Energy upconversion sensitized by a platinum(II) terpyridyl acetylide complex†
Pingwu Du and Richard Eisenberg*
Department of Chemistry, University of Rochester, RC Box 270216, Rochester, NY 14627-0216, USA. E-mail: eisenberg@chem.rochester.edu; Fax: +001-585-276-0205
First published on 9th July 2010
Abstract
The platinum(II) terpyridine acetylide complex, [Pt(ttpy)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)]ClO4 (1, where ttpy = 4′-p-tolylterpyridine), sensitizes the 3π–π* excited state of 9,10-diphenylanthracene (DPA) that in turn produces upconverted fluorescence via triplet–triplet annihilation (TTA). The photoluminescence of 1 is readily quenched by DPA with a rate constant close to the diffusion limit via energy transfer. Selective excitation of 1 leads to the upconverted fluorescence with a near quadratic dependence of the DPA fluorescence intensity on incident light power. This is the first time that the 3MLCT charge transfer excited state of a platinum polypyridine complex has been used to promote photon upconversion.
CPh)]ClO4 (1, where ttpy = 4′-p-tolylterpyridine), sensitizes the 3π–π* excited state of 9,10-diphenylanthracene (DPA) that in turn produces upconverted fluorescence via triplet–triplet annihilation (TTA). The photoluminescence of 1 is readily quenched by DPA with a rate constant close to the diffusion limit via energy transfer. Selective excitation of 1 leads to the upconverted fluorescence with a near quadratic dependence of the DPA fluorescence intensity on incident light power. This is the first time that the 3MLCT charge transfer excited state of a platinum polypyridine complex has been used to promote photon upconversion.
Introduction
In energy upconversion, the absorption of two lower energy photons results in the emission of a single higher energy photon. One well recognized way in which this process occurs involves a sequence of (1) low energy photon absorption by two sensitizers, (2) intersystem crossing to the triplet manifold, (3) triplet energy transfer to two energy acceptor molecules and (4) triplet–triplet annihilation (TTA) to generate one acceptor molecule in its lowest singlet excited state together with the second acceptor in its electronic ground state.1,2 Attractive features of TTA for upconversion are its independence of coherence in the excitation radiation and the low power of the radiation that can drive it (on the order of 1 mW cm−2 or even lower).3 The efficiency of upconversion increases as the population of emitter triplet states gets larger through triplet sensitization by the initial light absorber. In this regard, the use of metal complexes as triplet sensitizers has long been recognized because of the large spin–orbit coupling that the complexed metal ions possess which in turn leads to rapid intersystem crossing from the complex's singlet excited state upon absorption to its lowest triplet excited state.The advantages of TTA for upconversion using transition metal complexes have attracted much attention for the development of efficient systems and devices for generating higher energy light from visible and/or near-infrared excitation. In 2003, Wegner and co-workers reported a bimolecular system involving a Pd(II) porphyrin complex as the sensitizer and polyfluorene as the triplet annihilator to generate blue light.4 The intensity of the blue light was found to be substantially enhanced by using a silver matrix surface5 or employing a novel design of an end-capped matrix polymer with the Pd(II) porphyrin sensitizer.6 In order to utilize low-energy photons for upconversion, excitation wavelengths of the sensitizer have been tuned to the near-infrared region by increasing the conjugation of the unsaturated coordinating ligand.3,7 Likewise, the singlet emission of the triplet acceptor/annihilator can also be moved to lower energy to generate blue-green light,7 yellow light3,8 and even white light.9
Castellano and co-workers have reported another representative example using [Ru(dmb)3]2+ (dmb = 4,4′-dimethyl-2,2′-bipyridine) as the sensitizer.10 Selective excitation of [Ru(dmb)3]2+ yields upconverted singlet blue or yellow emission at lower excitation power.1b,9,11 Other metal complex sensitizers have also been reported recently by Castellano et al., including a Pd(II) phthalocyanine complex,8 and an Ir(III) cyclometalated complex.12
In the past decade, luminescent Pt(II) bipyridine and terpyridine complexes have received increasing attention as a result of their rich photochemical and photophysical properties, i.e., their long triplet excited state lifetimes (>1 μs), good emission quantum yields, electron transfer quenching reactions and potential applications in fields such as chemosensors,13 photoinduced hydrogen production,14 and photocatalytic devices.15 However, to date, there have been no reports about using these Pt(II) bi- or terpyridyl complexes as triplet sensitizers for energy upconversion. Herein we report the first example of doing so with the Pt(II) acetylide complex [Pt(ttpy)(CCPh)]ClO4 (1) that serves to sensitize upconversion in 9,10-diphenylanthracene (DPA) via TTA. The selective excitation of 1 is possible because its metal-to-ligand charge transfer (MLCT) absorption band lies in the visible region where DPA does not absorb.
Experimental section
Materials
The Pt sensitizer 1 was synthesized following the procedure reported previously14a involving the reaction of [Pt(ttpy)Cl]Cl with phenylacetylene in the presence of CuI as the catalyst in triethylamine–DMF. The perchlorate salt of [Pt(ttpy)(CCPh)]+ was obtained by metathetical exchange of perchlorate for chloride using KClO4. Warning: perchlorate salts of metal complexes are known to be explosive and should therefore be handled with extreme care and proper protection. 9,10-Diphenylanthracene (DPA) was used as purchased (Aldrich), as were all other reagents and solvents purchased from commercial sources.Physical measurements
Absorption spectra were recorded using a Hitachi U2000 scanning spectrophotometer (200–1100 nm). Emission spectra were obtained using a Spex Fluoromax-P fluorometer corrected for instrument response. Monochromators were positioned with a 5 nm band-pass, and solution samples were degassed by at least three freeze–pump–thaw cycles.Quenching experiments
For the quenching experiments, a stock solution of the sensitizer 1 was made in methylene chloride. Samples with known quencher concentrations were prepared by adding an appropriate amount of quencher to 10 mL volumetric flasks containing the diluted Pt sensitizer stock solution (2.2 × 10−5 M). Prior to measurement, the solution samples were deaerated through three freeze–pump–thaw cycles. Steady-state luminescence spectra were then collected for each sample, while UV-vis spectra were monitored for each experiment to determine potential photochemical products and/or photodegradation. In the simple bimolecular quenching reactions, there was no evidence of any change in the absorption spectra under all experimental conditions.Upconversion emission experiments
All samples were deaerated with nitrogen gas for at least 30 min prior to measurements and were maintained under a N2 atmosphere by a simple purge throughout the experiments. Excitation radiation was obtained with an argon ion laser (Coherent Innova 300), the multi-line output of which was split into selected wavelength components (514.5 nm) using a diffraction grating in concert with several optics. The excitation laser wavelength was effectively suppressed by using suitable edge filters.Results and discussion
Steady-state absorption and emission properties
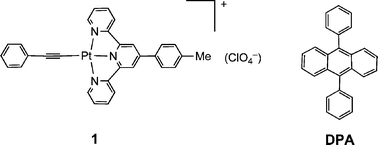
Fig. 1a shows the steady-state absorption spectra of sensitizer 1, DPA and a mixture of 1 and DPA. The sensitizer exhibits broad low energy absorption features in methylene chloride between 375–550 nm with λmax at 429 nm (molar extinction coefficient ε of ∼8570 dm3 mol−1cm−1) and 476 nm (ε∼7600 dm3 mol−1cm−1), corresponding to the dπ(Pt)–π(ttpy) metal-to-ligand charge transfer (MLCT) transitions in agreement with literature values.16 DPA has a strong vibronically structured absorption with maxima at 396, 374, 356 and 340 (sh) nm corresponding to a π–π* transition. The absorption spectrum of a mixture of 1 and DPA in methylene chloride corresponds to a simple sum of the individual components, indicating the absence of measurable electronic interactions between 1 and DPA in their respective ground states. Significantly, the absorption of sensitizer 1 extends to longer wavelength than that of DPA in the visible region, indicating the possibility of selective excitation of 1 in the presence of DPA in a two-component system.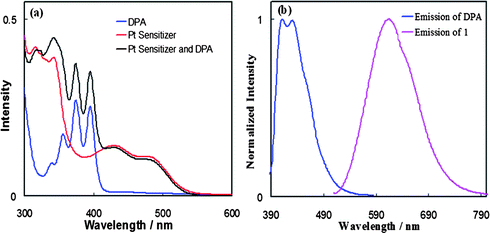 | ||
| Fig. 1 (a) Absorption spectra of sensitizer 1 (red plot, 2.2 × 10−5 M), DPA (blue plot, 2.2 × 10−5 M), a mixture of sensitizer 1 and DPA (black plot, 2.2 × 10−5 M for both) in degassed methylene chloride solution; (b) photoluminescence spectra of sensitizer 1 (pink plot, λex = 500 nm) and DPA (blue plot, λex = 350 nm) in degassed methylene chloride solution. | ||
The emission spectra of 1 and DPA are shown in Fig. 1b. Complex 1 exhibits a strong photoluminescence in the range of 500–800 nm with λmax at 613 nm in methylene chloride and a relative luminescence quantum yield (Φ) of 0.049 based on the degassed acetonitrile solution of [Ru(bpy)3](PF6)2 as the reference (Φ = 0.062).17 DPA has strong blue emission maximized at 414 nm and 432 nm, with a slight but discernible shoulder at 460 nm and a fluorescence quantum yield close to unity.18
Luminescence quenching of sensitizer 1 by DPA
We previously reported that 1 undergoes oxidative quenching by MV2+ with a rate constant kq of 3.3 × 109 M−1 s−1 and reductive quenching by triethanolamine (TEOA) with a rate constant kq of 1.4 × 109 M−1 s−1 in acetonitrile–water (v/v, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) via photoinduced electron transfer.14 These results were in accord with earlier reported quenching studies of related Pt diimine bis(acetylide) complexes by selected amines, which were also close to the diffusion-controlled limit (∼109–1010 M−1 s−1).19
:2) via photoinduced electron transfer.14 These results were in accord with earlier reported quenching studies of related Pt diimine bis(acetylide) complexes by selected amines, which were also close to the diffusion-controlled limit (∼109–1010 M−1 s−1).19In the presence of DPA, the photoluminescence of 1 selectively excited at 500 nm (Fig. 2a) is readily quenched. Quantitative quenching studies were conducted by measurement of steady-state luminescence intensities of 1 in the presence of varying amounts of DPA. The quenching in eqn (1) is dynamic in nature, and is modeled satisfactorily by the Stern–Volmer equation20 in eqn (2), where I0 is the integrated MLCT emission intensity in the absence of quencher, τ0 is the excited state lifetime in the absence of quencher, I and τ are the corresponding values in the presence of quencher, KSV is the Stern–Volmer quenching constant, kq is the bimolecular quenching rate constant, and [Q] is the quencher concentration. Linear plots of I0/I vs. [Q], shown in Fig. 2b, yield a value of KSV = 1.14 × 104 from which the quenching rate constant kq of 2.5 × 109 M−1 s−1 is obtained using the previously published value of τ0 (4.6 μs);16 the value of kq is close to the diffusion-controlled limit in methylene chloride. This value is comparable to those reported earlier for electron transfer quenching of [Pt(ttpy)(CCR)]+ and related Pt(diimine)(CCR)2 complexes mentioned above.14b,19a
| 3Pt(II)* + DPA → Pt(II) + 3DPA* | (1) |
| I0/I = τ0/τ + KSV[Q] (KSV = kqτ) | (2) |
 | ||
| Fig. 2 (a) Luminescent quenching study of sensitizer 1 (2.2 × 10−5 M) measured as the function of the concentration of DPA in deaerated CH2Cl2; (b) Stern–Volmer plot generated from the luminescent intensity quenching of sensitizer 1 by DPA in deaerated CH2Cl2. | ||
However, while the quenching rate constant kq for 1 by DPA is similar to those obtained using electron transfer quenchers, the quenching in eqn (1) must proceed by energy transfer and not by electron transfer based on the following analysis. The free energy change (ΔG) involved in a photoinduced electron transfer process can be estimated from the Rehm–Weller equation21 (eqn (3)) where E00 is the 3MLCT excited state energy of 2.48 eV,14b and Eox(D) and Ered(A) are the oxidation and reduction potentials of electron donor and acceptor, respectively. The quantities ωp and ωr represent the coulombic work terms required to bring together the products and reactants to form the ion pair or encounter complex, respectively. In the present case, the quantity ωp−ωr is small with an estimate of 26 mV.24 With this value and the redox potentials Eox(D) = 1.34 V (vs NHE in DMF) for 114b and Ered(A) = −1.55 V (vs NHE in PhCN) for DPA,22eqn (3) indicates that electron transfer from excited sensitizer 1 to DPA (oxidative quenching) is thermodynamically untenable (ΔG = ∼ +10.1 kcal mol−1). As for reductive quenching, the redox potentials are Ered(A) = −1.07 V (vs NHE in DMF) for 114b and Eox(D) = +1.61 V (vs NHE in PhCN) for DPA.22 Similarly, electron transfer from DPA to excited sensitizer 1 is also thermodynamically not viable (ΔG = ∼ +5.2 kcal mol−1).
| E0 = −ΔG = Ered(A) −Eox(D) + E00− (ωp−ωr) | (3) |
With respect to energy transfer for luminescence quenching, this must proceed via the 3MLCT state of 1. Intersystem crossing in complexes such as 1 is generally extremely rapid with near unit efficiency. Additionally, the energy of the singlet excited state of 1 (∼2.90 eV) is much lower than that of 1DPA* (∼3.06 eV22), as estimated from absorption spectra, meaning that singlet–singlet energy transfer is thermodynamically unfavorable. In contrast, the triplet energy of DPA (ET = 1.80 eV22) is lower than the triplet energy of 1 (ET = 2.48 eV14b), making triplet–triplet energy transfer thermodynamically viable. Because only 1 is excited upon irradiation, and the other possible quenching pathways are excluded on thermodynamic grounds, it is evident that in the system of 1 + DPA, triplet energy transfer is occurring as the means of quenching the emission of 1.
This conclusion of energy upconversion by complex 1 is further supported by lifetime measurements (see ESI Fig. S1†). The lifetime of complex 1 is ∼4.8 μs in the absence of DPA, which decreases to 2.1 μs and 1.2 μs in the presence of 0.11 mM and 0.24 mM DPA, respectively. These observations are consistent with the results reported previously for the quenching of the [Ru(dmb)3]2+ sensitizer in an upconversion system.9
Upconversion fluorescence at steady state
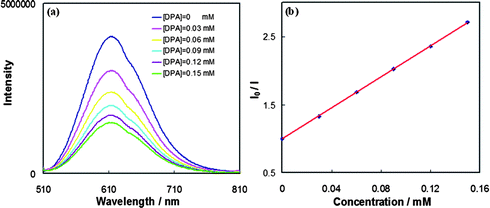
When the Pt sensitizer 1 (2.2 × 10−5 M) is selectively excited at 500 nm in the presence of DPA, the emission spectrum shows a blue, higher energy fluorescence (Fig. 3) that possesses the same shape as DPA fluorescence obtained by direct excitation (Fig. 1b). Relative to the emission of 1 in the absence of DPA (Fig. 3, black plot), the same concentration of 1 in the presence of 2.2 × 10−5 M DPA yields a 25% decrease in the emission intensity of 1 along with appearance of the higher energy emission of DPA (Fig. 3, blue plot). Sensitizer 1 possesses no emission at wavelengths below 500 nm. When the concentration of DPA is increased further to 2.4 × 10−4 M, the intensity of the upconverted blue emission becomes greater, along with additional diminution of the emission of 1. The latter result indicates that the observed upconversion is not yet saturated at the concentrations employed. A control experiment in the presence of only DPA (2.2 × 10−5 M) demonstrates no appreciable fluorescence upon excitation at 500 nm, indicating that interaction between the excited state of 1 and DPA is essential for the upconversion seen by the DPA fluorescence. | ||
| Fig. 3 Photoluminescence spectra in deaerated CH2Cl2 (λex = 500 nm) of (a) sensitizer 1 (black plot, 2.2 × 10−5 M); (b) 1:1 mixture of 1 and DPA (blue plot, 2.2 × 10−5 M and 2.2 × 10−5 M); (c) 1:11 mixture of 1 and DPA (red plot, 2.2 × 10−5 M and 2.4 × 10−4 M). | ||
The relative emission quantum yields for upconversion vary depending on the concentration of DPA. For a DPA concentration of 2.2 × 10−5 M, it is estimated as 1.1%, whereas for a DPA concentration of 2.4 × 10−4 M, the upconversion quantum yield decreases to ∼0.2%. These quantum yields are estimated relative to the emission of the [Pt(ttpy)(CCPh)]ClO4 standard obtained from the plot shown in Fig. 3. The decrease in upconversion efficiency with increasing DPA concentration in the range given is consistent with DPA self-quenching.9
Laser driven upconversion
Selective excitation of [Pt(ttpy)(CCPh)]ClO4 (1) in deaerated methylene chloride solution containing DPA at room temperature using an argon ion-pumped dye laser also generates the upconverted blue fluorescence of DPA in the 400–470 nm region. With the laser excitation wavelength fixed at 514.5 nm, only 1 is selectively excited. Fig. 4a shows the upconverted fluorescence spectra with peak maxima at 414 nm, 432 nm and 460 nm (sh) as a function of laser power. The observed emission spectra are identical with emission obtained from DPA directly by continuous irradiation at 350 nm in solution (Fig. 1b and Fig. 3). The spectra in Fig. 4 confirm again that the blue fluorescence is that of DPA generated through upconversion sensitized by 1. At the same time, the photoluminescence of 1 between 500 nm and 800 nm becomes increasingly weaker relative to that in the absence of DPA as the laser power is increased.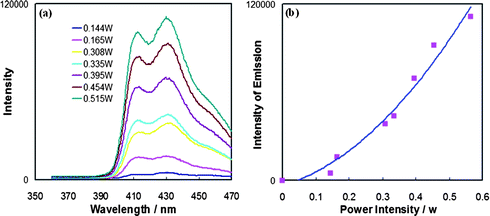 | ||
| Fig. 4 (a) Upconverted luminescence intensity as a function of incident laser power density (λex = 514.5 nm) from the mixture of the Pt sensitizer 1 (2.2 × 10−5 M) and DPA (2.4 × 10−4 M) in deaerated CH2Cl2; (b) upconverted emission signal intensity as a function of incident laser power density. | ||
As shown in Fig. 4a, an increase in the incident excitation power from 0.144 to 0.515 W cm−2 yields an increase in the upconverted fluorescence intensity of DPA. The intensity of the upconverted fluorescence is plotted as a function of the laser power density in Fig. 4b without normalization (emission intensity vs. laser power). The absence of a linear increase of emission intensity with laser power indicates that the DPA fluorescence from 390 to 470 nm occurs via a nonlinear process, which is in accord with previous analyses of TTA-produced upconversion.8,9,10a The solid line shown in Fig. 4b corresponds to a nearly quadratic fit of the emission intensity vs. power data with an exponential dependence of b = 1.9 for y = axb. The nearly quadratic dependence, as affirmed in a corresponding log(emission intensity) vs. log(laser power) plot, establishes that the upconverted emission signal is essentially proportional to the square of the incident power and hence to the square of the DPA triplet concentration. These results confirm the nonlinear photochemistry in TTA leading to photon upconversion.
In related studies, similar quadratic dependencies between the upconversion fluorescence intensity and the excitation power have been noted for the general mechanism of triplet sensitization by a heavy metal complex of the upconverted emission from an organic fluorophore.5,10a It is also noteworthy that the excitation power used in the present system, which is quite low (<1 W cm−2), is comparable to that used for the upconversion system based on the [Ru(dmb)3]2+ system (<1 W cm−2),9,10 but is higher than that used for the Pd(II) porphyrin–DPA system (<0.01 W cm−2),23 because the latter sensitizer has a much higher extinction coefficient at the excitation wavelength and a much longer excited state lifetime.
Scheme 1 is an energy level diagram of the upconversion process for the 1–DPA bimolecular system, showing the energies of the different species formed and the sequence of selective light absorption by 1, intersystem crossing, triplet sensitization of DPA and subsequent triplet–triplet annihilation to give 1DPA*. The net upconversion energy shift from the 3MLCT state of 1 to 1DPA* is ∼0.58 eV. The upconversion signal from DPA is quite stable for several hours upon continuous laser irradiation at 514.5 nm in deaerated solution.
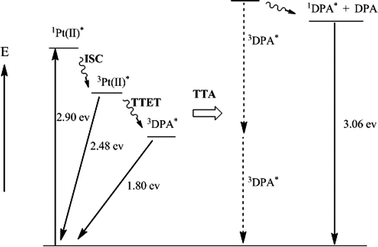 | ||
| Scheme 1 Energy level diagram of upconversion process. ISC = intersystem crossing; TTET = triplet–triplet energy transfer; TTA = triplet–triplet annihilation. | ||
Conclusions
The present work demonstrates for the first time the use of a platinum(II) terpyridine acetylide complex to sensitize the upconverted singlet blue fluorescence from 9,10-diphenylanthracene (DPA). The photoluminescence of Pt sensitizer 1 is readily quenched by DPA in the steady state and the upconverted blue emission results from a triplet–triplet annihilation (TTA) process, as confirmed by a quadratic dependence of the upconverted fluorescence intensity measured as a function of the laser power intensity. In addition to studies employing [Ru(dmb)3]2+, Ir(ppy)3, Pd(II) phthalocyanine and Pd(II) porphyrin complexes as triplet sensitizers, the current work demonstrates that platinum terpyridyl complexes can also promote energy upconversion in organic fluorophores through a triplet–triplet annihilation mechanism.Acknowledgements
We wish to thank the Division of Basic Sciences, U.S. Department of Energy for financial support of this research (DE-FG02-90ER14125). Dr Weih Zhao is greatly acknowledged for assistance with the laser experiments and we thank Mrs Shuang Liu for assistance with lifetime experiments.References
- (a) N. J. Turro, Modern Molecular Photochemistry, University Press, Menlo Park, CA, 1978 Search PubMed; (b) T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010 DOI:10.1016/j.ccr.2010.01.003 , 18th ISPPCC Issue.
- (a) C. A. Parker, Photoluminescence of Solutions, Elsevier, Amsterdam, 1968 Search PubMed; (b) J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970 Search PubMed.
- V. Yakutkin, S. Aleshchenkov, S. Chernov, T. Miteva, G. Nelles, A. Cheprakov and S. Baluschev, Chem.–Eur. J., 2008, 14, 9846 CrossRef CAS.
- P. E. Keivanidis, S. Baluschev, T. Miteva, G. Nelles, U. Scherf, A. Yasuda and G. Wegner, Adv. Mater., 2003, 15, 2095 CrossRef CAS.
- S. Baluschev, F. Yu, T. Miteva, S. Ahl, A. Yasuda, G. Nelles, W. Knoll and G. Wegner, Nano Lett., 2005, 5, 2482 CrossRef CAS.
- S. Baluschev, J. Jacob, Y. Avlasevich, P. E. Keivanidis, T. Miteva, A. Yasuda, G. Nelles, A. C. Grimsdale, K. Müllen and G. Wegner, ChemPhysChem, 2005, 6, 1250 CrossRef CAS.
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2008, 112, 3550 CrossRef CAS.
- T. N. Singh-Rachford, R. R. Islangulov and F. N. Castellano, J. Phys. Chem. A, 2008, 112, 3906 CrossRef CAS.
- (a) R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776 RSC; (b) R. R. Islangulov and F. N. Castellano, Angew. Chem., Int. Ed., 2006, 45, 5957 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 9266 CrossRef CAS.
- W. Zhao and F. N. Castellano, J. Phys. Chem. A, 2006, 110, 11440 CrossRef CAS.
- (a) V. W. W. Yam, R. P. L. Tang, K. M. C. Wong and K. K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS; (b) V.W.-W. Yam, R.P.-L. Tang, K.M.-C. Wong, X.-X. Lu, K.-K. Cheung and N. Zhu, Chem.–Eur. J., 2002, 8, 4066 CrossRef CAS.
- (a) P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726 CrossRef CAS; (b) P. Du, J. Schneider, P. Jarosz, J. Zhang, W. W. Brennessel and R. Eisenberg, J. Phys. Chem. B, 2007, 111, 6887 CrossRef CAS; (c) P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576 CrossRef CAS; (d) D. Zhang, L. Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440 CrossRef CAS.
- (a) S. Chakraborty, T. J. Wadas, H. Hester, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6865 CrossRef CAS; (b) A. Islam, H. Sugihara, K. Hara, L. P. Singh, R. Katoh, M. Yanagida, Y. Takahashi, S. Murata and H. Arakawa, Inorg. Chem., 2001, 40, 5371 CrossRef CAS.
- Q.-Z. Yang, L.-Z. Wu, Z.-X. Wu, L.-P. Zhang and C.-H. Tung, Inorg. Chem., 2002, 41, 5653 CrossRef CAS.
- J. M. Calvert, J. V. Caspar, R. A. Binstead, T. D. Westmoreland and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 6620 CrossRef CAS.
- G. Heinrich, S. Schoof and H. Gusten, J. Photochem., 1975, 3, 315 CrossRef.
- (a) M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447 CrossRef CAS; (b) T. J. Wadas, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2003, 42, 3772 CrossRef CAS.
- L. Vincze, F. Sandor, J. Pem and G. Bosnyak, J. Photochem. Photobiol., A, 1999, 120, 11 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CAS.
- F. E. Beideman and D. M. Hercules, J. Phys. Chem., 1979, 83, 2203 CrossRef CAS.
- (a) S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS; (b) R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652 CrossRef CAS.
- (a) M. S. Chan and J. R. Bolton, Sol. Energy, 1980, 24, 561 CrossRef CAS; (b) A. Launikonis, J. W. Loder, A. W.-H. Mau, W. H. F. Sasse, L. A. Summers and D. Wells, Aust. J. Chem., 1982, 35, 1341.
Footnote |
| † Electronic supplementary information (ESI) available: Luminescent lifetime measurements. See DOI: 10.1039/c0sc00244e |
| This journal is © The Royal Society of Chemistry 2010 |