Pushing the limits of steric demand around a biaryl axis: synthesis of tetra-ortho-substituted biaryl naphthalenes†‡
Adam C.
Glass
,
Sam
Klonoski
,
Lev N.
Zakharov§
and
Shih-Yuan
Liu
*
Department of Chemistry, University of Oregon, Eugene, Oregon 97403, USA. E-mail: lsy@uoregon.edu; Fax: +1 (541) 346-0487; Tel: +1 (541) 346-5573
First published on 23rd August 2010
Abstract
The synthesis of tetra-ortho-substituted biaryl naphthalenes, including examples bearing multiple ortho-isopropyl groups, has been developed via a catalytic rearrangement process.
Substituted biaryls are important substructures of many biologically active compounds,1 organic materials,2 and ligands in metal catalysts.3 The development of methods for the synthesis of biaryls has mostly been dominated by metal-mediated coupling chemistry (i.e., cross-coupling,4 direct arylation,5 and oxidative coupling6) due to its versatility. However, the assembly of highly substituted biaryls, in particular tetra-ortho-substituted derivatives, has generally been challenging for coupling-based strategies.7,8 This is arguably due to the steric demand imposed by the coupling of two bis-ortho-substituted biaryl precursors. Alternative approaches toward the construction of tetra-ortho-substituted biaryls have recently been developed that include annulation-based strategies, e.g., Diels–Alder9 and [2+2+2] cycloadditions.10
Despite the significant advances that have been made to date, novel strategies for the generation of tetra-ortho-substituted biaryls are still highly desired. We have been pursuing a rearrangement-based approach for the synthesis of substituted biaryl naphthalenes.11–13Scheme 1 illustrates our strategy in which the addition of an arene nucleophile to the starting material A is followed by a ring-expansion rearrangement to furnish the desired biaryl naphthalene Cvia a cyclopropyl carbinol intermediate B. The distinguishing feature of our approach is that the key C–C bond-forming step is accomplished through a simple addition of a nucleophile to a carbonyl.14 Given the strong thermodynamic driving force for this nucleophilic addition,15 we envision that our method could serve as a potential method for the construction of tetra-ortho-substituted biaryl naphthalenes. In this communication, we demonstrate that a range of tetra-ortho-substituted biaryl naphthalenes can be produced via our method, including a very hindered tetra-ortho-substituted biaryl naphthalene bearing three isopropyl groups in the ortho positions.
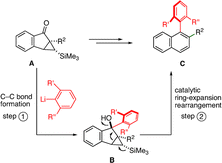 | ||
| Scheme 1 Ring-expansion rearrangement strategy for the synthesis of tetra-ortho-substituted biaryl naphthalenes. | ||
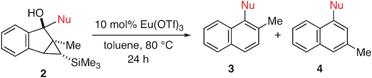
We first investigated the feasibility of the nucleophilic addition to starting material A (Step 1 in Scheme 1). We chose cyclopropyl carbonyl compound 1 (Table 1) as a representative electrophile for our survey. As can be seen from Table 1, the isolated yields of the addition products 2 are relatively independent of the electronic nature (entries 1–5) as well as the steric demand (entry 1 vs. entry 8, and entries 5–7) of the nucleophiles. A consistent yield of ∼70% yield was observed. Somewhat surprisingly, the highest isolated yield (81%) for the generation of 2 resulted from the addition of the largest nucleophile, 2,4,6-triisopropylphenyllithium (entry 7).
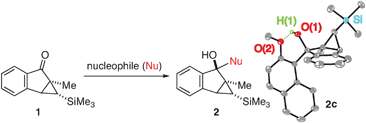







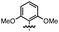
This nucleophilic addition reaction is highly diastereoselective. Only one single diastereomer was observed for each of the products 2 (as analyzed by 1H NMR). We have structurally characterized the adduct between 2 and 2-methoxy-1-naphthyllithium, i.e., 2c, viasingle crystal X-ray crystallography. The relative stereochemistry of the structure is consistent with an approach of the nucleophile opposite the blocking silylcyclopropane group (see Table 1). Interestingly, compound 2c adopts a conformation in the solid state where the carbinol proton H(1) engages in hydrogen bonding with the oxygen O(2) of the methoxynaphthalene (O(2)–H(1) = 1.83(2) Å, sum of van der Waals radii = 2.72 Å).16
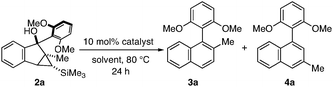
Having established the synthesis of an array of rearrangement precursors 2, we turned our attention to the catalytic ring-expansion rearrangement. We determined that the rearrangement of cyclopropyl carbinol 2a in the presence of a catalytic amount of Lewis acid furnished the desired tetra-ortho-substituted biaryl 3a, however as a 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27 mixture with its undesired tri-ortho-substituted isomer 4a (Table 2, entry 1).11 We discovered that the choice of solvent has a dramatic influence on regioselectivity. When the reaction is performed in toluene instead of 1,2-dichloroethane, the ratio of 3a to 4a improves to 93:7 (Table 2, entry 2). Other solvents that we screened did not improve the yield and/or the regioselectivity significantly (Table 2, entries 3–8). Interestingly, a reversal of regioselectivity was observed when acetonitrile was used as the solvent (Table 2, entry 8). We also screened a number of Lewis acids (Table 2, entries 9–12) and found europium triflate (entry 2) to be the most efficient catalyst under our conditions.
:27 mixture with its undesired tri-ortho-substituted isomer 4a (Table 2, entry 1).11 We discovered that the choice of solvent has a dramatic influence on regioselectivity. When the reaction is performed in toluene instead of 1,2-dichloroethane, the ratio of 3a to 4a improves to 93:7 (Table 2, entry 2). Other solvents that we screened did not improve the yield and/or the regioselectivity significantly (Table 2, entries 3–8). Interestingly, a reversal of regioselectivity was observed when acetonitrile was used as the solvent (Table 2, entry 8). We also screened a number of Lewis acids (Table 2, entries 9–12) and found europium triflate (entry 2) to be the most efficient catalyst under our conditions.
| Entry | Catalyst | Solvent | Yield %a |
3a:4ab |
|---|---|---|---|---|
| a GC yield (3a + 4a) vs. a calibrated internal standard, average of two runs. b Determined by GC. | ||||
| 1 | Eu(OTf)3 | 1,2-Dichloroethane | 68 | 73:27 |
| 2 | Eu(OTf)3 | Toluene | 78 |
93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :7 :7
|
| 3 | Eu(OTf)3 | THF | 54 | 92:8 |
| 4 | Eu(OTf)3 | DMF | 29 | 76:24 |
| 5 | Eu(OTf)3 | t-BuOH | 42 | 82:18 |
| 6 | Eu(OTf)3 | PhCl | 53 | 96:4 |
| 7 | Eu(OTf)3 | 1,3-Dichlorobenzene | 52 | 93:7 |
| 8 | Eu(OTf)3 | MeCN | 15 | 25:75 |
| 9 | Sm(OTf)3 | Toluene | 76 | 85:15 |
| 10 | Er(OTf)3 | Toluene | 79 | 81:19 |
| 11 | SnCl4 | Toluene | 31 | 40:60 |
| 12 | BF3·Et2O | Toluene | 29 | 35:65 |
With an optimized rearrangement protocol established, we subjected the various rearrangement precursors 2 from Table 1 to these conditions to test the scope of our method. We were pleased to see that a variety of substrates rearranged to furnish the desired biaryl naphthalenes in moderate to good yields (Table 3). With the exception of entry 1 (Nu = 2,6-dimethoxyphenyl), which gave a 93:7 mixture of regioisomers, all other substrates that we tested gave the tetra-ortho-substituted isomer exclusively. Noteworthy is the rearrangement of 2g, which produced a tetra-ortho-substituted biaryl bearing two ortho-isopropyl groups in 86% yield.







Encouraged by these results, we sought to push the limits of steric demand around the biaryl axis by replacing the α-methyl group in 1 with an isopropyl group. The corresponding precursor 5 (Scheme 2) can be synthesized in a few steps from commercially available indanone.17Scheme 2 shows that a series of bis-ortho-substituted phenyllithium nucleophiles add to the carbonyl electrophile 5 in modest yield.
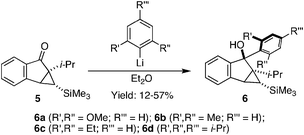 | ||
| Scheme 2 Nucleophilic addition to 5. | ||
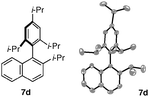
We were very pleased to observe that intermediates 6a–6d underwent catalytic ring opening rearrangement under our optimized conditions to yield hindered tetra-ortho-substituted biaryl naphthalenes. As can be seen from Table 4, the isolated yields are independent of the steric demand around the biaryl axis. “Nucleophiles” (Nu in Table 4) containing methoxy, methyl, ethyl, and isopropyl groups in the 2,6-positions rearrange to produce the desired biaryl in reasonable yield (entries 1–4). We were also very pleased to see that the undesired regioisomer 8 is not observed under our reaction conditions. Noteworthy is the synthesis of 7d, which is a tetra-ortho-substituted biaryl containing three ortho-isopropyl substituents. To the best of our knowledge, it is the most sterically encumbered biaryl naphthalene that has been synthesized to date. We have structurally characterized 7dviasingle crystal X-ray diffraction, thus unambiguously establishing its identity.
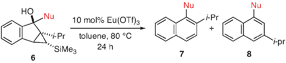





We have initiated preliminary studies toward an asymmetric version of this process. To this end, we successfully isolated optically pure 1via semi-preparatory chiral HPLC. Treatment of optically pure 1 with 2-methoxynaphthyllithium and subsequent catalytic rearrangement with Eu(OTf)3 furnished the desired tetra-ortho-substituted biaryl naphthalene 3c in 54% isolated yield (over two steps) in 52% ee. Current efforts are geared toward optimizing the efficiency of this asymmetric process and developing a better understanding of its mechanism.
Support has been provided by the University of Oregon. This material is based upon work supported by the National Science Foundation under Grant No. DGE-0742540 (A.C.G.).
Notes and references
- For examples, see: G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615–624 Search PubMed.
- For recent examples, see: (a) K. Takaishi, M. Kawamoto and K. Tsubaki, Org. Lett., 2010, 12, 1832–1835 CrossRef CAS; (b) Y. Ueno, S. Komatsuzaki, K. Takasu, S. Kawai, Y. Kitamura and Y. Kitade, Eur. J. Org. Chem., 2009, 4763–4769 CrossRef CAS.
- For an overview of biaryl phosphine, BINAP, and BINOL ligands in catalysis applications, see: (a) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS; (b) M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801–1836 CrossRef CAS; (c) J. M. Brunel, Chem. Rev., 2005, 105, 857–897 CrossRef CAS.
- For an overview, see: Modern Arylation Methods, ed. L. Ackermann, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed.
- For an overview, see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS; (b) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC.
- For an overview, see: (a) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC; (b) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540–548 RSC.
- For recent successful examples, see: (a) M. G. Organ, S. Calimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383–2387 CAS; (b) L. Ackermann, H. K. Potukuchi, A. Althammer, R. Born and P. Mayer, Org. Lett., 2010, 12, 1004–1007 CrossRef CAS.
- For pioneering work in the synthesis of tetra-ortho-substituted biaryls via cross-coupling, see: (a) S. Miyano, S. Okada, T. Suzuki, S. Handa and H. Hashimoto, Bull. Chem. Soc. Jpn., 1986, 59, 2044–2046 CAS; (b) C. Dai and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 2719–2724 CrossRef CAS; (c) A. F. Littke, L. Schwarz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343–6348 CrossRef CAS; (d) J. Yin, M. P. Rainka, X. X. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 1162–1163 CrossRef CAS; (e) J. E. Milne and S. L. Buchwald, J. Am. Chem. Soc., 2004, 126, 13028–13032 CrossRef CAS.
- For leading references, see: (a) J. R. Perkins and R. G. Carter, J. Am. Chem. Soc., 2008, 130, 3290–3291 CrossRef CAS; (b) M. Hapke, A. Gutnov, N. Weding, A. Spannenberg, C. Fischer, C. Benkhäuser-Schunk and B. Heller, Eur. J. Org. Chem., 2010, 509–514 CrossRef CAS.
- For leading references, see: (a) K. Tanaka, Chem.–Asian J., 2009, 4, 508–518 CrossRef CAS; (b) G. Nishida, K. Noguchi, M. Hirano and K. Tanaka, Angew. Chem., Int. Ed., 2007, 46, 3951–3954 CrossRef CAS.
- A. C. Glass, B. B. Morris, L. N. Zakharov and S.-Y. Liu, Org. Lett., 2008, 10, 4855–4857 CrossRef CAS.
- For a comprehensive review on the synthesis of substituted naphthalenes, see: C. B. de Koning, A. L. Rousseau and W. A. L. van Otterlo, Tetrahedron, 2003, 59, 7–36 Search PubMed.
- For leading references of biaryl naphthalene synthesis using rearrangement-based methods, see: (a) Y. Nishii, T. Yoshida, H. Asano, K. Wakasugi, J. Morita, Y. Aso, E. Yoshida, J. Motoyoshiya, H. Aoyama and Y. Tanabe, J. Org. Chem., 2005, 70, 2667–2678 CrossRef CAS; (b) G. S. Viswanathan, M. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2002, 41, 2138–2141 CrossRef CAS; (c) T. Shibata, Y. Ueno and K. Kanda, Synlett, 2006, 411–414 CrossRef CAS; (d) T. Hamura, T. Suzuki, T. Matsumoto and K. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 6294–6296 CrossRef CAS; (e) Z. B. Zhu, Y. Wei and M. Shi, Chem.–Eur. J., 2009, 15, 7543–7548 CrossRef CAS; (f) A. Padwa, U. Chiacchio, D. J. Fairfax, J. M. Kassir, A. Litrico, M. A. Semones and S. L. Xu, J. Org. Chem., 1993, 58, 6429–6437 CrossRef CAS . To the best of our knowledge, there were no examples of tetra-ortho-substituted biaryl naphthalenes in these reports.
- M. Hatano, T. Miyamoto and K. Ishihara, Curr. Org. Chem., 2007, 11, 127–157 CrossRef CAS.
- For example, the enthalpy of the addition of MeMgBr to di-t-butyl ketone has been determined to be −150 kJ mol−1, see: T. Holm, Acta Chem. Scand., Ser. B, 1976, 30, 985–990 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- See Supporting Information for details.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data. CCDC 778408 and CCDC 778409 contain the supplementary crystallographic data for complex 2c and 7d, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02170a |
| § Correspondence concerning X-ray crystallography should be directed to Lev Zakharov (E-mail: lev@uoregon.edu). |
| This journal is © The Royal Society of Chemistry 2011 |