Multiple approaches to enantiopure spirocyclic benzofuranones using organocatalytic cascade reactions†‡
Carlo
Cassani§
a,
Xu
Tian§
a,
Eduardo C.
Escudero-Adán
a and
Paolo
Melchiorre
*ab
aICIQ-Institute of Chemical Research of Catalonia, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: pmelchiorre@iciq.es; Tel: +34 977920208
bICREA-Institució Catalana de Recerca i Estudis Avançats, Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 19th August 2010
Abstract
Three distinct aminocatalytic cascade reactions leading to enantiomerically pure spirocyclic benzofuranones have been devised, highlighting the ability of organocascade to generate high degrees of stereochemical and architectural complexity in a single chemical transformation.
Finding cost-effective and sustainable synthetic ways to reproduce the rich structural diversity and complexity of natural molecules has always captured the attention of chemists,1 especially in relation to biologically active compounds.2 The emerging field of aminocatalytic cascade reactions3 has recently provided a way of achieving stereochemical and molecular complexity while addressing the requests for atom and step economy or protecting-group-free synthesis.4 The synthetic potential of this bio-inspired approach has been validated by recent applications of asymmetric aminocascade reactions to the total synthesis of natural products.5 Here, we report on complementary approaches to accessing enantiomerically pure spiro-benzofuranone cyclohexane derivatives with multiple stereocenters. Thus, we further illustrate the potential and versatility of aminocascade catalysis for facing challenging synthetic problems using disparate tactics. The three cascade reactions are based on distinct catalytic machineries and provide straightforward access to natural-product-inspired compound collections2b that would be difficult to synthesise by other catalytic methods.6
Our ongoing efforts7 to design novel aminocatalytic cascade reactions to access valuable complex compounds prompted us to investigate the preparation of spirocyclic benzofuranones. This scaffold is featured in a number of natural products8 that exhibit biological activities ranging from antioxidant properties to potential application in anticancer and Alzheimer's therapy (Fig. 1).
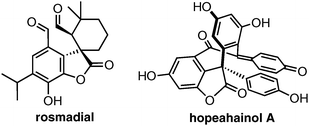 | ||
| Fig. 1 Naturally occurring and biologically active benzofuranones. | ||
We decided to attack the synthetic problem by exploiting the established ability of chiral secondary amine A9 to integrate orthogonal activation modes of aldehydes (enamine and iminium ion catalysis) into more elaborate cascade sequences.10 Based on the relatively low pKa of benzofuranone 1,11 we thought we might exploit its ambident nucleophilic profiles12 to design a cascade reaction with α,β-unsaturated aldehydes 2. As planned in Table 1, we anticipated that benzofuranone 1 should first intercept the iminium ion generated by catalyst A's condensation with enal 2. The resulting prochiral carbon nucleophile I should be reactive enough to initiate a second, intermolecular, iminium catalyzed conjugate addition with another molecule of 2 to afford intermediate II. The last intramolecular aldolization–dehydration sequence, driven by enamine catalysis, should afford the desired spiro-derivative 3.
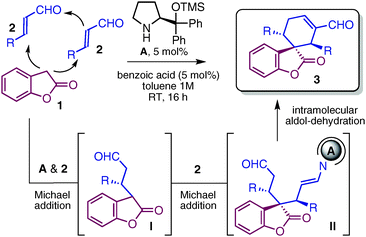
| Entry | R | 3 | Yieldb (%) | drc | eed |
|---|---|---|---|---|---|
| a The organocascade proceeds under the catalysis of A by way of an iminium-catalysed Michael addition of 1 to 2, followed by a second iminium-mediated Michael addition of the chiral nucleophilic intermediate I to another molecule of enal 2 and an enamine-driven intramolecular aldol reaction of II followed by a dehydration step, leading to the spiro compounds 3. Reactions were carried out at room temperature using 5 mol% of catalyst A and 3 equiv. of enals 2 on a 0.2 mmol scale. b Yield of isolated product. c A single diastereoisomer was always detected by 1H NMR analysis of the crude mixture. d ee of 3 determined by HPLC analysis. e Carried out with 1 mol% of catalyst A at 40 °C. | |||||
| 1 | Ph | a | 75 | >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 ∶1 |
>99 |
| 2e | Ph | a | 68 | >19∶1 |
>99 |
| 3 | p-NO2–C6H4 | b | 59 | >19∶1 |
>99 |
| 4 | p-MeO–C6H4 | c | 54 | >19∶1 |
>99 |
| 5 | p-Cl–C6H4 | d | 80 | >19∶1 |
99 |
| 6 | 2-Furyl | e | 49 | >19∶1 |
99 |
| 7 | Me | f | 51 | 19∶1 |
>99 |
| 8 | CO2Et | g | 38 | >19∶1 |
99 |
Our organocascade strategy was evaluated conducting the reactions in toluene at room temperature for 16 hours, under an aerobic atmosphere. Optimization experiments revealed that 5 mol% of catalyst A in combination with an acidic co-catalyst, such as benzoic acid (5 mol%), fully converted benzofuranone 2 into the spiro-adducts 3 with perfect control over the stereochemistry (a single stereoisomer out of the possible 8 being detected, de and ee >99%). Even a catalyst loading as low as 1 mol% afforded the enantiopure spirobenzofuranone 3a with good chemical yield and a practical reaction time of 16 hours (entry 2). As highlighted in Table 1, there appears to be significant tolerance toward structural and electronic variations of enals enabling access to a variety of complex spiro-compounds 3a–g with three stereocenters. Different substituents at the aromatic moiety, regardless of their electronic properties, and a hetero-aromatic substituent were well-tolerated (entries 1–6). Crotonaldehyde and ethyltrans-4-oxo-2-butenoate were also suitable substrates of the cascade (entries 7 and 8).
The ability to address a particular synthetic problem through disparate approaches is a general validation for the versatility and reliability of a chemical strategy. A central goal of our organocascade catalysis studies has been to demonstrate the potential of accessing enantiomerically pure spiro-benzofuranone cyclohexane derivatives using complementary organocascade reactions, based on distinct catalytic machineries. Inspired by previous studies from our laboratory7a–b and others,10a we recognized that identifying a suitable compound 4, bearing the benzofuranone moiety, was key to designing novel organocascade reactions. Compound 4 seemed well-tailored to first act as a Michael acceptor and then to originate, after the conjugate addition, a nucleophilic intermediate able to continue the cascade. Given this reactivity profile, we included compound 4 as the third component of a three-component domino strategy that exploited the catalytic ability of secondary amine A to realize an enamine–iminium–enamine sequential activation of aldehydes 5 and α,β-unsaturated aldehydes 2.10a As portrayed in Table 2, the triple organocascade provides fast and easy access to spiro-benzofuranone cyclohexene carbaldehydes 6 with almost perfect control over stereochemistry. This reaction proceeds at 40 °C in the presence of the catalytic salt A·o-F–C6H4CO2H (20 mol%) in toluene by way of a catalysed Michael/Michael/aldol condensation sequence. A variety of substituents at the β-position of the benzofuranone-based compounds 4 are well tolerated, as are different aldehydic substrate combinations.
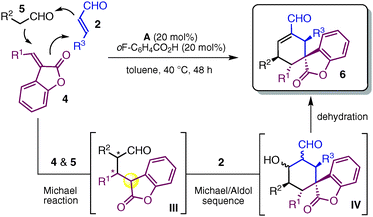
| Entry | R1 | R2 | R3 | 6 | Yieldb (%) | drc | eed |
|---|---|---|---|---|---|---|---|
|
a The triple organocascade proceeds by way of an enamine-catalysed Michael addition of 5 to 4, followed by an iminium-mediated Michael addition of the chiral nucleophilic intermediate III to 2 and an enamine-catalysed intramolecular aldol reaction to afford IV. The last dehydration step leads to the spirocyclic compounds 6. Reactions were carried out using 2 equiv. of aldehyde 5 and 1.5 equiv. of 4 on a 0.2 mmol scale.
b Yield of isolated product.
c A single diastereoisomer was always detected by 1H NMR analysis of the crude mixture.
d ee of 6 determined by HPLC analysis.
e Reaction carried out using isomerically pure 4 having an (E) geometry.
f Reaction carried out using a 3∶1 mixture of (E) and (Z) isomers of 4.
|
|||||||
| 1 | Phe | Me | Ph | a | 56 | >19∶1 |
>99 |
| 2 | Phf | Me | Ph | a | 58 | >19∶1 |
>99 |
| 3 | p-Cl–C6H4 | Me | Ph | b | 57 | >19∶1 |
99 |
| 4 | p-NO2–C6H4 | Me | Ph | c | 52 | >19∶1 |
>99 |
| 5 | Propyl | Me | Ph | d | 56 | >19∶1 |
>99 |
| 6 | CO2Et | Me | Ph | e | 54 | >19∶1 |
>99 |
| 7 | CO2Et | Me | Me | f | 70 | >19∶1 |
95 |
| 8 | CO2Et | Allyl | Ph | g | 57 | >19∶1 |
>99 |
Notably, using a 3∶1 mixture of E/Z isomers of compound 4 leads to the same stereochemical outcome as employing geometrically pure E isomer (compare entries 1 and 2, Table 2, a single stereoisomer, de and ee >99%).13 This stereo-convergent catalytic path provokes interesting mechanistic considerations,14 as well as delineating a more practical synthetic protocol (a mixture of 4 can be used). Detailed studies along this line are ongoing in our laboratories and will be reported in due course.15
Next, we wished to develop a complementary organocascade strategy based on the activation of ketone compounds, in which the spiro-benzofuranone cyclohexane architecture was constructed with the simultaneous creation of two bonds and three stereogenic centres in one single chemical step (Table 3). Central to the implementation of this strategy was the unique ability of the primary amine 9-amino(9-deoxy)epi-hydroquinine B16 to facilitate the formation of the nucleophilic dienamine intermediate V (influencing the equilibrium with the iminium ion generated in situ by condensation with enones 7) while selectively directing the reaction manifold toward a stepwise double-Michael addition sequence.7a,b The best results in terms of both yield and stereoselectivity were achieved using 20 mol% of amine A in combination with an acidic co-catalyst, such as o-fluoro benzoic acid (30 mol%). Results shown in Table 3 illustrate how this cascade enabled access to a variety of complex spiro-compounds 8 with a good diastereomeric and enantiomeric ratio.
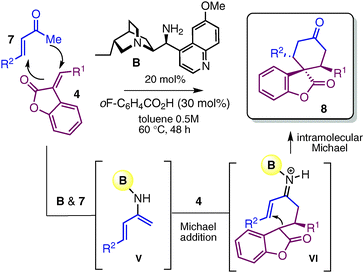
| Entry | R1 | R2 | 8 | Yieldb (%) | drc | eed |
|---|---|---|---|---|---|---|
|
a
4 first acts as a Michael acceptor, intercepting the nucleophilic dienamine intermediate V generated by catalyst B condensation with enones 7. The resulting prochiral carbon nucleophile II then selectively engages in an intramolecular, iminium catalyzed conjugate addition to afford spiro-derivatives 8. Reactions were carried out using 2 equiv. of enones 7 on a 0.2 mmol scale.
b Yield of isolated product, referred to as the sum of diastereoisomers.
c Determined by 1H NMR analysis of the crude mixture.
d ee of 8 determined by HPLC analysis.
e Reaction carried out using isomerically pure 4 having an (E) geometry.
f Reaction carried out using a 3∶1 mixture of (E) and (Z) isomers of 4.15
g Isolated yield of the single, major diastereoisomer.
|
||||||
| 1 | Phe | Ph | a | 70g | 5.5∶1 |
>99 |
| 2 | Phf | Ph | a | 71g | 6.3∶1 |
>99 |
| 3 | p-Cl–C6H4 | Ph | b | 75 | 3.5∶1 |
97 |
| 4 | p-NO2–C6H4 | Ph | c | 82 | 2∶1 |
98 |
| 5 | Propyl | Ph | d | 40 | >19∶1 |
91 |
| 6 | CO2Et | Ph | e | 77 | 2.2∶1 |
85 |
| 7 | Ph | p-Cl–C6H4 | f | 85 | 3.5∶1 |
97 |
| 8 | Ph | 3-Thienyl | g | 91 | 4∶1 |
96 |
The stereochemical outcomes for all three distinct cascade approaches were unambiguously determined by anomalous dispersion X-ray crystallographic analysis of the chlorine derivatives 3d, 6b, and 8b (CCDC 780474, 781708, and 781524, respectively).
In summary, we have presented three direct and easy approaches to accessing densely functionalized spirocyclic molecules in essentially enantiomerically pure form. In view of the abundance of important benzofuranone-derived natural products bearing a spiro-center in the 3-position of the heterocycle, we believe these methods could be useful in asymmetric synthesis.
This research was supported by the Institute of Chemical Research of Catalonia (ICIQ) Foundation, and Ministerio de Educación y Ciencia (grant Consolider Ingenio 2010-CSD2006-0003). We thank Dr J. Benet-Buchholz (X-ray Diffraction Unit, ICIQ) for the structures of 3d, 6b, and 8b.
Notes and references
- (a) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323–332 CrossRef CAS; (b) K. C. Nicolaou and S. A. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11929–11936 CrossRef CAS; (c) E. J. Sorensen and H. M. L. Davies (ed.), Special Issue on: Rapid Formation of Molecular Complexity in Organic Synthesis. Chem. Soc. Rev., 2009, 38, 2969–3276.
- (a) Stereochemical aspects of drug action and disposition, ed. M. Eichelbaum, B. Testa and A. Somogyi, Springer, 2003 Search PubMed; (b) K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2009, 48, 3224–3242 CrossRef CAS.
- (a) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570–1581 CrossRef CAS; (b) A. M. Walji and D. W. C. MacMillan, Synlett, 2007, 1477–1489 CAS; (c) B. List, Chem. Commun., 2006, 819–824 RSC; (d) B. Westermann, M. Ayaz and S. S. van Berkel, Angew. Chem., Int. Ed., 2009, 48, 846–849 CrossRef.
- (a) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS; (b) I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 Search PubMed; (c) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- For an inspiring review: C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 Search PubMed.
- To our knowledge, just one catalytic asymmetric method for the preparation of spiro-benzofuranones has been reported, based on an asymmetric palladium-catalyzed trimethylenemethane [3+2]-cycloaddition: B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396–12397 Search PubMed.
- (a) G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200–7203 CrossRef CAS; (b) L.-Y. Wu, G. Bencivenni, M. Mancinelli, A. Mazzanti, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7196–7199 CrossRef CAS; (c) P. Galzerano, F. Pesciaioli, A. Mazzanti, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7892–7894 CrossRef CAS. Ref. 7a describes organocascade approaches to spiro-oxindoles; see also: (d) X.-H. Chen, Q. Wei, S.-W. Luo, H. Xiao and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819–13825 CrossRef CAS.
- (a) A. A. Hussein, J. J. M. Meyer, M. L. Jimeno and B. Rodríguez, J. Nat. Prod., 2007, 70, 293–295 CrossRef CAS; for the isolation of hopeahainol A, see: (b) H. M. Ge, C. H. Zhu, D. H. Shi, L. D. Zhang, D. Q. Xie, J. Yang, S. W. Ng and R. X. Tan, Chem.–Eur. J., 2008, 14, 376–381 CrossRef CAS; (c) L. Pérez-Fons, M. T. Garzón and V. Micol, J. Agric. Food Chem., 2010, 58, 161–171 CrossRef CAS; (d) M. W. Pertino, C. Theoduloz, J. A. Rodríguez and V. Lazo, J. Nat. Prod., 2010, 73, 639–643 CrossRef CAS. For the synthesis and biological evaluation of hopeahainol A, see: (e) K. C. Nicolaou, T. R. Wu, Q. Kang and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2009, 48, 3440–3443 CrossRef CAS; (f) K. C. Nicolaou, Q. Kang, T. R. Wu, C. S. Lim and D. Y.-K. Chen, J. Am. Chem. Soc., 2010, 132, 7540–7548 CrossRef CAS.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS; (b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS.
- For leading examples: (a) D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861–863 CrossRef CAS; (b) A. Carlone, S. Cabrera, M. Marigo and K. A. Jorgensen, Angew. Chem., Int. Ed., 2007, 46, 1101–1104 CrossRef CAS.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1991, 56, 4218 CrossRef CAS.
- O. A. Reutov, I. P. Beletskaya and A. L. Kurts, Ambident Anions, Plenum, NY, 1983 Search PubMed. For the use of ambident anions in aminocatalytic cascade reactions with enals, see ref. 10b.
- Partial equilibration of the double-bond geometry of compound 4 bearing a β-phenyl group has been observed under the employed reaction conditions.
- The stereo-convergence of the triple cascade depicted in Table 2 might be rationalized on the basis of a reversible first enamine-catalysed Michael addition step between aldehydes 5 and 4, leading to intermediate III: a rapid equilibrium may account for a dynamic kinetic resolution process driven by the perfectly suited matched-pair combination of the chiral iminium ion (generated by condensation of catalyst B and enals 2) and only one out of the possible eight stereoisomers of intermediate III. Unfortunately, to date we were not able to isolate the key intermediate III. We are actively pursuing this target. See ref. 15 for another possible stereo-convergent path.
- At present, we cannot rule out a stereo-convergence path driven by a different reactivity of the E and Z isomers of 4, where the double-bond geometry scrambling continuously regenerates the fast reacting isomer.
- For recent reviews, see: (a) G. Bartoli and P. Melchiorre, Synlett, 2008, 1759–1771; (b) Y.-C. Chen, Synlett, 2008, 1919–1930 CrossRef CAS and references therein.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental, NMR and HPLC data. CCDC 780474 (3d), 781708 (6b), and 781524 (8b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc01957g |
| § C.C. and X.T. contributed equally to this research. |
| This journal is © The Royal Society of Chemistry 2011 |