Brønsted acid differentiated metal catalysis by kinetic discrimination†‡
Magnus
Rueping
* and
René M.
Koenigs
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, D-52074 Aachen, Germany. E-mail: magnus.rueping@rwth-aachen.de; Fax: +49 241 8092665
First published on 17th August 2010
Abstract
A Brønsted acid differentiated metal catalyzed hydrogenation has been developed. A combinatorial variation of chiral triflylamides with achiral metal complexes results in a highly active catalyst for the asymmetric reduction.
Over the past few decades, asymmetric catalysis has become a key feature of modern organic synthesis. Typically transition metal complexes bearing different chiral mono- or multidentate ligands were employed to perform reactions in a highly enantioselective fashion. Following the recent renaissance of organocatalysis numerous applications of metal-free reactions have additionally been reported.
The combination of organo- and transition metal catalysis is a contemporary concept and gives way to innovative reaction protocols that rely on the beneficial interplay of an organocatalyst and a metal catalyst.1 The potential of combined Brønsted acid and metal catalysis has been demonstrated by initial applications.2–4 However, in all the reactions developed only chiral phosphoric acid diesters or amino acids were utilized; the application of the more acidic N-triflylphosphoramides in combination with a metal catalyst has not been reported to date.
Given that achiral metal-triflimides and metal-triflates are highly reactive catalysts the analogous chiral metal–N-triflylphosphoramides should act as potent chiral catalysts for enantioselective transformations.
From a structural point of view, the anion of N-triflylphosphoramides can act similarly to the triflimide anion, either as a bidentate5a (Fig. 1a) or a monodentate ligand5b,c (Fig. 1b) or as a noncoordinating anion5d,6 (Fig. 1c). Furthermore, protonation of the basic ligand by the chiral N-triflylphosphoramide may result in ion-pair formation in close proximity to the metal centre (Fig. 1d).5e
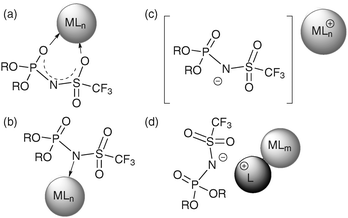 | ||
| Fig. 1 Coordination properties of chiral N-triflylphosphoramides and a cationic metal complex. | ||
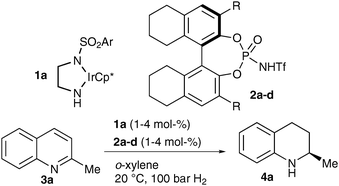
We decided to examine the combination of an achiral iridium(III) diamine complex7 and a chiral N-triflylphosphoramide. The Ir–diamine complexes are inactive in hydrogenation reactions; however, upon protonation by a strong Brønsted acid, they turn into highly active hydrogenation catalysts. If a chiral, strongly acidic Brønsted acid is applied, the acid not only activates the catalyst but also renders it chiral. Thus, enantioselectivities should be dependent on the chiral activating acid.
We started our investigations using an achiral ethylenediamine derived iridium(III) complex, different chiral N-triflylphosphoramides and quinaldine as a model substrate.8 To our delight, selectivities strongly depended on the nature of the substituents in the 3,3′ position of the chiral N-triflylphosphoramide, demonstrating that the chiral Brønsted acid fulfils both before mentioned purposes.
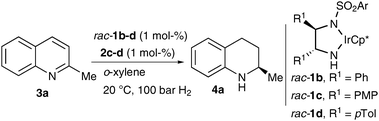
The validity of this assumption is further underlined by the interesting observation that the absolute configuration of the reaction product depends on the substituents in the 3,3′ position of the chiral N-triflylphosphoramide (Table 1, entries 3 and 4). To further improve the selectivities of the reaction we decided to vary the ethylenediamine backbone. Thus, the selectivities and reactivities of the hydrogenation of quinaldine were efficiently increased (Table 2). Further reaction optimization included the evaluation of different sulfonylated DPEN ligands and Brønsted acids. Moreover, the variation of solvents and hydrogen pressure was investigated.
| Entrya | Mol% | R | HA | Time | Conv.b | e.r.b |
|---|---|---|---|---|---|---|
| 1 1: Ar = 2-Naphthyl, TRIP = 2,4,6-(iPr3)Phenyl. a Reaction conditions: 0.15 mmol 3a, 0.8 mL o-xylene, 1 mol% [Ir], 1 mol% additive. b Determined by GC on chiral stationary column. | ||||||
| 1 | 4 | Phenyl | 2a | 24 | 25 | rac |
| 2 | 4 | 4-tBu-Phenyl | 2b | 24 | 25 | 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶57 ∶57 |
| 3 | 4 | TRIP | 2c | 24 | 50 | 31∶69 |
| 4 | 1 | 9-Phenanthryl | 2d | 60 | 40 | 66∶34 |
| Entrya | R1 | R | HA | Time | Conv.b | e.r.b |
|---|---|---|---|---|---|---|
| 1 1: Ar = 2-Naphthyl, TRIP = 2,4,6-(iPr3)Phenyl. a Reaction conditions: 0.15 mmol 3a, 0.8 mL o-xylene, 1 mol% [Ir], 1 mol% additive. b Determined by GC on chiral stationary column. | ||||||
| 1 | Ph | TRIP | 2c | 14 | 80 | 58∶42 |
| 2 | Ph | 9-Phenanthryl | 2d | 24 | >95 | 91∶9 |
| 3 | PMP | 9-Phenanthryl | 2d | 24 | 80 | 87∶13 |
| 4 | p-Tol | 9-Phenanthryl | 2d | 24 | >95 | 86∶14 |
These experiments showed that elevated hydrogen pressures and apolar, aromatic solvents are optimal for obtaining high selectivities. The latter finding supports the fact that non-protic solvents are beneficial in chiral ion-pair catalysis.
With the optimal conditions in hand, we examined the substrate scope of this newly developed asymmetric hydrogenation (Table 3). In general, a variety of 2′-substituted quinolines can be reduced to the corresponding tetrahydroquinolines in good yields and with good enantioselectivities if a racemic iridium(III) complex is used in combination with the chiral N-triflylphosphoramide 2d.

| Entrya | R1 | R2 | Yieldb | e.r.c | |
|---|---|---|---|---|---|
| a Reaction conditions: 0.3 mmol 3a–h, 1.6 mL o-xylene, 1 mol% catalyst. b Yield of isolated product. c Determined by GC on chiral stationary column. d 2 mol% 1b, 2 mol% 2d. | |||||
| 1 | 4a | H | Me | 96 | 91∶9 |
| 2 | 4b | 6-F | Me | 92 | 87∶13 |
| 3 | 4c | 6-Cl | Me | 93 | 86∶14 |
| 4 | 4d | 6-Br | Me | 97 | 84∶16 |
| 5d | 4e | 8-Cl | Me | 90 | 85∶15 |
| 6d | 4g | H | n-Butyl | 99 | 85∶15 |
| 7d | 4h | H | n-Pentyl | 95 | 83∶17 |
With regard to the reaction mechanism we were interested in explaining the observed selectivities. Therefore, we prepared the enantiopure iridium(III) complexes (R,R)-1b and (S,S)-1b and tested both in combination with 2d in the hydrogenation reaction of 2-methylquinoline. Interestingly it was observed that the two complexes were remarkably different with regard to both reactivity and selectivity. The combination including (S,S)-1b was less reactive than rac-1b. The opposite enantiomer (R,R)-1b exhibited slightly increased selectivities and further improvement was observed on cooling the reaction mixture. However, it is important to note that in order to perform this reaction in a highly enantioselective fashion chiral acidic additives are required as treatment of (R,R)-1b with the achiral N-triflylphosphoramide (TPA) leads to diminished selectivity.
Thus, the asymmetric induction of complex rac-1b/2d can only be rationalized by a kinetic differentiation of the two enantiomeric iridium catalysts.
Rac-1b is protonated by the chiral acid 2d, resulting in two different complexes of which the combination (R,R)-1b–H+/2d− is much more reactive than the combination of (S,S)-1b–H+/2d−. However, these diastereomeric complexes not only differ in their catalytic reactivity but also in the intrinsic selectivity (Fig. 2; Table 4, entries 3 and 5).9,10
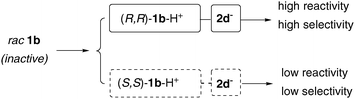 | ||
| Fig. 2 Chiral Brønsted acid differentiated metal catalysis. | ||
| Entrya | 1b | HA | Mol% | T/°C | Time/h | Conv.b | e.r.b |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 0.15 mmol 3a, 0.8 mL o-xylene. b Determined by GC on chiral stationary column. TPA = (PhO)2P(O)NHTf. | |||||||
| 1 | (S,S)-1b | — | 1 | 20 | 24 | no rct | — |
| 2 | (R,R)-1b | TPA | 1 | 20 | 24 | >95 | 72∶28 |
| 3 | (S,S)-1b | 2d | 1 | 20 | 40 | 75 | 16∶84 |
| 4 | rac-1b | 2d | 1 | 20 | 24 | >95 | 91∶9 |
| 5 | (R,R)-1b | 2d | 1 | 20 | 24 | >95 | 94∶6 |
| 6 | (R,R)-1b | 2d | 2 | −10 | 36 | >95 | 97∶3 |
Finally, we examined the substrate scope of the Brønsted acid differentiated iridium catalyzed hydrogenation of quinolines (Table 5). In general, a variety of different 2′-substituted quinolines, as well as different quinoline cores, can be reduced in good yields and with excellent enantioselectivities (up to 94% ee).
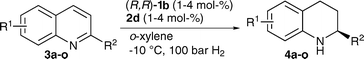
| Entrya | T/°C | R1 | R2 | Yieldb | e.r.c | |
|---|---|---|---|---|---|---|
| a Reaction conditions: 0.2 mmol 3a–n, 1.0 mL o-xylene, 1–4 mol% 1b and 2d, reaction time 24–48 h, reaction temperature as indicated, 100 bar H2 pressure. b Yield of isolated product. c Determined by GC or HPLC on chiral stationary column. | ||||||
| 1 | −10 | 4a | H | Me | 92 | 97∶3 |
| 2 | −10 | 4b | 6-F | Me | 85 | 94∶6 |
| 3 | 20 | 4c | 6-Cl | Me | 96 | 93∶7 |
| 4 | 20 | 4d | 6-Br | Me | 98 | 92 ∶8 |
| 5 | 20 | 4e | 8-Cl | Me | 83 | 92∶8 |
| 6 | −10 | 4f | H | n-Propyl | 77 | 95∶5 |
| 7 | −10 | 4g | H | n-Butyl | 82 | 94∶6 |
| 8 | −10 | 4h | H | n-Pentyl | 84 | 95∶5 |
| 9 | −10 | 4i | H | i-Butyl | 71 | 94∶6 |
| 10 | −10 | 4j | H | –(CH2)2–Ph | 74 | 95.5∶4.5 |
| 11 | −10 | 4k | H | –(CH2)2–(3,4-(OMe)2–Ph) | 66 | 95∶5 |
| 12 | −10 | 4l | H | –(CH2)2–(3,4-OCH2O–Ph) | 72 | 96∶4 |
| 13 | −10 | 4m | H | –(CH2)2–(3-OMe–Ph) | 81 | 96∶4 |
| 14 | −10 | 4n | H | –(CH2)2–(4-(Me)–Ph) | 76 | 96∶4 |
In summary, we herein report the first Brønsted acid differentiated metal catalyzed hydrogenation by kinetic discrimination. Applying this concept, we were able to show for the first time, that skillful combination of chiral N-triflylphosphoramides and cheap, racemic iridium complexes can be used to obtain good enantioselectivities. Furthermore, the optimal catalyst combination can be readily determined by utilizing a fast combinatorial approach in which racemic metal complexes are simply combined with chiral acids in order to identify the ideal catalyst system. To date, only chiral phosphoric acid diesters have been utilized in combination with metal complexes. Based on the different coordination properties of N-triflylphosphoramides (see Fig. 1) the repertoire of combined Brønsted acid–metal catalysis is not only enhanced by an additional Brønsted acid, but also by the possibility of designing substantially different chiral complexes bearing mono- or bidentate or even non-coordinating chiral anions that could conceivably exhibit distinct activation modes.
We thank Evonik Degussa for generous support and the Fonds der chemischen Industrie (FCI) for a scholarship to RMK.
Notes and references
- (a) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (b) C. Zhong and X. Shi, Eur. J. Org. Chem., 2010, 2999–3025 CrossRef CAS; (c) M. Rueping, R. M. Koenigs and J. Atodiresei, Chem. Eur. J., 2010 DOI:10.1002/chem.201001140.
- M. Rueping, A. P. Antonchick and C. Brinkmann, Angew. Chem., Int. Ed., 2007, 46, 6903–6906 CrossRef CAS.
- Selected articles: (a) M.-C. Lacasse, C. Poulard and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 12440–12441 CrossRef CAS; (b) V. Komanduri and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 16448–16449 CrossRef CAS; (c) G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496–499 CrossRef CAS; (d) S. Mukherjee and B. List, J. Am. Chem. Soc., 2007, 129, 11336–11337 CrossRef CAS; (e) W. Hu, X. Xu, J. Zhou, W.-J. Liu, H. Huang, J. Hu, L. Yang and L.-Z. Gong, J. Am. Chem. Soc., 2008, 130, 7782–7783 CrossRef CAS; (f) X. Xu, J. Zhou, L. Yang and W. Hu, Chem. Commun., 2008, 6564–6566 RSC; (g) K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2008, 130, 14452–14453 CrossRef CAS; (h) G. L. Hamilton, T. Kanai and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 14984–14985 CrossRef CAS; (i) Z.-Y. Han, H. Xiao, X.-H. Chen and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 9182–9183 CrossRef CAS; (j) X.-Y. Liu and C.-M. Che, Org. Lett., 2009, 11, 4204–4207 CrossRef CAS; (k) M. Terada and Y. Toda, J. Am. Chem. Soc., 2009, 131, 6354–6355 CrossRef CAS; (l) Y. Lu, T. C. Johnstone and B. A. Arndtsen, J. Am. Chem. Soc., 2009, 131, 11284–11285 CrossRef CAS; (m) Q. Cai, Z.-A. Zhao and S.-L. You, Angew. Chem., Int. Ed., 2009, 48, 7428–7431 CrossRef CAS; (n) Q.-W. Zhang, C.-A. Fan, H.-J. Zhang, Y.-Q. Tu, Y.-M. Zhao, P. Gu and Z.-M. Chen, Angew. Chem., Int. Ed., 2009, 48, 8572–8574 CrossRef CAS; (o) B. Zhao, H. Du and Y. Shi, J. Org. Chem., 2009, 74, 8392–8395 CrossRef CAS; (p) S. Liao and B. List, Angew. Chem., Int. Ed., 2010, 49, 628–631 CAS; (q) R. L. Lalonde, Z. J. Wang, M. Mba, A. D. Lackner and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 598–601 CAS; (r) L. Yang, Q. Zhu, S. Guo, B. Qian, C. Xia and H. Huang, Chem.–Eur. J., 2010, 16, 1638–1645 CrossRef CAS.
- (a) C. Li, C. Wang, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2008, 130, 14450–14451 CrossRef CAS; (b) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967–6969 CrossRef CAS; (c) B. Villa-Marcos, C. Li, K. R. Mulholland, P. J. Hogan and J. Xiao, Molecules, 2010, 15, 2453–2472 Search PubMed. In all cases only the corresponding enantiopure iridium complexes were utilized. Racemic complexes were not investigated. Also see: (d) M. Klussmann, Angew. Chem., Int. Ed., 2009, 48, 7124–7125 CrossRef CAS.
- (a) A. Babai and A.-V. Mudring, Inorg. Chem., 2006, 45, 3249–3255 CrossRef CAS; (b) L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704–4707 CrossRef CAS; (c) O. G. Polyakov, S. M. Ivanova, C. M. Gaudinski, S. M. Miller, O. P. Anderson and S. H. Strauss, Organometallics, 1999, 18, 3769–3771 CrossRef CAS; (d) U. Hintermair, T. Gutel, A. M. Z. Slawin, D. J. Cole-Hamilton, C. C. Santini and Y. Chauvin, J. Organomet. Chem., 2008, 693, 2407–2414 CrossRef CAS; (e) C. S. Letko, Z. M. Heiden and T. B. Rauchfuss, Eur. J. Inorg. Chem., 2009, 4927–4930 CrossRef CAS.
- Selected important articles on chiral non-coordinating anions in asymmetric catalysis: (a) J. Lacour and V. Hebbe-Viton, Chem. Soc. Rev., 2003, 32, 373–382 RSC; (b) J. Lacour and D. Moraleda, Chem. Commun., 2009, 7073–7089 RSC; (c) D. B. Llewellyn, D. Adamson and B. A. Arndtsen, Org. Lett., 2000, 2, 4165–4168 CrossRef CAS; (d) D. Chen, B. Sundararaju, R. Krause, J. Klankermeyer, P. H. Dixneuf and W. Leitner, ChemCatChem, 2010, 2, 55–57 Search PubMed; (e) S. P. Smidt, N. Zimmermann, M. Studer and A. Pfaltz, Chem.–Eur. J., 2004, 10, 4685–4693 CrossRef CAS.
- Selected articles on iridium(III)–diamine catalysts in asymmetric reductions: (a) K. Mashima, T. Abe and K. Tani, Chem. Lett., 1998, 1199–1200 CrossRef CAS; (b) K. Murata, T. Ikariya and R. Noyori, J. Org. Chem., 1999, 64, 2186–2187 CrossRef CAS; (c) A. Ros, A. Magriz, H. Dietrich, M. Ford, R. Fernandez and J. M. Lassaletta, Adv. Synth. Catal., 2005, 347, 1917–1920 CrossRef CAS; (d) T. Ohkuma, N. Utsumi, M. Watanabe, K. Tsutsumi, N. Arai and K. Murata, Org. Lett., 2007, 9, 2565–2567 CrossRef; (e) X. Wu, J. Liu, X. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718–6722 CrossRef CAS; (f) Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2007, 129, 14303–14310 CrossRef CAS; (g) S.-Y. Shirai, H. Nara, Y. Kayaki and T. Ikariya, Organometallics, 2009, 28, 802–809 CrossRef CAS; (h) O. Soltani, M. A. Ariger and E. M. Carreira, Org. Lett., 2009, 11, 4196–4198 CrossRef CAS.
- Selected articles on organocatalytic reductions from our group: (a) M. Rueping, T. Theissmann and A. P. Antonchick, Synlett, 2006, 1071–1074 CrossRef CAS; (b) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683–3686 CrossRef; (c) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 6751–6755 CrossRef CAS; (d) M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2007, 46, 4562–4565 CrossRef CAS; (e) M. Rueping, T. Theissmann, S. Raja and J. W. Bats, Adv. Synth. Catal., 2008, 350, 1001–1006 CrossRef CAS; (f) M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2008, 47, 5836–5838 CrossRef CAS; (g) M. Rueping, F. Tato and F. R. Schoepke, Chem.–Eur. J., 2010, 16, 2688–2691 CrossRef CAS; (h) M. Rueping, E. Sugiono and F. R. Schoepke, Synlett, 2010, 852–865 CrossRef CAS; (i) M. Rueping and T. Theissmann, Chem. Sci., 2010 10.1039/c0sc00206b.
- Selected review articles regarding kinetic discrimination: (a) K. Mikami, M. Terada, T. Korenaga, Y. Matsumoto, M. Ueki and R. Angelaud, Angew. Chem., Int. Ed., 2000, 39, 3532–3556 CrossRef CAS; (b) K. Muñiz and C. Bolm, Chem.–Eur. J., 2000, 6, 2309–2316 CrossRef; (c) P. J. Walsh, A. E. Lurain and J. Balsells, Chem. Rev., 2003, 103, 3297–3344 CrossRef CAS; (d) J. W. Faller, A. R. Lavoie and J. Parr, Chem. Rev., 2003, 103, 3345–3368 CrossRef CAS; (e) K. Mikami and M. Yamanaka, Chem. Rev., 2003, 103, 3369–3400 CrossRef CAS.
- Selected articles regarding kinetic discrimination: (a) K. Mikami and S. Matsukawa, Nature, 1997, 385, 613–615 CrossRef CAS; (b) K. Mikami, T. Korenaga, M. Terada, T. Ohkuma, T. Pham and R. Noyori, Angew. Chem., Int. Ed., 1999, 4, 495–497 CrossRef; (c) J. Long and K. L. Ding, Angew. Chem., Int. Ed., 2001, 40, 544–547 CrossRef CAS; (d) A. M. Costa, C. Jimeno, J. Gavenonis, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2002, 124, 6929–6941 CrossRef CAS; (e) M. T. Reetz and X. Li, Angew. Chem., Int. Ed., 2005, 44, 2962–2964 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and spectra. See DOI: 10.1039/c0cc02167a |
| This journal is © The Royal Society of Chemistry 2011 |