Iodine-containing radio-opaque polyphosphazenes†
Mark D.
Hindenlang
a,
Anton A.
Soudakov
a,
Gregory H.
Imler
a,
Cato T.
Laurencin
bc,
Lakshmi S.
Nair
bc and
Harry R.
Allcock
*a
aDepartment of Chemistry, The Pennsylvania State University, 104 Chemistry Research Building. University Park, PA 16802, USA. E-mail: hra@chem.psu.edu
bDepartment of Orthopedic Surgery, University of Connecticut Health Center, Farmington, CT 06030, USA
cDepartment of Chemical, Materials and Biomolecular Engineering, University of Connecticut, Storrs, CT 06269-3222, USA
First published on 30th July 2010
Abstract
The first poly(organophosphazenes) with iodinated side groups for possible use as radio-opaque biomaterials are reported. Depending on the types of side groups present, these polymers may be biostable or bioerodible. Single-substituent polymers with 4-iodophenoxy or 4-iodophenylanaline ethyl ester units as the only side groups were prepared. Although a single-substituent polymer with 3,5-diiodotyrosine ethyl ester groups was difficult to synthesize, probably because of steric hindrance, mixed-substituent polymers that contained the non-iodinated ethyl esters of glycine, alanine, or phenylalanine plus a corresponding iodinated substituent could be synthesized. 31P, 1H, and 13C NMR spectroscopy techniques were used to follow the linkage of the side groups to the phosphazene skeleton and to establish the ratios of the different side groups. Hydrolysis of the iodo-amino acid/non-iodinated amino acid side group species in deionized water at 37 °C followed a bulk hydrolysis profile, with the rates dependent on the structure of the side groups. The effectiveness of these polymers as X-ray opaque materials was examined by the use of the poly(organophosphazenes) and conventional organic polymers as filters for copper Kα or rhenium–tungsten–molybdenum radiation. The phosphazene polymers that contained iodine in the side groups were opaque to X-rays, whereas the conventional organic polymers were essentially transparent to the same radiation.
Introduction
The increasing use of polymers for implantation in the human body is accompanied by the challenge of monitoring these devices and their lifetimes by standard X-ray imaging techniques. Most polymers do not absorb significantly in the X-ray region of the electromagnetic spectrum, but recent reports have shown that polymers can be made opaque to X-rays. Initially, these examples were metal-oxide–polymer composites which require large amounts of inorganic salts often with more than 25 wt% of barium sulfate or zirconium dioxide incorporated as a composite. High inorganic salt loading has a detrimental effect on the physical properties of the composite, and cyto-toxicity issues arise resulting from long-term exposure and leaching.1–6 A second method for rendering polymers radio-opaque is via a chemical incorporation of a radio-contrast dye, typically containing bromine or iodine.4–13 Although the resultant polymers are substantially more biocompatible than composite polymer systems, they are hydrogels which lack the necessary physical properties,3,13 are too susceptible to hydrolytic degradation,7 or are too hydrolytically stable for implants which remodel over time.4,5,9 In this article we report the introduction of iodine into the side group structure of poly(organophosphazenes) to facilitate radiological imaging of these polymers.Poly(organophosphazenes) are a broad class of hybrid inorganic–organic polymers usually produced by chlorine-replacement reactions carried out on poly(dichlorophosphazene) (1). The nucleophiles used for chlorine replacement include alkoxides, aryloxides, or amines, and the different side groups control the properties and final uses of the polyphosphazene.14 It is also possible to generate mixed-substituent polyphosphazenes in which two or more different organic groups are linked to the phosphazene backbone. The emerging applications for polyphosphazenes include battery and solar cell electrolytes,15,16 optical materials,17,18 bioinert elastomers,19–21 and bioerodible materials.22,23
Previous work in our program showed that specific poly(organophosphazenes) are biocompatible while being either biostable or biodegradable. For example, the biostable polymer, poly(diphenoxyphosphazene) (2), is resistant to hydrolytic degradation at body pH.24 Similarly, poly[bis(2,2,2-trifluoroethoxy)phosphazene] (6), is biostable, hydrophobic, and borders on superhydrophobic.25 On the other hand, polyphosphazenes with amino acid ethyl ester side groups linked through the amino terminus are hydrolytically sensitive and degrade to biologically benign small molecules. The steric bulk and hydrophobicity of the group at the α-position of the amino acid govern the rate of hydrolysis of these polymers.26–29 Polyphosphazenes are an attractive choice for X-ray opaque applications. The presence of phosphorus atoms in the polymer backbone, the use of various side groups that contain heavy elements such as iodine, and the demonstrated tune-ability of hydrolytic degradation provide potential starting points for polyphosphazenes to be utilized in the field of radio-opaque polymers. This study has laid the groundwork for the development of biocompatible polyphosphazenes that are radio-opaque.
Poly[bis(4-iodophenoxy)phosphazene] (5), poly(diethoxyphosphazene) (7), and species 2 and 6, were investigated initially as model systems. These polymers were exposed to Cu Kα and Re–W–Mo medical X-rays to provide a baseline of radio-opacity. Information gathered from the synthesis and characterization of these polymers was then applied to the design and synthesis of iodine-containing radio-opaque amino acid ester polyphosphazenes.
Experimental
Reagents and equipment
All synthesis reactions were carried out under a dry argon atmosphere using standard Schlenk line techniques. Tetrahydrofuran (EMD), 1,4-dioxane (EMD), and triethylamine (EMD) were dried using solvent purification columns.30 Ethanol (EMD) and 2,2,2-trifluoroethanol (Aldrich) were distilled from CaH2 and were stored under dry nitrogen. The synthesis of 4-iodophenylalanine ethyl ester by a slight modification of a previously published method gave the desired product in quantitative yields.31 3,5-Diiodotyrosine ethyl ester hydrochloride (Senn Chemicals), glycine ethyl ester hydrochloride (Bachem), alanine ethyl ester hydrochloride (Chem-Impex), phenylalanine ethyl ester hydrochloride (Chem-Impex), and phenylalanine (Bachem) were used as received. Poly(dichlorophosphazene) was prepared by the thermal ring-opening polymerization of recrystallized and sublimed hexachlorocyclotriphosphazene (Fushimi Chemical Co., Japan) in evacuated Pyrex tubes at 250 °C. Chemicals and polymers not mentioned above were used as received without purification. 31P, 13C, and 1H NMR spectra were obtained with use of a Bruker 360 WM instrument operated at 145 MHz, 90 MHz, and 360 MHz, respectively. Glass transition temperatures were measured with a TA Instruments Q10 differential scanning calorimetry apparatus with a heating rate of 10 °C min−1 and a sample size of ca. 10 mg. Gel permeation chromatograms were obtained using a Hewlett-Packard HP 1100 gel permeation chromatograph equipped with two Phenomenex Phenogel linear 10 columns and a Hewlett-Packard 1047A refractive index detector. The samples were eluted at 1.0 mL min−1 with a 10 mM solution of tetra-n-butylammonium nitrate in THF. The elution times were calibrated with polystyrene standards.Synthesis of polymers 2–7
Polymers 2–7 were synthesized in a similar fashion, with the synthesis of polymer 4 described as an example. Poly(dichlorophosphazene) (5.00 g, 0.0431 mol) was dissolved in 500 mL of dry 1,4-dioxane. Sodium metal (1.55 g, 0.0647 mol) was suspended in 200 mL of dry 1,4-dioxane, and phenol (6.08 g, 0.0647 mol) was added. This suspension was stirred until the sodium metal had been consumed by the reaction. Half of this solution was added dropwise to the polymer solution. The mixture was refluxed for 24 hours as the progress of the reaction was monitored by 31P NMR spectroscopy. Sodium metal (0.99 g, 0.0431 mol) was suspended in 200 mL of dry 1,4-dioxane and allowed to react with 4-iodophenol (9.49 g, 0.0431 mol) until the sodium metal had reacted. This solution was added dropwise to the polymer reaction mixture and the solution was refluxed while the progress of substitution was monitored by 31P NMR spectroscopy. The reaction was completed by dropwise addition of the remaining sodium phenoxide solution. When complete chlorine replacement was determined by 31P NMR spectroscopy, most of the solvent was removed in vacuo, and the residue was precipitated into water. Polymer 4 was purified by repeated precipitations from THF into water (2×) and hexanes (2×) and was isolated as an off-white polymer (yield of 79%) soluble in THF and dioxane. Characterization data for polymers 2–7 are shown in Table 1.| Polymer | Side group | 31P NMR (ppm) | 13C NMR (ppm) | 1H NMR (ppm) | M w (×103 g mol−1) | T g (lit.14) (°C) |
|---|---|---|---|---|---|---|
| 2 | 100% OPh | −16.47 | 116.1, 120.8, 130.2, 151.8 | 6.87 (3H), 7.43 (2H) | 1042 | −5.63 (−8) |
| 3 | 75/25% OPh/OPh-I | −16.83 | 89.2, 121.5, 124.4, 125.6, 129.8, 140.1, 151.9 | 6.48 (1H), 6.75 (3H), 6.92 (4.5H), 7.14 (1H) | 3589 | −6.33 |
| 4 | 50/50% OPh/OPh-I | −16.64 | 88.8, 121.9, 124.2, 125.2, 130.2, 139.2, 152.3 | 6.49 (2H), 6.78 (2H), 6.90 (3H), 7.14 (2H) | 1912 | 18.90 |
| 5 | 100% OPh-I | −17.04 | 88.7, 116.9, 139.1, 152.1 | 6.52 (2H), 7.08 (2H) | 916 | 40.08 (43) |
| 6 | 100% Trifluoroethoxy | −7.42 | 59.1, 60.3, 62.0, 63.2 (CH2), 108.2, 119.2, 128.9, 140.8 (CF3) | 4.63 (2H) | 762 | −64.28 (−66) |
| 7 | 100% Ethoxy | −6.42 | 17.9 (CH3), 58.2 (CH2) | 4.12 (2H), 1.22 (3H) | 473 | −85.62 (−84) |
| 8 | 25/75% I2-Tyr/Gly | 3.34 | 13.5 (2CH3), 39.4 (1.5CH2), 55.8 (2CH2), 61.2 (0.5CH), 126.3, 127.9, 130.2, 137.8 (4Ar), 174.1 (2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
1.24 (6H), 3.27 (1H), 3.79 (3H), 3.94 (0.5H), 4.14 (4H), 6.89 (1H) | 123 | −2.31 |
| 9 | 25/75% I2-Tyr/Ala | 1.96 | 13.2 (2CH3), 19.5 (1.5CH3), 43.6 (2CH/CH2), 55.4 (2CH2), 60.9 (0.5CH) 126.6, 128.3, 129.5, 137.6 (4Ar), 174.1 (2CO) |
1.22 (6H), 1.39 (4.5H), 3.24 (1H), 3.56 (0.5H), 3.60 (1.5H), 4.14 (4H), 6.89 (1H) | 137 | 20.60 |
| 10 | 25/75% I2-Tyr/Phe | 0.72 | 14.1 (2CH3), 41.1 (2CH2), 55.7 (2CH2), 60.8 (2CH), 126.5, 128.2, 129.9, 137.8 (4Ar), 174.1 (2CO) |
0.88 (6H), 3.01 (4H), 3.82 (4H), 4.30 (2H), 7.11 (9.5H) | 199 | 52.52 |
| 12 | 50/50% I-Phe/Gly | −2.93 | 13.5 (2CH3), 39.4 (CH2), 56.2 (CH2), 61.2 (CH2), 92.6, 131.4, 137.1 (3Ar), 174.1 (2CO) |
1.24 (6H), 3.31 (2H), 3.59 (2H), 4.09 (4H), 6.93 (2H), 7.47 (2H) | 246 | 34.91 |
| 13 | 50/50% I-Phe/Ala | −1.86 | 13.2 (2CH3), 19.5 (CH3), 43.6 (CH/CH2), 55.4 (2CH2), 61.1 (CH), 92.6, 131.4, 137.1 (3Ar), 174.1 (CO) |
1.23 (6H), 1.42 (3H), 3.31 (2H), 3.59 (2H), 4.09 (4H), 6.93 (2H), 7.50 (2H) | 126 | 35.81 |
| 14 | 50/50% I-Phe/Phe | −2.31 | 14.1 (2CH3), 41.1 (2CH2), 55.7 (2CH2), 60.8 (2CH), 92.5, 128.2, 129.9, 137.8 (4Ar), 174.1 (2CO) |
1.25 (6H), 3.49 (4H), 3.74 (2H), 4.09 (4H), 7.02–7.80 (9H) | 102 | 37.65 |
| 15 | 100% I-Phe | −2.47 | 14.2 (CH3), 40.3.0 (CH2), 55.7 (CH2), 61.4 (CH), 92.6, 131.4, 137.1 (3Ar), 174.1 (CO) |
0.98 (3H), 3.00 (2H), 3.93 (3H), 6.92 (2H), 7.49 (2H) | 120 | 38.75 |
Synthesis of polymers 8–14
Polymers 8–14 were synthesized in a similar manner, with the preparation of polymer 9 described as an example. Poly(dichlorophosphazene) (5.00 g, 0.0431 mol) was dissolved in dry THF (500 mL). Alanine ethyl ester hydrochloride (11.58 g, 0.0754 mol) was suspended in 200 mL of dry THF, and triethylamine (15.26 g, 0.151 mol) was added. This suspension was refluxed for 24 hours, then filtered, and half of the reaction mixture was added dropwise to the polymer solution. The reaction mixture was refluxed and monitored by 31P NMR spectroscopy. 3,5-Diiodotyrosine ethyl ester hydrochloride (10.72 g, 0.0216 mol) was suspended in 200 mL of dry THF, and triethylamine (4.36 g, 0.0432 mol) was added. This suspension was refluxed for 24 hours, then filtered and added dropwise to the polymer solution. The mixture was refluxed for 24 hours, with the progress of the reaction again monitored by 31P NMR spectroscopy. Complete chlorine replacement was then achieved by addition of the remainder of the alanine ethyl ester solution followed by heating the reaction mixture at reflux. After confirmation of complete halogen replacement by 31P NMR spectroscopy, the reaction mixture was filtered, concentrated in vacuo, and the residue precipitated into hexanes. Polymer 9 was purified by dialysis against methanol (3 days) and, after solvent removal, was isolated as a pale yellow-orange polymer (yield of 82%). The resulting polymers are soluble in a variety of organic solvents such as methanol, ethanol, THF, and dioxane. Characterization data for polymers 8–14 are shown in Table 1.Hydrolytic degradation of polymers 8–14
Polymers 8–14 (200 mg) were dissolved in 2 mL of chloroform (10% solution g mL−1). Films were solution-cast and air-dried for 24 hours, and then dried under reduced pressure for 7 days. Each film had a thickness of approximately 500 µm. Samples were distributed into 18 different test tubes containing 10 mL of deionized water (pH = 6.0). The tubes were contained in a constant shaker bath at 37 °C. Three samples were removed at each of the time points: 7, 14, 21, 28, 35, and 42 days. The weight loss was monitored for each solid sample and the pH of each hydrolysis medium was analyzed. 31P and 1H NMR techniques were utilized to follow the formation of any small molecule hydrolysis products dissolved in the aqueous medium. Silver nitrate and ninhydrin tests were employed to qualitatively determine the presence of phosphates and ammonia or amino acids, respectively.Radio opacity testing
Results and discussion
The synthesis of bioerodible polyphosphazenes with iodo-aryloxy side groups presents a challenge. Other investigators32–34 reported a method for iodination of aryloxyphosphazenes using triflic acid and a pyridine–iodine complex, conditions that would lead to decomposition of the amino acid ester polyphosphazenes that are the focus of this present work. All the polymers used in the present work were prepared by the replacement of chlorine atoms in poly(dichlorophosphazene) (1) by organic units that already contained iodinated substituents, thus avoiding decomposition of the polymers during a subsequent iodination process. Three different classes of poly(organophosphazenes) were synthesized. The first included single-substituent and mixed-substituent polymers with phenoxy-, 4-iodophenoxy-, trifluoroethoxy-, or ethoxy side groups. These are relatively straightforward syntheses carried out to give non-bioerodible polymers for use as controls in the evaluations of X-ray opacity. The linkage of the aryl units to the skeleton via Ar–O–P linkages generates hydrolytically stable polymers. The second group included mixed-substituent polymers that contained diiodotyrosine side groups together with non-iodinated amino acid ester substituents, all linked to the skeleton through the amino terminus of the side units. The third group of polymers contained iodophenylalanine either as single-substituent species or co-substituted with a non-iodinated amino acid ester.Synthesis and structural characterization of polymers 2–7
In the first group, polymers 2–7 were synthesized by the process shown in Scheme 1. The progress of each reaction was monitored using 31P NMR spectroscopy via the disappearance of the PCl2 peak at −17 ppm and growth of the peak that corresponds to P(OR)2 (indicated in Table 1), which represents full chlorine replacement. Formation of the single-substituent polymers 2, 6, and 7 usually required only 48 hours or less in refluxing THF to complete the substitution. However, the introduction of 4-iodophenoxy groups to form polymer 5 required at least a week for complete halogen replacement to be achieved even after replacing the THF solvent by refluxing dioxane at 100 °C.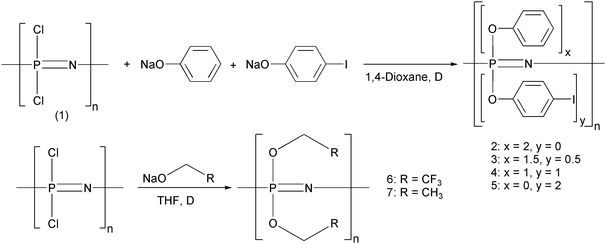 | ||
| Scheme 1 Synthesis and structures of polymers 2–7. | ||
The initial attempts to produce the mixed-substituent polymers 3 and 4 involved the dropwise addition of sodium 4-iodophenoxide to poly(dichlorophosphazene) in refluxing dioxane as the first substituent, followed by sodium phenoxide as the second substituent. However, loss of the 31P NMR signal after 24 hours of refluxing in dioxane indicated that the partially substituted polymer had precipitated from solution. A change of solvent to THF or benzene did not improve the solubility of this partly substituted polymer. Therefore, a three-step addition of the substituents was utilized for the synthesis of polymers 3 and 4 using boiling dioxane as a solvent. Half of the stoichometrically required sodium phenoxide was added first in a dropwise manner to ensure that a wide distribution of phenoxy groups was obtained and to maintain the solubility of the polymer during the subsequent addition of 4-iodophenoxide. The 31P NMR spectrum was monitored over the duration of the reaction to determine when substitution of each addition was complete. For example, after 24 hours of refluxing at the first stage, substitution by the added reagent was complete and all of the stoichometrically required 4-iodophenoxide was then added dropwise. The remaining sodium phenoxide was added after stabilization of the chemical shifts in the 31P NMR spectrum indicated consumption of the 4-iodophenoxide. The final 31P NMR spectrum showed only one peak for polymers 2–7, which indicated complete chlorine atom replacement and a random distribution of the two side groups in the co-substituted polymers (3 and 4).
Thermal characterization of polymers 2–7
The glass transition temperatures (Tg) of the iodine-bearing aryloxyphosphazene polymers 2–5 illustrate the influence of the iodine atoms. Polymers 2 and 3 share similar glass transition temperatures, but the values for 4 and 5 are much higher than those of 2 and 3, presumably due to restriction of free motion in the polymer brought about by their high loading of iodine atoms.14,35Synthesis and structural characterization of polymers 8–15
For polymers in the second group, attempts to synthesize the single-substituent polymer, poly[bis(ethyl 3,5-diiodotyrocinato)phosphazene] (11) in boiling THF or dioxane resulted in precipitation of partly substituted derivatives and an inability to replace the remaining chlorine atoms. Moreover, the initial attempts to synthesize the mixed-substituent diiodotyrosine polymers 8–11, which involved an initial incorporation of 3,5-diiodotyrosine ethyl ester (R2) in boiling THF, followed by completion of the substitution with the second co-substituent (glycine ethyl ester, alanine ethyl ester, or phenylalanine ethyl ester, R1) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, also resulted in a loss of the 31P NMR signal before the second substituent was added. Again, this indicated that the partially substituted polymers had precipitated from solution during the first phase of substitution. The use of boiling dioxane in place of THF did not change this behavior. Therefore a three-step addition of the reagents, similar to the syntheses used for polymers 3 and 4, was adopted (Scheme 2). The chlorine replacement was monitored by 31P NMR spectroscopy as the first portion of R1, followed by all of R2, and ending with the remaining quantity of R1 was added to the polymer solution. For polymers 8–11, the final ratio of substituents was 0.5:1.5 (R2:R1).
:1 ratio, also resulted in a loss of the 31P NMR signal before the second substituent was added. Again, this indicated that the partially substituted polymers had precipitated from solution during the first phase of substitution. The use of boiling dioxane in place of THF did not change this behavior. Therefore a three-step addition of the reagents, similar to the syntheses used for polymers 3 and 4, was adopted (Scheme 2). The chlorine replacement was monitored by 31P NMR spectroscopy as the first portion of R1, followed by all of R2, and ending with the remaining quantity of R1 was added to the polymer solution. For polymers 8–11, the final ratio of substituents was 0.5:1.5 (R2:R1).
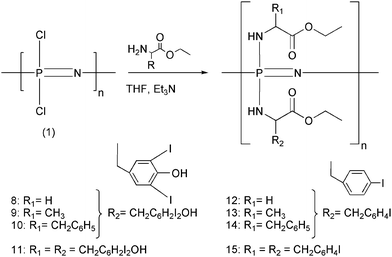 | ||
| Scheme 2 Synthesis and structures of polymers 8–15. The ratio of 3,5-diiodotyrosine ethyl ester to glycine ethyl ester, alanine ethyl ester, or phenylalanine ethyl ester was maintained at a 0.5:1.5 ratio. The ratio of 4-iodophenylalanine ethyl ester to glycine ethyl ester, alanine ethyl ester, or phenylalanine ethyl ester was held at a 1:1 ratio. | ||
The 31P NMR spectra for polymers 8–10 showed only one broad peak for the final product. This was attributed to the presence of amino acid residues that generated skeletal 31P signals in the 1 ppm to −5 ppm range and the presence of both geminal and non-geminal substitution patterns. 1H NMR spectroscopy was used to confirm the presence of both types of organic side groups in approximately a 0.5:1.5 ratio based on shifts unique to each substituent.
For the third group of polymers that contained the ethyl ester of 4-iodophenylalanine, the restrictions to the syntheses were not so severe. Even the single-substituent polymer 15 could be formed in refluxing THF. Although the mixed-substituent iodophenylalanine-based polymers, 12–14, were more soluble than 8–11, the three-step method was used to ensure complete substitution. Thus, half of the non-iodinated R1 reagent was added first. When 31P NMR spectroscopy indicated that all of the R1 nucleophile had reacted, R2 (4-iodophenylalanine ethyl ester) was added to the reaction mixture. When R2 had reacted completely, as determined by NMR, the remaining portion of R1 was added to the reaction mixture. The final 31P and 1H NMR spectra for polymers 12–15 showed a single peak or peak integrations which indicated that all these polymers contained a random distribution of the R1 and R2 substituents in approximately a 1:1 ratio.
Thermal characterization of polymers 8–15
The glass transition temperatures (Tg) of polymers 8–15 are shown in Table 1. With one exception (50%/50% (ethyl diiodotyrosinyl/ethyl glycinyl)) the glass transition temperatures are at or above room temperature. This provides a range of properties for different medical applications. The values for polymers 8–10 cover a range from −2.31 °C (8) to 52.52 °C (10). The Tg's of these polymers are governed by the high level of incorporation of R1 rather than the minority 3,5-diiodotyrosine ethyl ester. The Tg range is wide because the steric size of the substituents directly influences the thermal properties of the polymer. Poly[(ethyl glycinato)1.62(ethyl tyrosinato)0.38phosphazene] has a glass transition temperature of 5.4 °C which is similar to the Tg value of 8 (−2.31 °C).36With polymers 12–15, the Tg is dependent on both the 4-iodophenylalanine ethyl ester and R1 because the substituents are present in roughly equal amounts. The Tg's of 12 and 13 are much higher than the values for poly[bis(ethyl glycinato)phosphazene] or poly[bis(ethyl alaninato)phosphazene] (−20 °C and −15 °C respectively), but lower than the Tg of poly[bis(ethyl phenylalaninato)phosphazene] (40–68 °C).14,26 However, with 14 and 15, the measured Tg's are lower than for the phenylalanine single-substituent polymer as the result of the increased free volume from 4-iodophenylalanine ethyl ester units and the elimination of crystallinity which had previously been detected.
Hydrolytic degradation of polymers 8–10 and 12–15
Solution cast films of 8–10 are insoluble in water. Previously reported studies have shown that complete hydrolytic degradation of amino acid substituted polyphosphazenes yields ammonia, phosphates, free amino acid, and ethanol.26–29,37–39 The high solubility of these products in water allows “sink” conditions to be achieved using the procedure described in the Experimental section. Thus, a heterogeneous hydrolytic degradation profile was generated over a period of 6 weeks. The initial mass of each film was recorded before the film was immersed in water. Each week, a set of samples was removed and the remaining portion of the film was freeze-dried for at least one week. Fig. 1A shows the mass retention for polymers 8–10. Despite polymers 8 and 9 losing 25% of their original mass after only 7 days, by the end of 6 weeks, these polymers retained 65% and 62% of their initial weight. Polymer 10 showed very little mass loss over the duration of the study. The incorporation of 25% of the bulky, hydrophobic 3,5-diiodotyrosine ethyl ester as the side group is not sufficient to slow the rate of degradation. Thus, the co-substituent (glycine, alanine, or phenylalanine ethyl ester) controls the rate of hydrolysis. The rates of hydrolysis are consistent with the general trend that the hydrolytic half-life of polyphosphazenes substituted with bulky, hydrophobic amino acid residues (phenylalanine ethyl ester) is much longer than the half-life of polyphosphazenes that contain less sterically hindered amino acids, such as glycine or alanine residues. When the mass loss of polymers 12–15 (Fig. 1B) is compared to that of polymers 8–10, the 4-iodophenylalanine polymers retained significantly more of their initial mass over the 6 week period with the maximum mass loss of 30%. Although polymers 14 and 15 might be expected to have slower rates of hydrolysis than polymers 12 and 13, they were in fact found to hydrolyze faster. The 1:1 ratio of side groups in polymers 12–15 allows the 4-iodophenylalanine to influence the rate of hydrolysis, resulting in the similar mass loss profiles for the polymers.
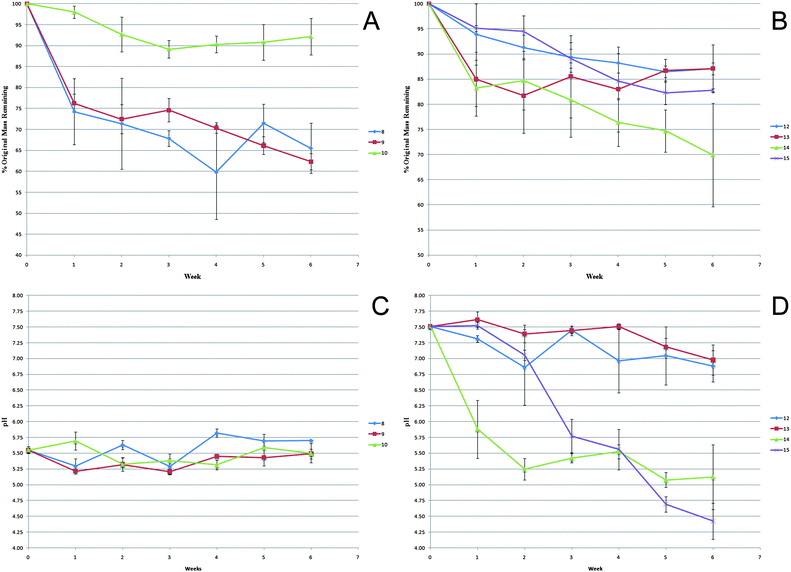 | ||
| Fig. 1 (A) Mass loss profile of polymers 8–10 during a 6 week hydrolysis study. (B) Mass loss profile of polymers 12–15 during a 6 week hydrolysis study. (C) pH measurements of the hydrolysis media for polymers 8–10. (D) pH measurements of the hydrolysis media for polymers 12–15. | ||
The hydrolysis media were analyzed in an attempt to assess the degradation mechanism for polymers 8–15. The presence of phosphates was first detected after 2 weeks as a sharp peak at 0 ppm in the 31P NMR spectrum. The addition of silver nitrate to the hydrolysis medium yielded a yellow precipitate of silver phosphate, confirming the presence of phosphates. Free amino acid residues of glycine, alanine, and phenylalanine and ethanol were present in the 1H NMR spectrum of the hydrolysis medium after 1 week. The free 3,5-diiodotyrosine or 4-iodophenylalanine residues were not detected by 1H NMR spectroscopy until week three. The presence of ammonia or the free amine termini of the amino acids was further confirmed by a positive ninhydrin test.
These results are consistent with a bulk erosion mechanism where the least sterically hindered amino acid ethyl ester is cleaved from the polymer backbone. The order in which the ester functionality is hydrolyzed and the side group is cleaved from the phosphazene backbone cannot be determined from these results. However, the presence of phosphates in the aqueous medium is consistent with a complete hydrolytic degradation of the phosphazene backbone.
The pH of each hydrolysis medium (Fig. 1C and D) remained constant for the duration of the 6 week study, with the exception of polymers 14 and 15. Hydrolysis of the polymer backbone yielded ammonia and phosphates which form a buffered solution that causes little change in the pH of the hydrolysis medium. The decrease in the pH of the hydrolysis medium for polymers 14 and 15 is caused by the liberation of trace amounts of hydrochloric acid during hydrolytic degradation. Small molecule model studies have shown that two competing equilibria exist for the hydrogen chloride byproduct of the macromolecular substitution of poly(dichlorophosphazene) with an amino group. The hydrogen chloride can either be absorbed by the triethylamine hydrochloride acceptor or complexed with the basic nitrogen atoms in the phosphazene, the latter making complete removal of the hydrochloride salt during purification difficult.14 The concentration of trapped hydrochloride salt necessary to affect the observed changes in pH is well below the limit of detection by 1H NMR spectroscopy. The acidic hydrolysis medium also explains the accelerated hydrolysis of polymers 14 and 15. Previous studies have shown that when phosphazenes are exposed to an acidic solution, the degradation is accelerated when compared to hydrolysis in deionized water or basic solutions.37–39
The 3,5-diiodotyrosine and 4-iodophenylalanine residues released as a result of hydrolysis are expected to have minimal effect on biocompatibility. 3,5-Diiodotyrosine is a natural product which is used in the synthesis of thyroxine, a thyroid hormone, and regulator of thyroid peroxidase.40 4-Iodophenylalanine residues have been used in numerous studies as a non-natural amino acid for fluorescence and bioluminescence modification.41–43 During its use as a radiological tag in brain tumor imaging, animal testing showed no acute physiological response to doses of 10 mg·kg−1.44 Despite this evidence, further studies are needed to confirm the biocompatibility of these polymers.
Radio opacity testing and medical imaging
Small samples of the thermally fabricated polymers 2–5 were characterized after fabrication utilizing 1H and 31P NMR, GPC, and DSC methods. The results were identical to the initial characterization, which confirmed that no degradation occurred as a result of the processing. As a preliminary test, polymers 2, 4–6, 8–10, and 12–15 were exposed to a copper Kα X-ray source. Their ability to block these “soft” X-rays was judged relative to the source being completely blocked or unblocked. Although all the biostable polymers (2–6) showed some ability to block this radiation, those polymers with iodinated side groups were the most opaque. Similarly, the thin films of the biocompatible polymers which averaged one iodine per repeat unit were also opaque to the X-rays. All the polymers were then characterized a final time to confirm that short term exposure to “soft” X-rays, such a copper Kα, does not cause detectable degradation of the polymer.X-Ray images were then obtained using a rhenium–tungsten–molybdenum target and a protocol employed for the imaging of a broken finger. The X-ray image of polymers 2, 4, and 5 at two different thicknesses [(A) 4.80 mm and (B) 1.97 mm] was obtained. Polymer 2, which contained no iodine, was transparent at both thicknesses. Incorporation of one or more iodine atoms per repeating unit (polymers 4 and 5) is sufficient to render the polymers opaque to tungsten X-rays at either thickness. This series of polymers also shows that image contrast is proportional to the amount of iodine incorporated into the polymer backbone and the thickness of the polymer sample. The radiologic opacity of the biocompatible polymers 8–10 and 12–15 was evaluated as both solution cast thin films and pressed pellets. The thin films and pellets were clearly visible in the image, which demonstrates the possibility for these polymers to be used on their own as a bulk material or applied as a coating to a compatible substrate. To further illustrate the uniqueness of the biostable and biocompatible polymer systems, Fig. 2 shows an X-ray image of selected polymers (5, 10, 14, and 15) alongside several common organic polymers. Only two of the polymers imaged, poly(vinyl chloride) and polydimethylsiloxane, come close to the opacity of the reference poly(organophosphazenes) and neither are as good a candidate for most biomedical applications due to the need for plasticizers and cross-linkers, the toxicity of the additives, or their mechanical properties. The polymer samples were again tested several times to ensure that no degradation occurred as a result of the processing or exposure to “hard” X-rays.
![Medical X-ray photograph of selected common polymers thermoformed into 4.80 mm thick pellets for comparison to radio opaque polyphosphazenes. The position of each pellet is outlined for clarity. (A) Poly[bis(4-iodophenoxy)phosphazene] (5), (B) poly[(ethyl phenylalanato)(ethyl 4-iodophenylalanato)phosphazene] (14), (C) poly[bis(ethyl 4-iodophenylalanato)phosphazene] (15), (D) poly[(ethyl phenylalanato)1.5(ethyl 3,5-diiodotyrocinato)0.5phosphazene] (10), (E) polyethylene, (F) poly(methyl methacrylate), (G) polyvinyl chloride, (H) polystyrene, (I) poly(ethylene oxide), (J) poly(lactic-co-glycolic acid) (50/50), and (K) polydimethylsiloxane.](/image/article/2010/PY/c0py00126k/c0py00126k-f2.gif) | ||
| Fig. 2 Medical X-ray photograph of selected common polymers thermoformed into 4.80 mm thick pellets for comparison to radio opaque polyphosphazenes. The position of each pellet is outlined for clarity. (A) Poly[bis(4-iodophenoxy)phosphazene] (5), (B) poly[(ethyl phenylalanato)(ethyl 4-iodophenylalanato)phosphazene] (14), (C) poly[bis(ethyl 4-iodophenylalanato)phosphazene] (15), (D) poly[(ethyl phenylalanato)1.5(ethyl 3,5-diiodotyrocinato)0.5phosphazene] (10), (E) polyethylene, (F) poly(methyl methacrylate), (G) polyvinyl chloride, (H) polystyrene, (I) poly(ethylene oxide), (J) poly(lactic-co-glycolic acid) (50/50), and (K) polydimethylsiloxane. | ||
Conclusions
To the best of our knowledge this is the first reported synthesis of poly(organophosphazenes) that contain 3,5-diiodotyrosine ethyl ester or 4-iodophenylalanine ethyl ester side groups for use as X-ray opaque materials. The synthesis of single-substituent 4-iodophenoxy polymer had been previously reported,45 but had not been evaluated for X-ray opacity. The new mixed-substituent polymers were obtained through a three-step replacement of the chlorine atoms in poly(dichlorophosphazene) by sodium phenoxide or an amino acid ethyl ester and a similar iodinated derivative. A series of biostable poly[bis(aryloxy)phosphazene] derivatives with an increasing amount of iodine incorporated into the polymer were synthesized. 4-Iodophenoxy side groups have a marked influence on increasing the Tg values. Two sets of amino acid ester polymers were synthesized with 3,5-diiodotyrosine ethyl ester or 4-iodophenylalanine ethyl ester and glycine ethyl ester, alanine ethyl ester, or phenylalanine ethyl ester as the co-substituents. The physical and thermal properties of polymers 8–10 are controlled by the non-iodinated co-substituent due to the low incorporation of the iodine-containing side group. Conversely, polymers 12–15 have very similar physical and thermal properties resulting from the 1:1 ratio of 4-iodophenylalanine and the co-substituent. All polymers with an average of one iodine per repeat unit were opaque to both copper Kα and rhenium–tungsten–molybdenum X-rays.
Acknowledgements
This work was supported by NIH grant number RO1EB004051. We thank Mark Angelone at The Pennsylvania State University Materials Research Institute for his help in testing synthesized polymers with the copper Kα X-ray source. We also thank Melanie Harris and the Radiology Center at The Pennsylvania State University Student Health Center for their assistance obtaining images on the Kodak 7500 DR X-ray unit.Notes and references
- C. Kasperk, J. Hillmeier, G. Noldge, I. A. Grafe, K. Da Fonseca, D. Raupp, H. Bardenheuer, M. Libicher, U. M. Liegibel, U. Sommer, U. Hilscher, W. Pyerin, M. Vetter, H.-P. Meinzer, P.-J. Meeder, R. S. Taylor and P. Nawroth, J. Bone Miner. Res., 2005, 20, 604–612 CrossRef.
- M. P. Ginebra, L. Albuixech, E. Fernandez-Barragán, C. Aparicio, F. J. Gil, J. San Román, B. Vázques and J. A. Planell, Biomaterials, 2002, 23, 1873–1882 CrossRef CAS.
- M. A. B. Kruft, F. H. van der Veen and L. H. Koole, Biomaterials, 1997, 18, 31–36 CrossRef CAS.
- E. J. H. Boelen, G. Lewis, J. Xu, T. Slots, L. H. Koole and C. S. J. van Hooy-Corstjens, J. Biomed. Mater. Res., Part A, 2008, 86, 76–88 CrossRef.
- G. Lewis, L. H. Koole and C. S. J. van Hooy-Corstjens, J. Biomed. Mater. Res., Part B, 2009, 91, 537–544 CrossRef.
- B. Nottelet, J. Coudane and M. Vert, Biomaterials, 2006, 27, 4948–4954 CrossRef CAS.
- A. L. Carbone, M. J. Song and K. E. Uhrich, Biomacromolecules, 2008, 9, 1604–1612 CrossRef CAS.
- Y. Yang, S. M. Dorser, M. L. Becker, S. Lin-Gibson, G. E. Schumacher, G. M. Flaim, J. Kohn and C. G. Simon, Biomaterials, 2008, 29, 1901–1911 CrossRef CAS.
- D. Mawad, L. A. Poole-Warren, P. Martens, L. H. Koole, T. L. B. Slots and C. S. J. van Hooy-Corstjens, Biomacromolecules, 2008, 9, 263–268 CrossRef CAS.
- M. Okamura, T. Yamanobe, T. Arai, H. Uehara, T. Komoto, S. Hosoi and T. Kumazaki, J. Mol. Struct., 2002, 602, 17–28 CrossRef.
- F. Mottu, D. A. Rufenacht, A. Laurent and E. Doelker, Biomaterials, 2001, 23, 121–131.
- M. Okamura, H. Uehara, T. Yamanobe, T. Komoto, S. Hosoi and T. Kumazaki, J. Mol. Struct., 2000, 554, 35–45 CrossRef CAS.
- M. A. B. Kruft, A. Benzina, R. Blezer and L. H. Koole, Biomaterials, 1996, 17, 1803–1812 CrossRef CAS.
- H. R. Allcock, Chemistry and Applications of Polyphosphazenes, Wiley-Interscience, Hoboken, NJ, 2003, pp. 460–465 Search PubMed.
- H. R. Allcock and R. M. Wood, J. Polym. Sci., Part B: Polym. Phys., 2006, 18, 2358–2368 CrossRef.
- D. T. Welna, D. A. Stone and H. R. Allcock, Chem. Mater., 2006, 18, 4486–4492 CrossRef CAS.
- Z. Li, C. Huang, J. Hua, J. Qin, Z. Yang and C. Ye, Macromolecules, 2004, 37, 371–376 CrossRef CAS.
- Z. Li, J. Qin, X. Deng and Y. Cao, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3428–3433 CrossRef CAS.
- L. Gettleman, J. M. Vargo, P. H. Gebert, C. L. Farris, R. J. LeBouef and H. R. Rawls, Polym. Sci. Technol., 1987, 35, 55–61 Search PubMed.
- L. Gettleman, J. M. Vargo, P. H. Gebert, C. L. Farris, R. J. LeBouef and H. R. Rawls, in Advances in Biomedical Polymers, ed. C. G. Gebelein, Plenum, New York, 1987, pp. 55–61 Search PubMed.
- L. Gettleman, Phosphorus, Sulfur Silicon Relat. Elem., 1999, 144, 205–208 Search PubMed.
- S. G. Kumbar, S. Bhattacharyya, S. P. Nukavarapu, Y. M. Khan, L. S. Nair and C. T. Laurencin, J. Inorg. Organomet. Polym. Mater., 2006, 16, 365–385 CAS.
- A. K. Andrianov and J. Chen, J. Appl. Polym. Sci., 2006, 101, 414–419 CrossRef CAS.
- H. R. Allcock, Phosphorous Nitrogen Compounds, Academic Press, Inc., New York, NY, 1972, pp. 354–361 Search PubMed.
- H. R. Allcock, L. Steely, A. Singh and M. D. Hindenlang, J. Adhes. Sci. Technol., 2009, 23, 435–445 CrossRef.
- (a) H. R. Allcock, S. R. Pucher and A. G. Scopelianos, Macromolecules, 1994, 27, 1071–1075 CrossRef CAS; (b) The exact Tg for this polymer is dependent on crystallinity and molecular weight of the sample.
- H. R. Allcock, T. J. Fuller, D. P. Mack, K. Matsumura and K. M. Smeltz, Macromolecules, 1977, 10, 824–830 CrossRef CAS.
- A. Singh, N. R. Krogman, S. Sethuraman, L. S. Nair, J. Sturgeon, P. Brown, C. T. Laurencin and H. R. Allcock, Biomacromolecules, 2006, 7, 914–918 CrossRef CAS.
- N. R. Krogman, A. Singh, L. S. Nair, C. T. Laurencin and H. R. Allcock, Biomacromolecules, 2007, 8, 1306–1310 CrossRef CAS.
- A. Pangborn, M. Giardello, R. Grubbs, R. Rosen and F. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- H. Lei, M. S. Stoakes, K. P. B. Herath, J. Lee and A. W. Schwabacher, J. Org. Chem., 1994, 59, 4206–4210 CrossRef CAS.
- Polyphosphazenes for Biomedical Applications, ed. A. K. Andrianov, John Wiley & Sons, Hoboken, NJ, 2009, pp. 379–410 Search PubMed.
- G. A. Carriedo, A. Presa-Soto and M. L. Valenzula, Macromolecules, 2008, 41, 6972–6976 CrossRef CAS.
- G. A. Carriedo and M. L. Valenzula, Macromolecules, 2009, 43, 126–130.
- G. L. Hagnauer and B. R. LaLiberte, J. Polym. Sci., Part B: Polym. Phys., 1976, 14, 367–371 Search PubMed.
- H. R. Allcock, A. Singh, A. M. A. Ambrosio and W. Laredo, Biomacromolecules, 2003, 4, 1646–1653 CrossRef CAS.
- A. K. Andrianov and A. Marin, Biomacromolecules, 2006, 7, 1581–1586 CrossRef CAS.
- J. Crommen, J. Vandorpe and E. Schacht, J. Controlled Release, 1993, 24, 167–180 CrossRef CAS.
- A. L. Weikel, N. R. Krogman, N. Q. Nguyen, L. S. Nair, C. T. Laurencin and H. R. Allcock, Macromolecules, 2009, 42, 636–639 CrossRef CAS.
- D. Deme, E. Fimiani, J. Pommier and J. Nunez, Eur. J. Biochem., 1975, 51, 329–336 CrossRef CAS.
- P. G. Schultz and L. Wang, Angew. Chem., Int. Ed., 2005, 44, 34–66 CrossRef CAS.
- V. De Filippis, A. Draghi, R. Frasson, C. Grandi, V. Musi, A. Fontana and A. Pastore, Protein Sci., 2007, 16, 1257–1265 CrossRef CAS.
- L. Rowet, M. Ensort, R. Mehl and S. Daunert, ACS Chem. Biol., 2010, 5, 455–460 CrossRef.
- S. Samnick, D. Hellwig, J. B. Bader, B. F. Romeike, J. R. Moringlane, W. Feiden and M. C-Kirsch, Nucl. Med. Commun., 2002, 23, 121–130 CrossRef CAS.
- M. A. Olshavsky and H. R. Allcock, Macromolecules, 1995, 28, 6188–6197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Example NMR spectra of select polymers. See DOI: 10.1039/c0py00126k |
| This journal is © The Royal Society of Chemistry 2010 |