Hydroamination as a route to nitrogen-containing oligomers†
Sharonna
Greenberg
and
Douglas W.
Stephan
*
Department of Chemistry, University of Toronto, 80 St George St., Toronto, Ontario, Canada M5S3H6. E-mail: dstephan@chem.utoronto.ca
First published on 3rd June 2010
Abstract
Hydroamination of H2NC6H4-p-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh (1) with 10 mol% Ti(NR2)4 (R = Me and Et) at 70 °C for 3.5 days affords oligomers (2) ((2a) R = Me and (2b) R = Et), characterized by NMR, IR, and UV/Vis spectroscopy, and by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry and gel permeation chromatography (GPC). These data indicate 10 to 15 repeat units in the chain. Model reactions, performed using phenylacetylene or diphenylacetylene and aniline or 2,6-diisopropylaniline, generated a variety of enamines and imines (compounds (3)–(11)), three of which were characterized by X-ray crystallography. Evidence suggests the hydroamination oligomerization follows the widely accepted [2 + 2] cycloaddition mechanism, although chain capping with NR2 (R = Me and Et) appears to occur via alkyne insertion in a Ti–N σ-bond.
CPh (1) with 10 mol% Ti(NR2)4 (R = Me and Et) at 70 °C for 3.5 days affords oligomers (2) ((2a) R = Me and (2b) R = Et), characterized by NMR, IR, and UV/Vis spectroscopy, and by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry and gel permeation chromatography (GPC). These data indicate 10 to 15 repeat units in the chain. Model reactions, performed using phenylacetylene or diphenylacetylene and aniline or 2,6-diisopropylaniline, generated a variety of enamines and imines (compounds (3)–(11)), three of which were characterized by X-ray crystallography. Evidence suggests the hydroamination oligomerization follows the widely accepted [2 + 2] cycloaddition mechanism, although chain capping with NR2 (R = Me and Et) appears to occur via alkyne insertion in a Ti–N σ-bond.
Introduction
Hydroamination of carbon–carbon multiple bonds represents an atom-economical methodology for the synthesis of amines.1–7 Though thermodynamically favourable,8,9 the direct reaction is plagued by a high activation energy barrier. As a result, a variety of hetero- and homogeneous catalysts have been reported to carry out catalytic hydroamination.1,8,10,11 These catalysts include acids11,12 and bases,11,13 heavier group 2 complexes,14–18 lanthanides and actinides,5 several group 4 metal species,2–4,6,7 other early transition metals such as vanadium19 and tantalum,20 late transition metals (Ru, Rh, Pd, Pt, Ag, and Au),8,10 and heavy metals including mercury and thallium.8The most well-studied catalysts are the group 4 transition metal compounds,7 as they are inexpensive, nontoxic, and commercially available or easily synthesized. Bergman and coworkers were first to describe the mechanism of group 4 metal-mediated hydroamination of an alkyne (Scheme 1).21–23 This widely accepted mechanism involves the generation of a group 4 metal imide, which undergoes [2 + 2] cycloaddition with an alkyne affording an azametallacyclobutene. Subsequent reaction of this species with amine generates an (amido)(enamido)metal species which releases enamine and regenerates the metal-imide species.
![Hydroamination of an alkyne using a group 4 catalyst via a [2 + 2] cycloaddition pathway.](/image/article/2010/PY/c0py00120a/c0py00120a-s1.gif) | ||
| Scheme 1 Hydroamination of an alkyne using a group 4 catalyst via a [2 + 2] cycloaddition pathway. | ||
Recently, some exceptions to this mechanism have been uncovered. For example, cationic24,25 and neutral26,27 group 4 complexes have been shown to catalyze intramolecular hydroamination cyclization of alkenes24–27 or alkynes27via a M–N σ-bond insertion mechanism. This pathway is analogous to that proposed by Marks et al.28 for group 3 transition metal and lanthanide catalysts (Scheme 2),5 and involves intramolecular insertion of a pendant alkene or alkyne into a M–N σ-bond. Reaction with additional amine releases enamine and regenerates the active metal amide catalyst. Interestingly, performing lanthanide-catalyzed olefin polymerization in the presence of certain bulky secondary amines affords amine-capped polyolefin.29
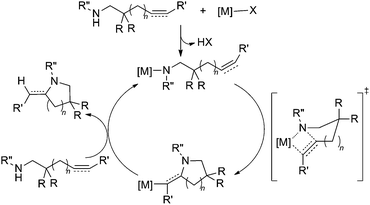 | ||
| Scheme 2 Hydroamination cyclization of an aminoalkene or aminoalkyne using a lanthanide catalyst via a σ-bond insertion pathway. | ||
In addition to mechanistic studies, many researchers have focused on applications in total synthesis,30 enantioselective catalysis,24,31,32 or the development of new or improved catalysts.2,4,6,24,25,33 Our goal is to employ hydroamination as a route to new oligomers and polymers. We rationalized that for a step-growth polymerization process, a maximal degree of polymerization necessitates precise control over the stoichiometry as expressed in the Carothers equation.34 Thus, bifunctional monomers were targeted, containing primary amine and alkyne moieties where the molecular geometry precludes intramolecular reaction. Herein, we report the catalytic hydroamination of such substrates as a route to oligomers, as well as the small molecule model studies designed to probe the structure of the oligomer and aspects of the oligomerization mechanism.
Experimental section
General considerations
All manipulations of air- and/or water-sensitive compounds were carried out under an atmosphere of dry oxygen-free nitrogen using standard Schlenk techniques or a Vacuum Atmospheres inert atmosphere glove box. 1H and 13C{1H} NMR spectra were acquired on a Bruker Avance 300 MHz spectrometer, a Bruker Avance 400 MHz spectrometer, a Bruker SpectroSpin 500 MHz spectrometer, a Varian Mercury 300 MHz spectrometer, or a Varian Mercury 400 MHz spectrometer. All NMR spectra were recorded in C6D6 at 25 °C, and 1H and 13C resonances were referenced to the solvent resonance. 1H–13C HSQC and HMBC experiments were carried out to aid in peak assignments. Infrared spectra were recorded using a Perkin-Elmer Spectrum One FT-IR spectrometer at 25 °C, either as a Nujol mull or deposited onto the NaCl plate from a CH2Cl2 or C6D6 solution. Elemental analyses were performed using a Perkin-Elmer 2400 C/H/N analyzer. UV/Vis spectra were acquired on a double-beam Lambda 12 UV-Visible spectrometer, using the solvent as the external standard. Mass spectra were recorded with a VG 70-250S mass spectrometer in positive ion electron impact (EI) mode.For gel permeation chromatography (GPC) analysis, samples were dissolved in THF (ca. 1–2 mg mL−1). Absolute polymer molecular weights were determined using a Waters GPC equipped with a refractive index detector, a laser light scattering detector, and a viscometer. Relative polymer molecular weights were determined using a Waters liquid chromatograph equipped with a differential refractometer. Polystyrene standards were purchased from Polymer Laboratories, with molecular weights varying between 580 and 283![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1. MALDI-TOF mass spectra were acquired using a Waters Micromass MALDI micro MX with the following conditions: positive polarity mode, reflectron flight path, 12 kV flight tube voltage, 10 Hz laser firing rate, 10 shots per spectrum, pulse 1950 V, detector 2350 V. The instrument was calibrated using polyethyleneglycol (PEG). Matrices were prepared using 6 mg of α-cyano-4-hydroxycinnamic acid (CHCA) in 1 mL of a 6 : 3 : 1 mixture of CH3CN : CH3OH : H2O plus one drop of CF3COOH. Analyte solutions consisted of 3–5 mg of oligomer in 1 mL of CH2Cl2. Samples were prepared using the layer method:35 1 µL of matrix was spotted onto the sample plate under an atmosphere of air, allowed to dry, then 1 µL of analyte was spotted onto the sample plate under an inert atmosphere,36 and the plate was allowed to dry again.
300 g mol−1. MALDI-TOF mass spectra were acquired using a Waters Micromass MALDI micro MX with the following conditions: positive polarity mode, reflectron flight path, 12 kV flight tube voltage, 10 Hz laser firing rate, 10 shots per spectrum, pulse 1950 V, detector 2350 V. The instrument was calibrated using polyethyleneglycol (PEG). Matrices were prepared using 6 mg of α-cyano-4-hydroxycinnamic acid (CHCA) in 1 mL of a 6 : 3 : 1 mixture of CH3CN : CH3OH : H2O plus one drop of CF3COOH. Analyte solutions consisted of 3–5 mg of oligomer in 1 mL of CH2Cl2. Samples were prepared using the layer method:35 1 µL of matrix was spotted onto the sample plate under an atmosphere of air, allowed to dry, then 1 µL of analyte was spotted onto the sample plate under an inert atmosphere,36 and the plate was allowed to dry again.
Anhydrous solvents were purchased from Aldrich and purified using Grubbs' column systems manufactured by Innovative Technology.37 C6D6 was purchased from Cambridge Isotopes Laboratories, vacuum distilled from Na/benzophenone, and freeze–pump–thaw degassed (×3). Unless otherwise noted, starting materials were purchased from Aldrich and used as received. Diethylamine and triethylamine were degassed by sonication; aniline, 2,6-diisopropylaniline and N-methylamine were degassed by sparging with N2. Hyflo Super Cel® (Celite) was dried in a vacuum oven for at least 12 h prior to use. Molecular sieves (4 Å) were dried at 100 °C under vacuum. Phenylacetylene was vacuum-distilled from CaH2 and stored in the dark at −35 °C. trans-Pd(PPh3)2Cl2, Ti(NMe2)4 and Ti(NEt2)4 were purchased from the Strem Chemical Co. Compound (1) has been previously reported.38
Synthesis of H2NC6H4-p-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CPh (1)
CPh (1)
To a NEt3 solution (50 mL) of 4-iodoaniline (2.190 g, 10.00 mmol) were added trans-Pd(PPh3)2Cl2 (70 mg, 0.010 mmol, 0.010 equiv.) and CuI (19 mg, 0.10 mmol, 0.010 equiv.). The brown mixture was stirred and freshly distilled HCCPh (1.175 g, 11.50 mmol, 1.15 equiv.) was added. The mixture turned red-orange in colour and was stirred overnight at room temperature. The solvent was removed in vacuo, and the residue extracted with Et2O and filtered through Celite. Removal of Et2O in vacuo resulted in a brown oil, which was then extracted with a 3 : 1 solution of CH2Cl2 : hexanes, filtered, and the solvent evaporated to give a brown solid. Yield: 1.785 g (92%). 1H NMR: 7.54 (m, 2H, o-C6H5), 7.40 (m, 2H, m-C6H4), 7.01–6.98 (m, 3H, m- and p-C6H5), 6.11 (m, 2H, o-C6H4), 2.73 (br s, 2H, NH2). 13C{1H} NMR: 147.4 (ipso-CN), 133.3 (ArH), 131.8 (ArH), 128.6 (ArH), 127.7 (ArH), 124.9 (quat-Ar), 114.7 (ArH), 112.9 (quat-Ar), 91.3 (CC), 88.1 (CC). EI-MS (m/z): 193.1 (100%) ([M]+). HRMS: C14H11N mass 193.0892, calcd mass 193.0891. FT-IR (from C6D6 soln, cm−1): ν(N–H) 3475, 3380 (med., sharp), ν(CC) 2211 (med., sharp). UV/Vis (CH3CN, ca. 10−5 M): λmax = 311 nm. Anal. Calcd for C14H11N: C, 87.01; H, 5.74; N, 7.25. Found: C, 86.88; H, 5.92; N, 6.82%.
Synthesis of H2N{[–C6H4-p-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CPhNH–]x/[–C6H4-p-CH2CPhN–]y}nC6H4CHCHNR2(2)
CPhNH–]x/[–C6H4-p-CH2CPhN–]y}nC6H4CHCHNR2(2)
Compound (1) (640 mg, 3.31 mmol), Ti(NR2)4 (R = Me, Et, 0.33 mmol, 0.10 equiv.), and 40 mL toluene were placed in a 100 mL bomb to give a brown mixture. After heating at 70 °C for 3.5 d, the mixture was evacuated to about 5–10 mL, then precipitated into a vortex of 75 mL hexanes. The solid brown precipitate was isolated on a frit.
Using Ti(NMe2)4 as the precatalyst, yield of (2a): 229 mg (36%). 1H NMR: 8.0–6.3 (br, 15H, ArH), 6.1 (d, 1.3H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, 3JH–H = 8 Hz), 3.8 (br, 1H, CH2), 2.8 (br, 1.1H, NH), 1.7 (br, 1.1H, N(CH3)2). The integration data suggest that the ratio of enamine : imine is ca. 2.7 : 1, and that the ratio of enamine + imine : NMe2 end group is ca. 10 : 1. 13C{1H} NMR (partial): 167.4 (CN), 138.7 (Ar), 133.3 (Ar), 132.9 (Ar), 131.9 (Ar), 131.8 (Ar), 128.9 (Ar), 128.6 (Ar), 128.2 (Ar), 124.8 (Ar), 101.7 (CH), 48.8 (N(CH3)2), 36.0 (CH2). FT-IR (from CH2Cl2 soln, cm−1): ν(N–H) 3384 (weak), no peaks detected from 2700 to 1650, ν(CN) 1620 (med., sharp), ν(Ph) 1592 (strong, sharp), ν(Ph) 1515 (med., sharp). UV/Vis (CH3CN, ca. 10−5 M): λmax = 311 nm. For GPC analysis, oligomer (2a) was placed in THF under air and filtered to remove insoluble particulates prior to acquiring GPC data. Since (2a) is partly soluble in THF, some of the sample was removed upon filtration. GPC (versus polystyrene standards): Mn 730, Mw 1540. GPC (laser light scattering detection): Mn 1230, Mw 1680. MALDI-TOF MS: highest molecular weight species 2944 = 15 × (193) + 45 m/z.
CH, 3JH–H = 8 Hz), 3.8 (br, 1H, CH2), 2.8 (br, 1.1H, NH), 1.7 (br, 1.1H, N(CH3)2). The integration data suggest that the ratio of enamine : imine is ca. 2.7 : 1, and that the ratio of enamine + imine : NMe2 end group is ca. 10 : 1. 13C{1H} NMR (partial): 167.4 (CN), 138.7 (Ar), 133.3 (Ar), 132.9 (Ar), 131.9 (Ar), 131.8 (Ar), 128.9 (Ar), 128.6 (Ar), 128.2 (Ar), 124.8 (Ar), 101.7 (CH), 48.8 (N(CH3)2), 36.0 (CH2). FT-IR (from CH2Cl2 soln, cm−1): ν(N–H) 3384 (weak), no peaks detected from 2700 to 1650, ν(CN) 1620 (med., sharp), ν(Ph) 1592 (strong, sharp), ν(Ph) 1515 (med., sharp). UV/Vis (CH3CN, ca. 10−5 M): λmax = 311 nm. For GPC analysis, oligomer (2a) was placed in THF under air and filtered to remove insoluble particulates prior to acquiring GPC data. Since (2a) is partly soluble in THF, some of the sample was removed upon filtration. GPC (versus polystyrene standards): Mn 730, Mw 1540. GPC (laser light scattering detection): Mn 1230, Mw 1680. MALDI-TOF MS: highest molecular weight species 2944 = 15 × (193) + 45 m/z.
Using Ti(NEt2)4 as the precatalyst, yield of (2b): 494 mg (77%). 1H NMR: 8.0–6.2 (br, 15H, ArH), 6.1 (d, 1H, CH, 3JH–H = 8 Hz), 3.8 (br, 1H, CH2), 3.0 (q, 0.5H, N(CH2CH3)2), 2.8 (br, 1H, NH), 1.0–0.8 (m, 0.8H, N(CH2CH3)2). The integration data suggest that the ratio of enamine : imine is ca. 2 : 1, and that the ratio of enamine + imine : NEt2 end group is ca. 12 : 1. 13C{1H} NMR (partial): 165.0 (CN), 133.3 (Ar), 131.9 (Ar), 131.8 (Ar), 128.9 (Ar), 128.6 (Ar), 126.5 (Ar), 101.7 (CH), 44.7 (N(CH2CH3)2), 31.5 (CH2), 21.8 (N(CH2CH3)2). FT-IR (from CH2Cl2 soln, cm−1): ν(N–H) 3384 (weak), ν(CN) 1620 (weak), ν(Ph) 1592 (weak), ν(Ph) 1515 (weak). MALDI-TOF MS: highest molecular weight species 2005 = 10 × (193) + 73 m/z.
Synthesis of PhCHCPh(NHPh) (3), PhCH2CPh(NPh) (4), PhCHCPh(NH(C6H3iPr2)) (5), and PhCH2CPh(N(C6H3iPr2)) (6)
These reactions were carried out under identical conditions. Diphenylacetylene (891 mg, 5.00 mmol) and Ti(NMe2)4 (112 mg, 0.500 mmol) were combined in a 50 mL bomb which was wrapped in aluminium foil. Freshly degassed aniline (0.46 mL, 0.47 mg, 5.0 mmol, for compounds (3) and (4)) or 2,6-diisopropylaniline (0.94 mL, 0.88 g, 5.0 mmol, for compounds (5) and (6)) was added via syringe. The reaction mixture was heated at 70 °C for 65 h, and volatile materials were removed in vacuo to afford a brown oil.
For (3) and (4),20 C20H17N. The ratio of (3) : (4) is 47 : 53. X-Ray quality crystals of (4) were obtained from the reaction mixture. 1H NMR: (3): 7.61–7.55 (m, C6H5), 7.17–7.12 (m, C6H5), 6.88–6.82 (m, C6H5), 6.69–6.65 (m, C6H5), 6.34 (s, C6H5), 5.61 (s, CHPh), 5.09 (s, NH); (4): 8.27–8.20 (m, C6H5), 7.73–7.67 (m, C6H5), 7.21–7.16 (m, C6H5), 3.83 (s, CH2). EI-MS (m/z): 271.1 (2%) ([M]+), 180.1 (17%) ([M]+ − CH2Ph), 178.1 (100%) (PhCCPh). HRMS: C20H17N mass 271.1366, calcd mass 271.1361. FT-IR (mixture of (3) and (4), deposited from C6D6 soln, cm−1): ν(N–H) 3392 (med., broad), ν(CN) 1626 (strong, sharp), ν(CC) 1600 (strong, sharp).
For (5)39 and (6), C26H29N. The ratio of (5) : (6) is 5: 95. X-Ray quality crystals of (6) grew from the reaction mixture. 1H NMR: (5): 7.53–7.51 (m, ArH), 7.1–6.9 (m, ArH), 5.30 (s, CHPh), 4.38 (br s, NH), 3.39 (septet, CH(CH3)2), 1.45 (d, CH(CH3)a(CH3)b), 1.30 (d, CH(CH3)a(CH3)b); (6): 8.07–8.05 (m, C6H5), 7.20–7.12 (m, NC6H3 and C6H5), 6.91–6.81 (m, C6H5), 3.86 (s, CH2), 2.88 (septet, CH(CH3)2), 1.21 (d, CH(CH3)a(CH3)b), 1.10 (d, CH(CH3)a(CH3)b). 13C{1H} NMR: (6) (partial): 165 (CN), 135 (N-ipso-C), 131.6 (ArH), 130.1 (ArH), 128.9 (ArH), 128.3 (ArH), 128.0 (ArH), 126.1 (ArH), 123.8 (ArH), 123.0 (ArH), 36.4 (CH2), 28.5 (CH(CH3)2), 23.6 (CH(CH3)a(CH3)b), 21.8 (CH(CH3)a(CH3)b). EI-MS (m/z): 355.2 (7%) ([M]+); 264.2 (100%) ([M]+ − CH2Ph), 178.1 (82%) (PhCCPh). HRMS: C26H29N mass 355.2307, calcd mass 355.2300. FT-IR (mixture of (5) and (6), deposited from C6D6 soln, cm−1): ν(N–H) 3399 (weak), ν(CN) 1627 (med., sharp), ν(CC) 1600 (med., sharp). FT-IR ((6) deposited from C6D6 soln, cm−1): ν(CN) 1627 cm−1 (strong, sharp).
Synthesis of CH2C(Ph)(NHPh) (7m), CHPhCH(NHPh) (7am), CH3C(Ph)(NPh) (8m), PhCH2CH(NPh) (8am), (PhCHCH)2(NPh) (9), CH2C(Ph)(NH(C6H3iPr2)) (10m), CHPhCH(NH(C6H3iPr2)) (10am), CH3C(Ph)(N(C6H3iPr2)) (11m), and PhCH2CH(N(C6H3iPr2)) (11am)
These reactions were carried out under identical conditions. Phenylacetylene (511 mg, 5.00 mmol) and Ti(NMe2)4 (112 mg, 0.500 mmol) were combined in a 50 mL bomb which was wrapped in aluminium foil. Freshly degassed aniline (0.46 mL, 0.47 mg, 5.0 mmol, for compounds (7), (8), and (9)) or 2,6-diisopropylaniline (0.94 mL, 0.88 g, 5.0 mmol, for compounds (10) and (11)) was added via syringe. The reaction mixture was heated at 70 °C for 60 h, and volatile materials were removed in vacuo to afford a brown oil.
For (7), (8),40 and (9). X-Ray quality crystals of (9) were obtained from the reaction mixture. The ratio of (7m) : (7am) : (8m) : (8am) : (9) is ca. 2 : 48 : 11 : 15 : 24. 1H NMR (partial): 6.50 (dd, 3JH–H = 14 Hz, 3JH–H = 7 Hz, CHNH, (7am)), 5.47 (d, 3JH–H = 14 Hz, CHPh, (7am)), 4.49 (d, 2JH–H = 2 Hz, CHaHb, (7m)), 3.46 (d, 3JH–H = 8 Hz, CH2, (8am)), 2.88 (s, CH3, (8m)). EI-MS for mixture of products (m/z): 297.2 (2%) ([(9)]+); 195.1 (92%) ([(7) and/or (8)]+), 147.1 (9%) ([H2CC(Ph)(NMe2) or PhCHCH(NMe2)]+). FT-IR (mixture of (7), (8), and (9), deposited from C6D6 soln, cm−1): ν(N–H) 3401 (med., broad), ν(CN) 1634 (v strong, sharp), ν(CC) 1595 (v strong, sharp), ν(C–N) 1275 (med., sharp). For crystals of (9): 1H NMR: 7.28–7.25 (m, 4H, CC6H5), 7.03–6.99 (m, 4H, CC6H5), 7.01–6.95 (m, 2H, m-NC6H5), 6.72–6.69 (d, 2H, PhC(H)C(H)N, 3JH–H = 12 Hz), 6.25–6.20 (m, 2H, CC6H5), 6.21–6.18 (m, 2H, o-NC6H5), 5.75 (d, 2H, PhC(H)C(H)N, 3JH–H = 12 Hz). 13C{1H} NMR (partial): 142.2 (ipso-NC6H5), 131.6 (m-NC6H5), 130.8 (CC6H5), 129.7 (CC6H5), 128.9 (p-NC6H5), 127.8 (CC6H5), 126.4 (CC6H5), 126.0 (CC6H5), 124.0 (CC6H5), 120.5 (PhC(H)C(H)N), 118.6 (ipso-CC6H5), 114.3 (o-NC6H5), 105.2 (PhC(H)C(H)N). EI-MS (m/z): 297.2 (22%) ([M]+), 295.1 (100%) ([M]+ − 2H). HRMS: C22H19N mass 297.1513, calcd mass 297.1517. FT-IR (deposited from C6D6 soln): ν(CC) 1594 cm−1 (strong, sharp), ν(C–N) 1275 cm−1 (strong, sharp). UV/Vis (CH3CN, ca. 10−5 M): λmax = 357 nm. Anal. Calcd for C22H19N: C, 87.85; H, 6.44; N, 4.71. Found: C, 88.46; H, 6.48; N, 5.27%.
For (10) and (11). The ratio of (10m) : (10am)39 : (11m) : (11am)39 is 3 : 11 : 40 : 46. 1H NMR (partial): 7.41 (t, 3JH–H = 5 Hz, CHN, (11am)), 6.65 (dd, 3JH–H = 14 Hz, 3JH–H = 7 Hz, CHNH, (10am)), 5.36 (d, 2JH–H = 2 Hz, CHaHb, (10m)), 5.19 (d, 3JH–H = 14 Hz, CHPh, (10am)), 4.38 (d, 2JH–H = 2 Hz, CHaHb, (10m)), 4.16 (br, NH, (10am)), 3.51 (d, 3JH–H = 5 Hz, CH2, (11am)), 2.35 (s, CH3, (11m)). EI-MS (m/z): 279.2 (3%) ([M]+), 264.2 (5%) ([M]+ − Me), 188.1 (19%) ([M]+ − CH2Ph), 177.2 (29%) (2,6-diisopropylaniline), 162.1 (100%) (2,6-diisopropylaniline − Me). HRMS: C20H25N mass 279.1981, calcd mass 279.1987. FT-IR (mixture of (10m), (10am), (11m), and (11am), deposited from C6D6 soln, cm−1): ν(N–H) 3401 (weak), ν(CN) 1620 (strong, sharp), ν(C–N) 1264 (med., sharp).
X-Ray data collection and reduction
Crystals were manipulated and suspended in Paratone or mounted in capillaries in a glove box, thus maintaining a dry, O2-free environment for each crystal. Diffraction experiments were performed on a Siemens SMART System CCD diffractometer. The data (4.5° < 2θ < 45–50.0°) were collected in a hemisphere of data in 1329 frames with 10 second exposure times. The observed extinctions were consistent with the space groups in each case. A measure of decay was obtained by re-collecting the first 50 frames of each dataset. The intensities of reflections within these frames showed no statistically significant change over the duration of the data collections. The data were processed using the SAINT and SHELXTL processing packages. An empirical absorption correction based on redundant data was applied to each dataset. Subsequent solution and refinement were performed using the SHELXTL solution package.Structure solution and refinement
Non-hydrogen atomic scattering factors were taken from the literature tabulations.41 The atom positions were determined using direct methods employing the SHELXTL direct methods routine. The refinements were carried out by using full-matrix least squares techniques on F, minimizing the function ω (Fo − Fc)2 where the weight ω is defined as 4Fo2/2σ (Fo2) and Fo and Fc are the observed and calculated structure factor amplitudes, respectively. In the final cycles of each refinement, all non-hydrogen atoms were assigned anisotropic temperature factors in the absence of disorder or insufficient data. In the latter cases atoms were treated isotropically. For compounds (4) and (6), the H atoms on C(2) were located and refined. All other C–H atom positions were calculated and allowed to ride on the carbon to which they are bonded assuming a C–H bond length of 0.95 Å. H-Atom temperature factors were fixed at 1.10 times the isotropic temperature factor of the C-atom to which they are bonded, and the H-atom contributions were calculated, but not refined. The locations of the largest peaks in the final difference Fourier map calculation as well as the magnitude of the residual electron densities in each case were of no chemical significance. Crystallographic parameters for compounds (4), (6), and (9) are given in Table 1.| (4) | (6) | (9) | |
|---|---|---|---|
| a Data collected with Mo Kα radiation (λ = 0.71069 Å). R = Σ(Fo − Fc)/ΣFo. b R w={Σ[w(Fo2 − Fc2)2]/Σ[w(Fo)2]}½. | |||
| Formula | C20H17N | C26H29N | C22H19N |
| Formula weight | 271.35 | 355.50 | 297.38 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21/n | P21/c | Pbca |
| a/Å | 5.5976(11) | 11.2839(5) | 16.1137(8) |
| b/Å | 8.3168(6) | 11.8464(5) | 8.8958(4) |
| c/Å | 31.769(3) | 15.6094(7) | 22.9830(12) |
| α/deg | |||
| β/deg | 92.784(4) | 94.908(2) | |
| γ/deg | |||
| V/Å3 | 1477.2(2) | 2078.91(16) | 3294.5(3) |
| Z | 4 | 4 | 8 |
| Temp./°C | −100 | −100 | −100 |
| d(calc)/g cm−1 | 1.220 | 1.136 | 1.199 |
| Abs coeff., µ/cm−1 | 0.070 | 0.065 | 0.0418 |
| Data collected | 9575 | 44347 |
11649 |
| Data Fo2 > 3σ(Fo2) | 2584 | 6382 | 2900 |
| Variables | 198 | 252 | 208 |
| R | 0.0410 | 0.0461 | 0.0566 |
| R w | 0.1009 | 0.1320 | 0.1711 |
| GOF | 1.029 | 1.032 | 1.040 |
Results and discussion
Hydroamination oligomerization
Reaction of monomer (1) with 0.10 equiv. of Ti(NMe2)4 at 70 °C in toluene for ca. 80–90 h resulted in hydroamination oligomerization to give (2a) (Scheme 3). Oligomer (2a) was isolated in 36% yield upon precipitation into a vortex of hexanes. Solubility of low molecular weight species in hexanes may account for the low yield. The broad peaks in the 1H NMR spectrum of (2a) are indicative of an oligomer. Signals attributed to enamine CH and imine CH2 moieties are observed at 6.1 and 3.8 ppm, respectively, in a ratio of ca. 2.7 : 1. The corresponding peaks in the 13C NMR spectrum are observed at 167 (CN), 101 (PhCH), and 36 ppm (CH2), assigned by HSQC and HMBC experiments. The IR spectrum of oligomer (2a) shows the absence of peaks between 2700 and 1650 cm−1 consistent with the loss of CC fragments. The peaks at 3384 and 1620 cm−1 are attributed to the N–H stretch of a secondary amine and the CN stretch of an imine moiety, respectively.42,43 The UV/Vis spectrum of oligomer (2a) is very similar to that of monomer (1), suggesting minimal conjugation along the oligomer chain. Relative to polystyrene standards, GPC data for (2a) indicate Mn = 730 and Mw = 1540, corresponding to a number-average degree of polymerization (DPn) of 4. Using laser light scattering detection, GPC data indicate a higher DPn of 6 (Mn = 1230 and Mw = 1680).
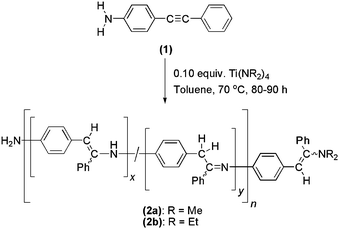 | ||
| Scheme 3 Hydroamination oligomerization of compound (1) to synthesize oligomers (2). | ||
According to the Carothers equation, the degree of polymerization depends on the stoichiometry of the functional groups and on the extent of the reaction. Since monomer (1) provides exact stoichiometry, the low degree of polymerization is likely to result from an incomplete reaction. However, it should be noted that the molecular weight values may be underestimated since GPC data were acquired under air in THF. Samples of (2a) were not completely soluble in THF and were filtered to remove insoluble material (possibly including higher molecular weight oligomers) prior to GPC analysis. Moreover, the residual water present in air may hydrolyze the backbone,43 leading to chain degradation. However, attempts at reducing the water-sensitive imine and enamine fragments of the oligomer backbone were inconclusive.
Literature precedent suggests that the mechanism of hydroamination for this group 4 catalyst follows a [2 + 2] cycloaddition pathway (Scheme 1).2–4,6,7,44 This mechanism accounts for chain growth because the hydroamination product after each turnover contains a primary amine functionality which can form a new titanium–imide species. However, this mechanism does not account for the observation of the major set of peaks in the MALDI-TOF mass spectrum of (2a) (Fig. 1), located at 193n + 45 m/z. While 193n corresponds to an integral number (n) of monomer units (mass = 193 Daltons), 45 corresponds to HNMe2, could result from insertion of alkyne into the Ti–NMe2 bond of the catalyst followed by protonolysis. Catalysis performed using Ti(NEt2)4 resulted in oligomer (2b), with peaks at 193n + 73 m/z in the MALDI-TOF mass spectrum. NMR data for oligomers (2a) and (2b) show resonances corresponding to the NR2 fragment and end group analysis by 1H NMR indicates approximately 10–12 repeat units in the chains. The incorporation of one secondary amine fragment is suggestive of a σ-bond insertion mechanism (Scheme 2).
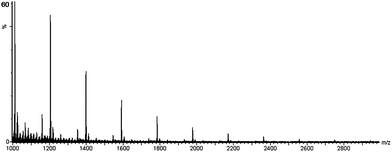 | ||
| Fig. 1 MALDI-TOF mass spectrum of (2a). Major peaks are located at 193n + 45 m/z, where 193 Daltons is the mass of the monomer, n is an integer, and 45 Daltons corresponds to addition of HNMe2. | ||
Model compounds for hydroamination oligomerization
To glean information regarding the mechanism of hydroamination, model reactions were performed using diphenylacetylene or phenylacetylene and aniline, 2,6-diisopropylaniline, N-methylaniline or diethylamine. No reaction was observed for either alkyne with N-methylaniline or diethylamine in the presence of Ti(NMe2)4 at 70 °C for 3–4 days. This lack of reactivity using a secondary amine is inconsistent with a mechanism that relies solely on σ-bond insertion, implying that the [2 + 2] cycloaddition mechanism is likely operative for the growth of the oligomer chain using this titanium(IV) catalyst.In the hydroamination reactions of diphenylacetylene with aniline or 2,6-diisopropylaniline (Scheme 4), a mixture of enamine (PhCHCPh(NHAr), (3) Ar = Ph or (5) Ar = C6H3iPr2) and imine (PhCH2CPh(NAr), (4) Ar = Ph or (6) Ar = C6H3iPr2) was formed (Scheme 3). The relative ratios of (3) : (4) and (5) : (6) are 47 : 53 and 5 : 95, respectively. The preferential formation of imines stands in contrast to several literature reports.21,39,45,46 Imines (4) and (6) were characterized by single-crystal X-ray diffraction studies (Fig. 2) and the hydrogen atoms on C(2) were unambiguously located and refined. Upon heating imine (6) to 70 °C for 4 days, with or without catalytic Ti(NMe2)4, no conversion to enamine (5) was detected by 1H NMR spectroscopy.
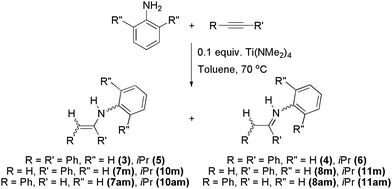 | ||
| Scheme 4 Synthesis of model compounds (3)–(8), (10) and (11). | ||
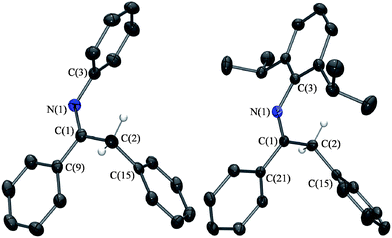 | ||
| Fig. 2 ORTEP drawings of (4) and (6), with ellipsoids drawn at the 50% probability level. All hydrogen atoms except those on C2 have been omitted for clarity. Selected bond lengths (Å) and angles (°) for (4): N(1)–C(1) 1.282(2), N(1)–C(3) 1.423(2), C(1)–C(2) 1.517(2), C(1)–C(9) 1.500(2), C(2)–C(15) 1.514(2), C(1)–C(2)–C(15) 114.3(1), C(2)–C(1)–N(1) 124.2(1), C(2)–C(1)–C(9) 118.6(1), N(1)–C(1)–C(9) 117.2(1), C(1)–N(1)–C(3) 120.9(1); (6): N(1)–C(1) 1.279(1), N(1)–C(3) 1.420(1), C(1)–C(2) 1.512(1), C(1)–C(21) 1.494(1), C(2)–C(15) 1.523(1), C(1)–C(2)–C(15) 115.81(8), C(2)–C(1)–N(1) 124.16(9), C(2)–C(1)–C(21) 116.86(8), N(1)–C(1)–C(21) 116.86(8), C(1)–N(1)–C(3) 124.43(8). | ||
Reactions using phenylacetylene and aniline or 2,6-diisopropylaniline (Scheme 4) afforded imines and enamines, which exhibited Markovnikov (M) or anti-Markovnikov (AM) regiochemistry.10 In general, the regiochemistry depends not only on the catalyst,39,45,47–52 but also on the amine49,50,52 and alkyne10,39,45,47,48,52 substrates. In the reaction of aniline and phenylacetylene, the 1H NMR and mass spectral data revealed a mixture of products, including CH2C(Ph)(NHPh) (7m), CHPhCH(NHPh) (7am), CH3C(Ph)(NPh) (8m), PhCH2CH(NPh) (8am) and the unexpected product (PhCHCH)2(NPh) (9). The ratio of these products in the reaction mixture was ca. 2 : 48 : 11 : 15 : 24. In the mass spectrum, H2CC(NMe2)(Ph) (or its anti-Markovnikov regioisomer CHPhCH(NMe2)) was also observed, as per literature precedent.40 Signals in the 1H NMR derived from the isomers of (7) and (8) were assigned on the basis of literature comparisons.39,40
Crystals of (9) were obtained from the reaction mixture, and their formulation was confirmed by two-dimensional NMR experiments and single crystal X-ray diffraction (Fig. 3). The bond distances and angles in (9) are typical of enamines.53,54 The N-atom is planar while the C3NC3 backbone is also very nearly planar. This planarity suggests donation of the N-lone pair into the extended π-system. This view is supported by λmax = 357 nm in the UV/Vis spectrum of (9), which is red-shifted dramatically from that of aniline (λmax = 230 nm). The formation of compound (9) most likely results from the reaction of (7am) or (8am) with an additional equivalent of phenylacetylene. The presence of compound (9) and H2CC(NMe2)(Ph) or CHPhCH(NMe2) implicates the insertion of phenylacetylene into a Ti–N σ-bond.
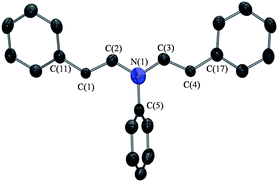 | ||
| Fig. 3 ORTEP drawing of (9), with ellipsoids drawn at the 50% probability level. All hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) for (9): N(1)–C(5) 1.506(4), N(1)–C(2) 1.393(3), N(1)–C(3) 1.415(3), C(1)–C(2) 1.357(3), C(3)–C(4) 1.346(3), C(1)–C(11) 1.423(3), C(4)–C(17) 1.436(3), C(2)–N(1)–C(5) 120.9(2), C(3)–N(1)–C(5) 119.9(2), C(2)–N(1)–C(3) 119.2(2), N(1)–C(2)–C(1) 126.3(3), N(1)–C(3)–C(4) 125.5(3), C(2)–C(1)–C(11) 125.0(2), C(3)–C(4)–C(17) 126.7(2). | ||
The related reactions employing 2,6-diisopropylaniline yielded CH2C(Ph)(NHAr) (10m), CHPhCH(NHAr) (10am), CH3C(Ph)(NAr) (11m) and PhCH2CH(NAr) (11am) (where Ar = C6H3iPr2), as judged by 1H NMR and mass spectral data.39 The product distribution of (10m) : (10am) : (11m) : (11am) was determined to be 3 : 11 : 40 : 46. In this case, no 2 : 1 addition product analogous to (9) was formed.
Conclusions
Oligomer (2), formed by hydroamination, contains up to 15 repeat units in the chain, capped by one molecule of dialkylamine which originates from the Ti(NR2)4 (R = Me and Et) catalyst. Model reactions yielding (3)–(11) suggest that oligomer (2) contains both imine and enamine moieties, with minimal regioselectivity. These model systems also suggest that propagation is dominated by the [2 + 2] cycloaddition mechanism, while the incorporation of a terminal secondary amine group proceeds via insertion of alkyne into Ti–N σ-bonds.Acknowledgements
We thank Prof. Mark Nitz and Dr Richard Jagt for their help with MALDI-TOF mass spectrometry, and Prof. Derek P. Gates and Dr Kevin J. T. Noonan for their assistance with GPC measurements. Financial support from NSERC of Canada is gratefully acknowledged. DWS is grateful for the award of a Canada Research Chair and a Killam Research Fellowship.Notes and references
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef.
- A. L. Odom, Dalton Trans., 2005, 225–233 RSC.
- N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS.
- A. V. Lee and L. L. Schafer, Eur. J. Inorg. Chem., 2007, 2245–2255 CrossRef.
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673–686 CrossRef CAS.
- S. Doye, Synlett, 2004, 1653–1672 CrossRef CAS.
- I. Bytschkov and S. Doye, Eur. J. Org. Chem., 2003, 935–946 CrossRef CAS.
- F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104–114 RSC.
- T. Straub, A. Haskel, T. G. Neyroud, M. Kapon, M. Botoshansky and M. S. Eisen, Organometallics, 2001, 20, 5017–5035 CrossRef CAS.
- R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407–1420 RSC.
- J.-J. Brunet and D. Neibecker, in Catalytic Heterofunctionalization: from Hydroamination to Hydrozirconation, ed. A. Togni and H. Grützmacher, Wiley-VCH Verlag GmbH, Weinheim, 2001 Search PubMed.
- T. Hamaya, Polym. Bull., 2000, 45, 199–206 CrossRef CAS.
- J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795–813 CrossRef CAS.
- M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042–2043 CrossRef CAS.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock, G. Kociok-Köhn and P. A. Procopiou, Inorg. Chem., 2008, 47, 7366–7376 CrossRef.
- M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670–9685 CrossRef CAS.
- S. Datta, P. W. Roesky and S. Blechert, Organometallics, 2007, 26, 4392–4394 CrossRef CAS.
- S. Datta, M. T. Gamer and P. W. Roesky, Organometallics, 2008, 27, 1207–1213 CrossRef CAS.
- C. Lorber, R. Choukroun and L. Vendier, Organometallics, 2004, 23, 1845–1850 CrossRef CAS.
- L. L. Anderson, J. Arnold and R. G. Bergman, Org. Lett., 2004, 6, 2519–2522 CrossRef CAS.
- P. J. Walsh, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1992, 114, 1708–1719 CrossRef CAS.
- A. M. Baranger, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 2753–2763 CrossRef CAS.
- S. Y. Lee and R. G. Bergman, Tetrahedron, 1995, 51, 4255–4276 CrossRef CAS.
- P. D. Knight, I. Munslow, P. N. O'Shaughnessy and P. Scott, Chem. Commun., 2004, 894–895 RSC.
- D. V. Gribkov and K. C. Hultzsch, Angew. Chem., Int. Ed., 2004, 43, 5542–5546 CrossRef CAS.
- S. Majumder and A. L. Odom, Organometallics, 2008, 27, 1174–1177 CrossRef CAS.
- B. D. Stubbert and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 6149–6167 CrossRef CAS.
- M. R. Gagné, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275–294 CrossRef CAS.
- S. B. Amin, S. Seo and T. J. Marks, Organometallics, 2008, 27, 2411–2420 CrossRef CAS.
- P. L. McGrane and T. Livinghouse, J. Am. Chem. Soc., 1993, 115, 11485–11489 CrossRef CAS.
- J. S. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 12220–12221 CrossRef CAS.
- G. Zi, L. Xiang and H. Song, Organometallics, 2008, 27, 1242–1246 CrossRef CAS.
- T. Hamann, E. Böhler and P. Swiderek, Angew. Chem., Int. Ed., 2009, 48, 4643–4645 CrossRef CAS.
- R. J. Young and P. A. Lovell, Introduction to Polymers, Nelson Thornes Ltd, Cheltenham, 2nd edn, 1991 Search PubMed.
- S. D. Hanton, Chem. Rev., 2001, 101, 527–569 CrossRef CAS.
- J. Libiszowski, A. Kowalski, R. Szymanski, A. Duda, J.-M. Racquez, P. Degée and P. Dubois, Macromolecules, 2004, 37, 52–59 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- J.-F. Létard, P. Guionneau, E. Codjovi, O. Lavastre, G. Bravic, D. Chasseau and O. Kahn, J. Am. Chem. Soc., 1997, 119, 10861–10862 CrossRef CAS.
- M. A. Esteruelas, A. M. López, A. C. Mateo and E. Oñate, Organometallics, 2005, 24, 5084–5094 CrossRef CAS.
- Y. Shi, J. T. Ciszewski and A. L. Odom, Organometallics, 2001, 20, 3967–3969 CrossRef.
- D. T. Cromer and J. T. Waber, International Tables of X-Ray Crystallography, 1974, vol. 4, pp. 71–147 Search PubMed.
- S. Eğe, Organic Chemistry: Structure and Reactivity, Houghton Mifflin Company, Boston, 4th edn, 1999 Search PubMed.
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- K. Weitershaus, B. D. Ward, R. Kubiak, C. Müller, H. Wadepohl, S. Doye and L. H. Gade, Dalton Trans., 2009, 4586–4602 RSC.
- M. A. Esteruelas, A. M. López, A. C. Mateo and E. Oñate, Organometallics, 2006, 25, 1448–1460 CrossRef CAS.
- V. Bertolasi, R. Boaretto, M. R. Chierotti, R. Gobetto and S. Sostero, Dalton Trans., 2007, 5179–5189 RSC.
- K. Takaki, S. Koizumi, Y. Yamamoto and K. Komeyama, Tetrahedron Lett., 2006, 47, 7335–7337 CrossRef CAS.
- M. L. Buil, M. A. Esteruelas, A. M. López and A. C. Mateo, Organometallics, 2006, 25, 4079–4089 CrossRef CAS.
- C. Li, R. K. Thomson, B. H. Gillon, B. O. Patrick and L. L. Schafer, Chem. Commun., 2003, 2462–2463 RSC.
- Z. Zhang and L. L. Schafer, Org. Lett., 2003, 5, 4733–4736 CrossRef CAS.
- A. Tillack, V. Khedkar, H. Jiao and M. Beller, Eur. J. Org. Chem., 2005, 5001–5012 CrossRef CAS.
- Y. Shi, C. Hall, J. T. Ciszewski, C. Cao and A. L. Odom, Chem. Commun., 2003, 586–587 RSC.
- K. L. Brown, L. Damm, J. D. Dunitz, A. Eschenmoser, R. Hobi and C. Kratky, Helv. Chim. Acta, 1978, 61, 3108–3135 CrossRef CAS.
- A. G. Cook, Enamines: Synthesis, Structure and Reactions, Marcel Dekker Inc., New York, 2nd edn, 1988 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra, UV/Vis spectra, and MALDI-TOF mass spectra. CCDC reference numbers 761296–761298. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0py00120a |
| This journal is © The Royal Society of Chemistry 2010 |