylate Michael addition reactions using amine and phosphine catalysts†
Guang-Zhao
Li
ab,
Rajan K.
Randev
a,
Alexander H.
Soeriyadi
c,
Gregory
Rees
a,
Cyrille
Boyer
c,
Zhen
Tong
b,
Thomas P.
Davis
c,
C. Remzi
Becer
a and
David M.
Haddleton
*a
aDepartment of Chemistry University of Warwick, Coventry, Warwickshire CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk; Fax: +44 (0)2476 528267; Tel: +44 (0)2476 523256
bResearch Institute of Materials Science, South China University of Technology, Guangzhou, 510640, China
cCentre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 7th June 2010
Abstract
This work describes a study into thiol–ene based Michael addition reactions. Different catalysts, primary and tertiary amines and phosphines, were investigated for the reaction of a range of thiols with dimers and oligomers of some (meth)acrylates. Primary and tertiary amines are efficient catalysts for the thiol–ene reaction, although these catalysts require several hours to reach high conversion. Moreover, the phosphine catalysts, dimethylphenylphosphine (DMPP) and tris-(2-carboxyethyl)phosphine (TCEP), were investigated in detail. DMPP is an efficacious catalyst yielding complete conversion in few minutes under optimized conditions. Importantly, the concentration of DMPP should be kept at catalytic levels to avoid the formation of by-products, originating from the addition of DMPP to the vinyl group. Furthermore, TCEP is an efficient catalyst for thiol–ene reactions in aqueous media when the pH of the medium is higher than 8.0 since at acidic pH the formation of by-products is observed.
Introduction
Over the last decade, click chemistry has opened new avenues to design and synthesize tailor-made macromolecules for different applications in chemistry as well as for the functionalization of nanoparticles and polymers.1–5 Although, numerous organic reactions have been described in the literature as “click reactions”, the copper catalyzed [3 + 2] Huisgen cycloaddition reaction has been widely employed in conjunction with metal-mediated controlled radical polymerization.6–9 However, the residual amount of copper and the ligand content in the final product give constraints and limit the application of Huisgen cycloaddition, especially for biological applications. This has directed research towards metal-free synthesis techniques.10–12 One of the most attractive and long time known organic reactions, thiol–ene addition, has become popular in polymer chemistry.13–18Thiol–ene addition reactions can proceed by two routes: (i) anti-Markovnikov radical addition and (ii) base or nucleophile catalyzed Michael addition reaction, Scheme 1.19–26 The thiol–ene/yne radical addition mechanism requires an external radical source and energy to generate radicals, which may be azo-initiators or photo-initiators using thermal or UV light energy.27–32 Moreover, it should be noted that thiol compounds may react non-selectively with any type of vinyl bond within the polymer. Despite the few drawbacks of thiol–ene radical addition reactions, it is possible to reach near quantitative conversions in relatively short periods by its high reactivity for low molecular weight compounds, as described recently.33
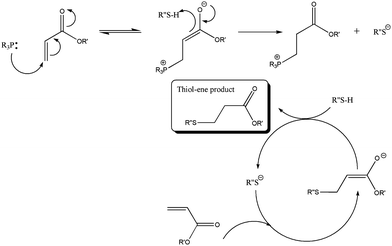 | ||
| Scheme 1 Proposed mechanism for the nucleophile mediated hydrothiolation of an acrylic carbon–carbon bond under phosphine catalysis. | ||
In the case of the Michael addition reaction, this is a facile reaction between nucleophilic species and activated olefins under basic conditions.34 The Michael reaction benefits from mild reaction conditions, minimal by-product formation, high functional group tolerance and high conversions when optimized. Various amines, enolates, thiols and phosphines can react with (meth)acrylates, (meth)acrylamides, maleimides, acrylonitriles and cyanoacrylates, which are Michael donors and Michael acceptors, respectively.35,36 There are a few parameters that should be taken into account when optimizing the synthetic protocol, such as the nucleophile type, solvent and substrate, all of which have been systematically investigated in this present study.
The synthesis of end functional polymers with high fidelity is challenging even though several controlled radical polymerization techniques have been developed.37 Metal mediated radical polymerization techniques have been widely employed to obtain bromide or chloride end-functionalized polymers that can be converted into azide groups to subsequently facilitate Cu-catalyzed azide–alkyne click reactions. Similarly, reversible addition-fragmentation chain transfer (RAFT) polymerization has been used to give thiol terminated polymers following the cleavage of the chain transfer agent.38–42 In general practice, amines have been used in excess to cleave the chain transfer agent from the chain end, however, the bulky structure of the polymer may reduce the cleavage efficiency. Additionally, the free thiol terminal-groups can be oxidized to disulfides. To avoid this side reaction, CAMD has developed a strategy proposing the in situ aminolysis of RAFT end-groups in the presence of pyridyl disulfide to yield a thiol protected polymer.43 In addition, RAFT polymerization requires dedicated chain transfer agents (CTA) designed according to the monomer structure and also optimized reaction conditions such as initiator/CTA ratio, solvent type and reaction temperature.44–51 Nevertheless, thiol terminated polymers have been successfully used in thiol–ene, thiol–yne, thiol–bromo, thiol–isocyanate and thiol–maleimide click reactions.52–60
Alternatively, vinyl terminated polymers have been prepared to react with thiol compounds.61–63 One of the most successful techniques to obtain vinyl-terminated polymers is catalytic chain transfer polymerization (CCTP) that proceeds via a free-radical mechanism in the presence of certain Co(II) catalysts.64–68 In particular, this is an ideal technique to synthesize very short oligomers, i.e. dimer, trimer and tetramer, with very high fidelity. This technique has been generally employed for the preparation of macromonomers and has been used successfully for a wide range of methacrylates. Advantageously, CCTP requires a very low amount of catalyst (in low ppm levels), yielding quasi-pure polymers with a very low amount of residual catalyst.
In this present work, the effect of different catalysts for the nucleophilic mediated thiol–ene reaction is described using model compounds, both monomers and oligomers obtained by CCTP. Different catalysts, including pentylamine and hexylamine (primary amines), triethyl amine (tertiary amine), and two different phosphines, dimethylphenylphosphine (DMPP) and tris(2-carboxyethyl)phosphine (TCEP), were investigated in the presence of different thiols, Scheme 2. This study had the objective to determine the optimum reaction conditions for nucleophile mediated thiol–ene click reactions.
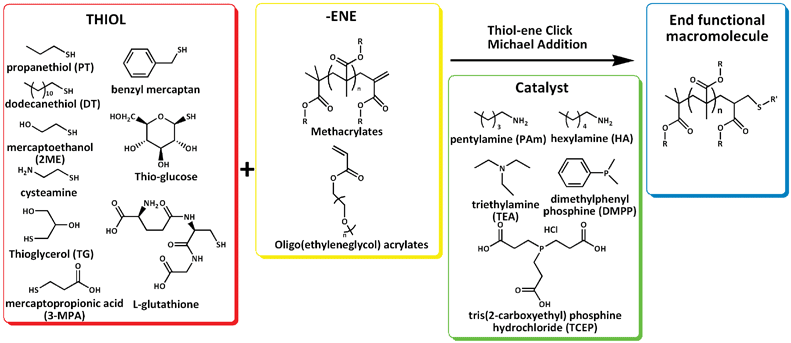
Experimental part
Materials
Methyl methacrylate (MMA, 99%, Sigma-Aldrich), 2-hydroxyethyl methacrylate (HEMA, 97%, Sigma-Aldrich), poly(ethylene glycol) methylether acrylate (PEGMEA454, Sigma-Aldrich), poly(ethylene glycol) methylether methyacrylate (PEGMEMA475, Sigma-Aldrich), 2,2′-azoisobutyronitrile (AIBN), 2-mercaptoethanol (2-ME, 99%, Fluka, Sigma-Aldrich), 1-thioglycerol (TG, 98%, Sigma-Aldrich), 1-propanethiol (PT, 99%, Sigma-Aldrich), 1-dodecanethiol (DT, 98+%, Sigma-Aldrich), benzyl mercaptan (BT, 99%, Sigma-Aldrich), glutathione (98%, Sigma-Aldrich), 1-thiol-β-D-glucose sodium salt (thiol–glucose, 99%, Sigma-Aldrich), 3-mercaptopropanoic acid (3-MPA, 99%, Sigma-Aldrich), dimethylphenylphosphine (DMPP, 99%, Aldrich), tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 98%, Alfa Aesar), triethylamine (TEA, 99%, Fisher Scientific UK Ltd.), pentylamine (PAm, 99%, Sigma-Aldrich), hexylamine (HA, 99%, Sigma-Aldrich), dimethyl sulfoxide-d6 (DMSO-d6) and acetone-d6 (99.9% atom %D, Sigma-Aldrich) and acetonitrile-d3 (ACN, 99.8% atom %D, Sigma-Aldrich) were used as received. PBS buffer (pH 7.0, 0.5 M) was made of NaH2PO4 (99.85%, Fisher Scientific UK Ltd.), carbonate–bicarbonate (CBB) buffer (pH 9.2, 0.2 M) was made of Na2CO3 (0.2 M) (99%, BDH, VWR International Ltd.) and NaHCO3 (0.2 M) (99%, Fisher Scientific UK Ltd.).Bis(boron difluorodimethylglyoximate) cobaltate(II) bis-(methanol) complex, (CH3OH)2Co-(dmgBF2)2 (CoBF), as prepared according to the method of Bakac et al.69 The activity of the synthesized CCT agent was tested with CCTP of MMA yielding a Cs value = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
Characterization
1H NMR spectroscopy was used to determine the monomer conversion. NMR spectra were recorded on a Bruker DPX-300 MHz and a Bruker DPX-400 MHz spectrometer at 25 °C on approximately 10% w/v solutions in deuterated NMR solvents from Sigma-Aldrich. All chemical shifts are reported in ppm (δ) relative to tetramethylsilane (TMS), referenced to the chemical shifts of residual solvent resonances (1H and 13C). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, m = multiplet.Mass spectra were recorded on either Bruker UHR-Q-TOF MaXis or Thermo Finnigan LCQ. ESI-MS was obtained using Thermo Finnigan LCQ Deca quadrupole ion trap mass spectrometer (Thermo Finnigan, San Jose, CA), equipped with an atmospheric pressure ionization source operating in the nebulizer assisted electrospray mode and was used in positive ion mode. Mass calibration was performed using caffeine, MRFA, and Ultramark 1621 in the m/z range of 195–1822 Da. All spectra were acquired within the m/z range of 150–2000 Da, and typical instrumental parameters were a spray voltage of 4.5 kV, a capillary voltage of 44 V, a capillary temperature of 275 °C and flow rate of 5 µL min−1. Nitrogen was used as sheath gas (flow: 50% maximum) and helium was used as auxiliary gas (flow: 5% maximum), 30 microscans, with maximum inject time of 10 ms per microscan. For each respective scan, approximately 35 scans were signal averaged to obtain the final spectrum. Solvent used was 3 : 1 mixture of DCM : methanol with sodium acetate concentration of 0.3 µM. Sodium acetate was added to the solvent prior to analysis to ensure that ionization would occur and to suppress potassium salt peaks. All data processing was done using Xcalibur™ included together with Thermoproducts. Mass spectra were recorded on a Bruker UHR-Q-TOF MaXis equipped with electrospray ionisation source in positive mode; samples were directly infused at 2 µL min−1 with a syringe pump. Nitrogen was used as nebulizer gas (0.4 bar) and dry gas (4 L min−1 at 180 °C). Capillary voltage was set at −3000 V. Data were acquired in the 50–2000 m/z range. Calibration was carried out with sodium formate solution (10 mM in 50% IPA). Mass spectra were acquired by MALDI-ToF (matrix-assisted laser desorption and ionization time-of-flight) mass spectrometry using a Bruker Daltonics Ultraflex II MALDI-ToF mass spectrometer, equipped with a nitrogen laser delivering 2 ns laser pulses at 337 nm with positive ion ToF detection performed using an accelerating voltage of 25 kV.
Catalytic chain transfer polymerization (CCTP)
Results and discussion
The optimal conditions for the thiol–ene reaction were investigated using primary amines, tertiary amine and phosphines at different temperatures and in different solvents using a broad range of monomers or oligomers. The synthesis of short oligomers bearing a terminal vinyl group was carried out using CCTP.23,61,62,70–75 This method has been successfully exploited for the synthesis of MMA and HEMA dimers (after separation from higher adducts) and OEGMA oligomers. The oligomers were tested using different thiol–ene reactions with a large range of thiol compounds and catalyst systems.Triethylamine catalyzed thiol–ene reactions
The Michael addition between a thiol and an activated vinyl group was carried out in the presence of triethylamine as catalyst at ambient temperature. Table 1 presents the results from using MMA dimer (dMMA), poly(ethylene glycol) acrylate (PEGMEA454) and poly(ethylene glycol) methacrylate (PEGMEMA475) in the presence of 2-mercaptoethanol.| Run | Monomer/dimer | Thiol | Ratios [M] : [T] : [cat]a | React. time/h | Conv. (%) |
|---|---|---|---|---|---|
| a [M] : [T] : [cat] = monomer/dimer : thiol : triethylamine. All reactions were performed at ambient temperature in d6-acetone. Conversion values were calculated by 1H NMR following the disappearance of the vinyl bond. | |||||
| T1 | dMMA | 2-ME | 1 : 1.5 : 0.1 | 15 | 42 |
| T2 | PEGMEA454 | 2-ME | 1 : 1.2 : 0.3 | 2 | 97 |
| T3 | PEGMEMA475 | 2-ME | 1 : 1.2 : 1.1 | 72 | 81 |
| T4 | PEGMEMA475 | 2-ME | 1 : 1.2 : 2.2 | 72 | 89 |
| T5 | PEGMEMA475 | 2-ME | 1 : 1.2 : 4.5 | 72 | 88 |
| T6 | PEGMEMA475 | 2-ME | 1 : 1.2 : 9.0 | 72 | 88 |
A relatively fast reaction rate was observed for PEGMEA454 with the reaction T2 catalyzed by a [TEA]/[thiol] ratio of 1.2/0.3 reaching quantitative conversions after 2 hours. However, in the case of methacrylates, even at extended reaction periods, i.e. 72 h, it was not possible to reach quantitative conversion values. Nevertheless, more than 80% conversion was measured for PEGMEMA475 using a [thiol]/[TEA] ratio equal to 1.2/1.1 after 3 days. To accelerate this reaction, the [monomer]/[thiol]/[TEA] ratio was varied from 1.0/1.2/1.1 to 1.0/1.2/9.0 (T3–T6) and the kinetics followed by 1H NMR, Fig. 1. There was no significant difference in the kinetic behavior as the ratios were varied. Thus, excess TEA does not accelerate the thiol–ene reaction with methacrylic vinyl groups. 1H NMR and ESI-MS does not show any formation of side products. The DMPP catalyzed reaction P13 is included as a comparison in Fig. 1, which reached quantitative conversions in few minutes; this will be discussed further in following sections.
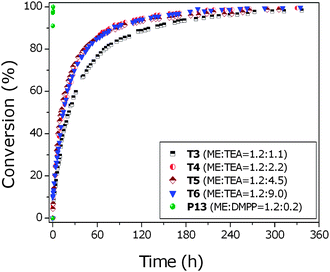 | ||
| Fig. 1 Conversion vs. time plot for the reaction of PEGMEMA475 with 2-ME in the presence of varying concentrations of TEA or DMPP. Conversion values were calculated by 1H NMR following the disappearance of the vinyl group. | ||
Primary amine catalyzed thiol–ene reactions
In order to accelerate the rate of reaction, we examined primary amines as catalysts at ambient temperature. Primary amines are more nucleophilic than tertiary amines. The formation of thiolate should be followed by a nucleophilic addition onto the vinyl group. PEGMEA454, PEGMEMA475 and PEGMEMA2080 monomers were reacted with 2-ME, 3-MPA and 1-dodecanethiol (DT), Table 2. Higher ratios of catalyst were used to compensate for the lower reactivity of methacrylates relative to acrylates.| Run | Monomer | Thiol | Ratios [M] : [T] : [cat]a | React. time/h | Conv. (%) |
|---|---|---|---|---|---|
| a [M] : [T] : [cat] = monomer : thiol : n-pentylamine. All reactions were performed at ambient temperature in d6-acetone as solvent. Conversion values were calculated by 1H NMR based on the consumption of the vinylic bonds. | |||||
| PAm1 | PEGMEA454 | DT | 1 : 1.2 : 0.1 | 24 | 10 |
| PAm2 | PEGMEA454 | 3-MPA | 1 : 1.2 : 0.1 | 24 | 20 |
| PAm3 | PEGMEA454 | BM | 1 : 1.2 : 0.1 | 24 | 78 |
| PAm4 | PEGMEA454 | 2-ME | 1 : 1.2 : 0.1 | 24 | 93 |
| PAm5 | PEGMEA454 | 2-ME | 1 : 1.2 : 0.3 | 0.5 | 93 |
| PAm6 | PEGMEMA475 | DT | 1 : 1.2 : 1.7 | 4 | 5 |
| PAm7 | PEGMEMA475 | 3-MPA | 1 : 1.2 : 1.4 | 4 | 3 |
| PAm8 | PEGMEMA475 | BM | 1 : 1.2 : 1.4 | 4 | 17 |
| PAm9 | PEGMEMA475 | TG | 1 : 1.2 : 1.4 | 4 | 77 |
| PAm10 | PEGMEMA475 | 2-ME | 1 : 1.2 : 1.3 | 4 | 33 |
| PAm11 | PEGMEMA475 | 2-ME | 1 : 1.2 : 2.7 | 4 | 49 |
| PAm12 | PEGMEMA475 | 2-ME | 1 : 1.2 : 5.4 | 4 | 74 |
| PAm13 | PEGMEMA475 | 2-ME | 1 : 1.2 : 10.8 | 4 | 95 |
| PAm14 | PEGMEMA2080 | 2-ME | 1 : 1.2 : 5.0 | 4 | 60 |
| PAm15 | PEGMEMA2080 | 2-ME | 1 : 1.2 : 10.0 | 4 | 69 |
| PAm16 | PEGMEMA2080 | 2-ME | 1 : 1.2 : 20.0 | 4 | 87 |
| PAm17 | PEGMEMA2080 | 2-ME | 1 : 1.2 : 40.0 | 4 | 99 |
Different thiol–ene reactions were conducted using 4 different thiols in the presence of a low concentration of n-pentylamine as the catalyst ([M]/[T]/[amine] = 1.0/1.1/0.1), Fig. 2. The reactions were monitored for up to 5 days and the vinyl bond conversion versus time was plotted, Fig. 2. The reaction rates differ depending on the thiol structure. For example, 1-dodecanethiol (DT) has the slowest rate (10% conversion in 24 h) in comparison to all other thiols used in this study. The reaction with 3-mercaptopropionic acid (3-MPA), PAm2, did not exceed 50% conversion, even after 4 days of reaction. However, benzyl mercaptan (BM) had a faster reaction rate PAm3 with 78% after 24 hours. One explanation of the low reactivity of 3-MPA is the presence of carboxylic acid functionality (PAm2) resulting in potentially the enolate quenching and the subsequent non-generation of thiolate.
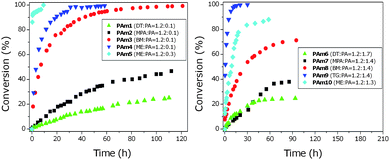 | ||
| Fig. 2 Vinyl bond conversion vs. time plots for PEGMEA454 (left) and PEGMEMA475 (right) reacting with different thiols in the presence of n-pentylamine as catalyst. Note: conversion values were calculated by 1H NMR based on the consumption of the vinylic bonds. | ||
It is evident that benzyl mercaptan (BM) and 2-ME react with PEGMEA454 relatively faster than DT and 3-MPA, PAm3–PAm5, PAm1 and PAm2, respectively. Quantitative conversions were obtained in less than 3 days by reacting BM in the presence of n-pentylamine, PAm3. The final product of PAm3 was characterized using MALDI-TOF MS confirming the expected structure, Fig. 3. All peaks are assigned and the main distribution (labeled as A and B ionized with Na+ or K+ salt, respectively) can be assigned to BM conjugated PEGMEA454 (a trace amount of un-reacted monomer (labeled as C) is also detected).
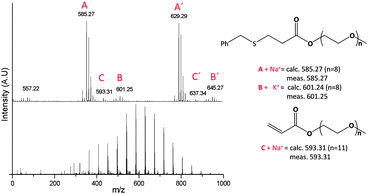 | ||
| Fig. 3 MALDI-TOF MS analysis of the final product of PEGMEA454 reacted with benzyl mercaptan PAm3 in the presence of n-pentylamine. | ||
The catalyst concentration was varied as 0.1 PAm4 and 0.3 PAm5 for the reaction of PEGMEA454 and 2-ME. The reactions reached ∼90% conversion after 24 hours and 0.5 hours using 0.1 and 0.3 equivalents of PA, respectively. There is a significant difference in the reaction rate by changing the catalyst concentration (in contrast to the TEA catalyzed reactions). The reaction kinetics with acrylates was followed by 1H NMR and all corresponding peaks of the starting material and the products are assigned, Fig. 4. Using relatively short oligomer chains has facilitated the characterization of both the products and the side products in detail.
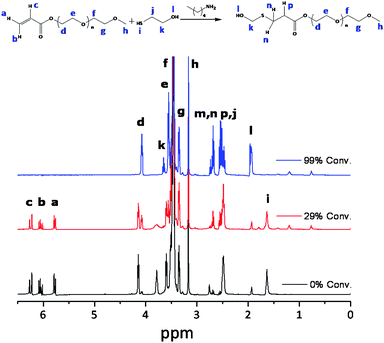 | ||
| Fig. 4 1H NMR spectra for Michael addition of PEGMEA454 and ME, using 0.1 eq. of n-pentylamine in d6-acetone, PAm4. | ||
The effect of the pentylamine concentration on the Michael addition reaction of 2-ME in the presence of PEGMEMA475 or PEGMEMA2080 was investigated in experiments, PAm6–PAm17, Table 2. The reaction kinetics of methacrylates with 2-ME PAm10–PAm17 was followed using 1H NMR, Fig. 5. A large excess of catalyst was used in these experiments for two reasons: (i) it has been shown that methacrylates react very slowly with thiols in the presence of amines, (ii) PEGMEMA2080 is a more sterically hindered molecule with reduced accessibility to the vinyl bonds. In these experiments, it is shown that the reaction proceeds much faster with a higher amount of catalyst present.
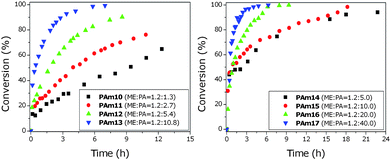 | ||
| Fig. 5 Michael addition of PEGMEMA475 (left) or PEGMEMA2080 (right) with 2-ME in different amounts of n-pentylamine. The reaction was performed in 0.5 mL d6-acetone and monitored via1H NMR. | ||
It is noted that using a large excess of n-pentylamine provides an observation of intermediate species in the nucleophile mediated hydrothiolation reaction. These intermediates were identified using MALDI-TOF MS from the reaction of PEGMEMA2080 and 2-ME, Fig. 6. The main distribution (labeled as A and A′) belongs to the product at 1521.9 Da (n = 30) and 1565.9 Da (n = 31). Moreover, starting material can be identified (labeled as B and B′, 1531.9 Da and 1575.9 Da) and the intermediates (labeled as C and C′, 1552.2 Da and 1596.3 Da) observed in the spectrum. The intermediate is assigned as a compound formed from the reaction of PEGMEMA2080 and n-pentylamine. Thiols are better nucleophiles than amines, however, in a large excess of amine (or for a higher [amine]/[thiol] ratio), such as in PAm12, these intermediates are visible by this technique. It is noted that MALDI-TOF MS does not provide quantitative information due to different ionization efficiencies of each compound. For instance, compound C is protonated and ionizes without forming a sodium adduct, as calculated from the spectra. It is interesting to note that MALDI-TOF analysis shows the presence of starting material, while almost full conversion was calculated from 1H NMR. Based on these results, it appears that vinyl terminated species ionizes more efficiently than the thiol or amine substituted species.
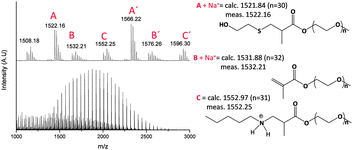 | ||
| Fig. 6 MALDI-TOF MS spectrum of PEGMEMA2080 reacted with 2-ME in the presence of 40.0 eq. of n-pentylamine PAm12. | ||
CCTP was used to synthesize oligo(PEGMEMA475) to give mixtures of dimers, trimers, tetramers and higher adducts, that were used directly for subsequent thiol addition reactions. The CCTP product mixture was reacted with six different thiols, 2-ME, BM, 3-MPA, cysteamine, glutathione and thiol–glucose, Table 3. The mixtures were reacted overnight at 40 °C. To improve the yield of the reaction using primary amine, reactions were carried out using [ene]/[thiol]/[amine] = 1/5/5 at 40 °C in organic solvents and aqueous solution. After 14 hours, a complete conversion of oligo(PEGMEMA475) was observed by 1H NMR, Table 3, and by ESI-MS, Fig. 7. ESI-MS spectra show the quantitative formation of expected product, i.e. thiol conjugated to oligo(PEGMEMA475), without the formation of by-products, Table 3.
| Run | Oligomer | Thiol | Ratio [M] : [T] : [cat]a | Solvent | Conv. (%) |
|---|---|---|---|---|---|
| a [M] : [T] : [cat] = oligo(PEGMEMA475) : thiol : hexylamine. Conversion values were calculated by 1H NMR with respect to vinyl peaks. All reactions were performed at 40 °C for 14 hours. | |||||
| HA1 | Oligo(PEGMEMA475) | 2-ME | 1 : 5 : 5 | MeCN | 100 |
| HA2 | Oligo(PEGMEMA475) | BM | 1 : 5 : 5 | MeCN | 100 |
| HA3 | Oligo(PEGMEMA475) | 3-MPA | 1 : 5 : 5 | MeCN | 100 |
| HA4 | Oligo(PEGMEMA475) | Cysteamine | 1 : 5 : 5 | D2O | 100 |
| HA5 | Oligo(PEGMEMA475) | Glutathione | 1 : 5 : 5 | D2O | 100 |
| HA6 | Oligo(PEGMEMA475) | HS-Glc | 1 : 5 : 5 | D2O | 100 |
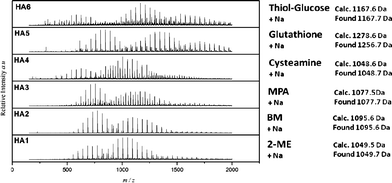 | ||
| Fig. 7 ESI-MS result of oligo(PEGMEMA475) reacted with various thiol compounds. Identified peaks of products are tabulated in Table S2, see ESI† (HA1–HA6). | ||
Characterization was also performed using ESI-MS, Fig. 7. The top spectra show the presence of monomer, dimer, trimer and also tetramer distributions modified by thiol–glucose (HA6). Similar results were obtained for the glutathione, cysteamine, BM, 2-ME and 3-MPA conjugated oligomers HA1–HA5. ESI-MS results confirm the synthesis of expected products. The presence of two main distributions observed for HA1, HA2, HA3 and HA6 can be attributed to the expected products. The two populations can be assigned to differently ionized forms (ionized with sodium or hydrogen). The calculated and measured molecular weight was found to have maximum error of 0.2 Da, well within the inherent 0.3 Da error of the instrument. Moreover, it can also be seen from the mass spectra that there is no starting material or side products (see ESI-MS spectrum, Fig. S4 in the ESI†). As explained above, mass spectroscopy is more sensitive than 1H NMR analysis, giving confidence that the reactions were complete, without detectable side products. The absence of addition of amine onto methacrylic bonds can be explained by the use of low [thiol]/[amine] ratio = 1 at 40 °C. In summary, hexylamine is an efficient catalyst for the Michael reaction of thiol/acrylates at ambient temperature without observation of any side products when the [amine]/[thiol] ratio is kept close to 1.0. The use of a higher ratio yields a side product which can be attributed to the addition of amine onto vinyl bonds. A slight increase of the temperature (at 40 °C) and the amount of thiol allow us to obtain a quantitative reaction with monomer or oligomer bearing a methacrylic double bond.
Phosphine catalyzed thiol–ene reactions
Lowe has reported that phosphine based catalysts are known as very reactive catalysts for Michael addition reactions.18 However, the highly reactive phosphine compound can also react with the vinyl group (methacrylate or acrylate) resulting in the formation of side reactions. Therefore, we initially performed some test reactions P1 and P2 by mixing MMA in the presence of different amounts of dimethylphenylphosphine (DMPP) in acetone, Table 4. The expected reaction is the nucleophilic addition of the phosphine to the vinyl group. After 4 days, the presence of vinyl group was monitored by 1H NMR and it was found that 4% and 23% of vinyl groups were reacted with a monomer to DMPP ratio equal to 0.1 and 1.0, respectively. Subsequently, a 1 : 1 ratio of [MMA]/[thiols] (TG or DT) were reacted in the presence of [vinyl group]/[thiol]/[DMPP] = 1/1/0.05 as catalyst in acetone solution. Full conversion was obtained in an hour for the reaction of TG (P3) and ≥93% conversion was calculated for the reaction of DT (P4). Similar reaction rates were observed when the dimer of MMA was used instead of the monomer (P5 and P6). As expected, solvent selection has a great influence on the reaction rates. Therefore, we tested acetonitrile (MeCN), acetone and DMSO with the reaction in DMSO displaying faster kinetics in comparison to MeCN and acetone (P7 and P8) attributable to the higher polarity of DMSO. The high polarity of DMSO allows a better stabilization of thiolate ion, and favors its formation. Solvent effects were also tested by using 1-propanethiol (PT) in all three solvents. The reaction in acetone was the slowest and in DMSO the fastest (P9, P11 and P12). In reactions P9 and P10, a higher ratio of thiol provides higher conversion values at the same reaction time. Finally, PEGMEMA475 monomer was reacted with 2-ME in the presence of DMPP and full conversion was observed after 30 minutes P13, Fig. 8.| Run | Monomer/oligomer | Thiol | Base | Ratios [M] : [T] : [cat]a | Solvent | React. time/h | Conv. (%) |
|---|---|---|---|---|---|---|---|
| a [M] : [T] : [cat] = monomer or oligomers : thiol : DMPP. Conversion values were calculated by 1H NMR. All reactions were performed at ambient temperature. | |||||||
| P1 | MMA | — | DMPP | 1 : 0 : 0.1 | Acetone | 96 | 4 |
| P2 | MMA | — | DMPP | 1 : 0 : 1.0 | Acetone | 96 | 23 |
| P3 | MMA | TG | DMPP | 1 : 1 : 0.05 | Acetone | 1 | 100 |
| P4 | MMA | DT | DMPP | 1 : 1 : 0.05 | Acetone | 3.5 | 93 |
| P5 | dMMA | TG | DMPP | 1 : 1 : 0.05 | Acetone | 1 | 97 |
| P6 | dMMA | DT | DMPP | 1 : 1 : 0.05 | Acetone | 4 | 81 |
| P7 | dMMA | DT | DMPP | 1 : 1.5 : 0.05 | MeCN | 2 | 83 |
| P8 | dMMA | DT | DMPP | 1 : 1.5 : 0.05 | DMSO | 2 | 95 |
| P9 | dMMA | PT | DMPP | 1 : 1.5 : 0.05 | Acetone | 9 | 72 |
| P10 | dMMA | PT | DMPP | 1 : 2.5 : 0.05 | Acetone | 8 | 85 |
| P11 | dMMA | PT | DMPP | 1 : 1.5 : 0.05 | MeCN | 9 | 98 |
| P12 | dMMA | PT | DMPP | 1 : 1.5 : 0.05 | DMSO | 1 | 100 |
| P13 | PEGMEMA475 | 2-ME | DMPP | 1 : 1.2 : 0.2 | Acetone | 0.5 | 100 |
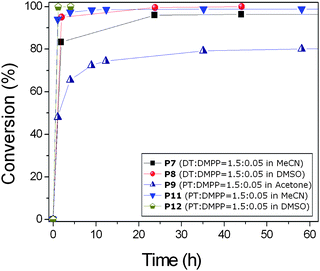 | ||
| Fig. 8 Vinyl bond conversion vs. time for MMA dimer in the presence of different thiols and solvents. | ||
Furthermore, the addition reaction on oligo(PEGMEMA475) was investigated using DMPP and TCEP as catalysts, Table 5. However, side reactions occur when an excess of catalyst is used and this should be taken into account when designing experimental conditions S1–S3. The presence of side reactions was investigated with ESI-MS, Fig. 9. In the presence of a large excess of DMPP or TCEP, by-products were clearly identified, corresponding to the presence of mono-addition of phosphine onto vinyl group for TCEP and DMPP.
| Run | Oligomer | Ratios [M] : [T] : [B] : [P]a | React. temp./°C | React. time/h | Cat. |
|---|---|---|---|---|---|
| a [M] : [T] : [B] : [P] = o(PEGMEMA475) : 2-ME : hexylamine : phosphine. | |||||
| S1 | o(PEGMEMA 475) | 1 : 5 : 0 : 2.5 | Ambient | 14 | TCEP (pH 8) |
| S2 | o(PEGMEMA 475) | 1 : 5 : 0 : 2.5 | Ambient | 14 | TCEP (pH 3) |
| S3 | o(PEGMEMA 475) | 1 : 5 : 2.5 : 2.5 | 40 | 14 | DMPP/HA |
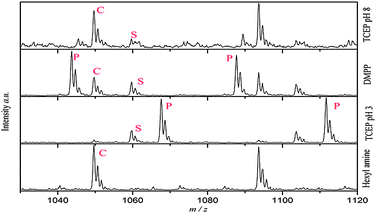 | ||
| Fig. 9 ESI-MS of oligo(PEGMEMA475) synthesized by CCTP used to identify the side reaction of phosphine catalyst. S = starting material, P = phosphine conjugated species, C = desired thiol–oligomer conjugate. Spectra were obtained with ESI-MS LCQ-Deca quadrupole. The list of corresponding compounds is given in the ESI†. | ||
In the case of TCEP, the pH has a large effect on the amount of side products formed. Indeed, at low pH, the major population observed corresponds to the presence of phosphine reacted polymers, while at high pH values the excepted product (thiol–oligomer conjugate) is present in large amounts. As expected, low pH does not favor the formation of thiolate anion (R–S−). These experiments show that the concentration of phosphine should be kept relatively low (or the [phosphine]/[thiol] ratio should be kept very low) to minimize side product formation.
Although the aim of this study was to investigate the effect of catalysts on thiol–ene click reactions, it was also observed that 2-hydroxyethyl methacrylate (HEMA, water soluble monomer) can react with 2-ME in water in the absence of catalyst, Table 6. However, it is crucial to control the pH of the solution in the right range.76 For instance, when PBS buffer was used and the pH was set to 7.1 and 9.2 for the reaction of HEMA and 2-ME in D2O after 4 hours 83% and 100% of conversion was observed for N1 and N2, respectively. Pure dimer of HEMA synthesized by CCTP77 was reacted with 2-ME under basic conditions in D2O and 60% of conversion was obtained in 30 minutes N3. However, the reaction with thiol–glucose took relatively longer time to reach more than 90% conversion N4.
| Run | Monomer/dimer | Thiol | Ratios [M] : [T]a | Solvent | React. time/h | Conv. (%) |
|---|---|---|---|---|---|---|
| a [M] : [T] = monomer or dimer : thiol. Conversion values were calculated by 1H NMR. All reactions were performed at ambient temperature. b PBS buffer was used at a pH value of 7.1. c PBS buffer was used at a pH value of 9.2. | ||||||
| N1 | HEMA | 2-ME | 1 : 1.5 | D2O | 4 | 83 |
| N2 | HEMA | 2-ME | 1 : 1.5 | D2O | 4 | 100 |
| N3 | dHEMA | 2-ME | 1 : 1.5 | D2O | 0.5 | 60 |
| N4 | dHEMA | HS-Glc | 1 : 1.5 | D2O | 44 | 91 |
Conclusion
We present here a detailed study on the effect of catalyst on nucleophilic thiol–ene click reactions. It has been shown that when triethylamine is used as catalyst, even in excess amounts, it does not give any noticeable side reactions since mechanistically it is operating differently from pentylamine, DMPP and TCEP. Moreover, primary amines, pentylamine and hexylamine, were tested as appropriate catalysts. However, these can react with the vinyl group and form stable species which can also be monitored by ESI-MS. Phosphine catalysts provide the fastest reaction, which also brings the highest amount of side reactions. Nevertheless, using the catalyst in catalytic amounts provide relatively fast reaction rates for the desired compounds. It should also be noted that the effect of solvent, type of thiol and the basicity of the medium have a great role on this reaction and should be selected with care based on these optimization reactions. These model reactions were performed on monomers, dimers and oligomers and will form the basis of our following studies on thiol-click reactions with vinyl-terminated polymers.Acknowledgements
This work was supported by the China Scholar Council (CSC). CB and TPD are thankful for the APD and Federation Fellowships from ARC, respectively. We also thank Mathew W. Jones, Dr Lijiang Song and Dr Giuseppe Mantovani for their help and advice. CRB is funded by European Union Marie Curie Fellowship scheme (proposal number 235999). GPC used in this research was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). We would like to thank the referees for their valuable comments.Notes and references
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- A. Dag, H. Durmaz, E. Demir, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6969–6977 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540–7545 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- N. Gupta, B. F. Lin, L. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138–145 Search PubMed.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- T. Clark, L. Kwisnek, C. E. Hoyle and S. Nazarenko, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 14–24 CrossRef CAS.
- L. A. Connal, C. R. Kinnane, A. N. Zelikin and F. Caruso, Chem. Mater., 2009, 21, 576–578 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- A. Geissler, M. F. Vallat, P. Fioux, J. S. Thomann, B. Frisch, J. C. Voegel, J. Hemmerle, P. Schaaf and V. Roucoules, Plasma Processes Polym., 2010, 7, 64–77 Search PubMed.
- Y. Li and T. Michinobu, Polym. Chem., 2010, 1, 72–74 RSC.
- J. W. Chan, H. Wei, H. Zhou and C. E. Hoyle, Eur. Polym. J., 2009, 45, 2717–2725 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS.
- N. ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef.
- A. Gress, A. Volkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS.
- Z. Jia, J. Liu, T. P. Davis and V. Bulmus, Polymer, 2009, 50, 5928–5932 CrossRef CAS.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344–1350 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenović, R. A. Prasath, A. J. Inglis, F. E. D. Prez, C. Barner-Kowollik, W. V. Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- C. Gimbert, M. Lumbierres, C. Marchi, M. Moreno-Manas, R. M. Sebastian and A. Vallribera, Tetrahedron, 2005, 61, 8598–8605 CrossRef.
- C. Gimbert, M. Moreno-Manas, E. Perez and A. Vallribera, Tetrahedron, 2007, 63, 8305–8310 CrossRef CAS.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133–1170 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- J. T. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302–4313 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad and S. H. Thang, Aust. J. Chem., 2009, 62, 1379–1381 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS.
- C. R. Becer, A. M. Groth, R. Hoogenboom, R. M. Paulus and U. S. Schubert, QSAR Comb. Sci., 2008, 27, 977–983 Search PubMed.
- R. M. Paulus, C. R. Becer, R. Hoogenboom and U. S. Schubert, Aust. J. Chem., 2009, 62, 254–259 CrossRef CAS.
- J. T. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2010, 43, 20–24 CrossRef CAS.
- M. Semsarilar, V. Ladmiral and S. Perrier, Macromolecules, 2010, 43, 1438–1443 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3931–3939 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- H. Li, B. Yu, H. Matsushima, C. E. Hoyle and A. B. Lowe, Macromolecules, 2009, 42, 6537–6542 CrossRef CAS.
- R. M. Hensarling, V. A. Doughty, J. W. Chan and D. L. Patton, J. Am. Chem. Soc., 2009, 131, 14673–14675 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- T. P. Davis, D. M. Haddleton and S. N. Richards, J. Macromol. Sci. Rev., Macromol. Chem. Phys., 1994, C34, 243–324 Search PubMed.
- T. P. Davis, D. Kukulj, D. M. Haddleton and D. R. Maloney, Trends Polym. Sci., 1995, 3, 365–373 CAS.
- D. Kukulj, T. P. Davis, K. G. Suddaby, D. M. Haddleton and R. G. Gilbert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 859–878 CrossRef CAS.
- K. G. Suddaby, D. R. Maloney and D. M. Haddleton, Macromolecules, 1997, 30, 702–713 CrossRef CAS.
- R. A. Sanayei and K. F. Odriscoll, J. Macromol. Sci., Part A: Pure Appl. Chem., 1989, 26, 1137–1149 Search PubMed.
- J. P. A. Heuts, D. Kukulj, D. J. Forster and T. P. Davis, Macromolecules, 1998, 31, 2894–2905 CrossRef CAS.
- A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753–1766 CrossRef CAS.
- J. P. A. Heuts, G. E. Roberts and J. D. Biasutti, Aust. J. Chem., 2002, 55, 381–398 CrossRef CAS.
- A. Bakac, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108–4114 CrossRef CAS.
- T. Y. J. Chiu, J. P. A. Heuts, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Chem. Phys., 2004, 205, 752–761 CrossRef CAS.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS.
- D. M. Haddleton, D. R. Maloney, K. G. Suddaby Adam Clarke and S. N. Richards, Polymer, 1997, 38, 6207–6217 CrossRef CAS.
- D. M. Haddleton, D. R. Maloney, K. G. Suddaby, A. V. G. Muir and S. N. Richards, Macromol. Symp., 1996, 111, 37–46 CAS.
- D. M. Haddleton, D. R. Maloney and K. G. Suddaby, Macromolecules, 1996, 29, 481–483 CrossRef CAS.
- D. M. Haddleton, M. C. Crossman, K. H. Hunt, C. Topping, C. Waterson and K. G. Suddaby, Macromolecules, 1997, 30, 3992–3998 CrossRef CAS.
- E. S. Read, K. L. Thompson and S. P. Armes, Polym. Chem., 2010, 1, 221–230 RSC.
- J. D. Biasutti, G. E. Roberts, F. P. Lucien and J. P. A. Heuts, Eur. Polym. J., 2003, 39, 429–435 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR and ESI-MS data. See DOI: 10.1039/c0py00100g |
| This journal is © The Royal Society of Chemistry 2010 |