Definite coordination arrangement of organometallic palladium complexes accumulated on the designed interior surface of apo-ferritin†‡§
Ziyue
Wang
a,
Yusuke
Takezawa
b,
Hiroki
Aoyagi
a,
Satoshi
Abe
c,
Tatsuo
Hikage
d,
Yoshihito
Watanabe
a,
Susumu
Kitagawa
ce and
Takafumi
Ueno
*cf
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Nagoya, 464-8602, Japan
bResearch Center for Material Science, Nagoya University, Furo-cho, Nagoya, 464-8602, Japan
cInstitute for Integrated Cell-Material Sciences (iCeMS), Funai Center, Kyoto University, Katsura, Nishikyou-ku, Kyoto, 615-8510, Japan. E-mail: taka@icems.kyoto-u.ac.jp
dHigh Intensity X-ray Diffraction Laboratory, Nagoya University, Furo-cho, Nagoya, 464-8602, Japan
eDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyou-ku, Kyoto, 615-8510, Japan
fPRESTO, Japan Science and Technology Agency (JST), Honcho, Kawaguchi, Saitama, 332-0012, Japan
First published on 23rd August 2010
Abstract
Apo-ferritin (apo-Fr) mutants are used as scaffolds to accommodate palladium (allyl) complexes. Various coordination arrangements of the Pd complexes are achieved by adjusting the positions of cysteine and histidine residues on the interior surface of the apo-Fr cage.
In the past several decades, the creation of functional molecules by incorporation of metal complexes into proteins has been an intriguing subject.1–6 This research effort, which stands at the convergence of chemistry, biology and material sciences, has led to promising achievements for new applications in catalysis,2 medicine,7,8 and bioimaging,9,10 among other areas. In many cases, the functions of these metal-containing proteins depend substantially on their molecular structures, particularly the structure of the protein environments in the vicinity of the bound metal ions. Thus, the development of techniques to enable design and provide accurate control of the binding structures of metal complexes within proteins has become one of the most attractive and significant research topics in the interdisciplinary fields of coordination chemistry and biology.1,3,4,11 Although comprehensive knowledge about the mechanisms for coordination of metal complexes and metal ions within proteins is needed, these mechanisms have remained obscure even for some widely researched protein templates such as myoglobin,1,4 streptavidin,12 and ferritin.13–15
Ferritin (Fr) is composed of 24 subunits and has an iron-storage cage with an internal diameter of 8 nm.16 Three-fold channels are formed at the positions where three adjacent subunits intersect. These channels provide the pathway for the penetration of foreign molecules or metal ions into the protein cavity.17 Although the subunits are assembled by non-covalent interactions, Fr is stable enough to retain an intact cage up to 70 °C and over the pH range of 2–11.18 These properties have allowed various nanoparticles of Au, Ag, Pd, CdS and CdSe to be synthesized within the apo-Fr cage.19–23 Moreover, apo-Fr has been used as a vehicle for diverse metal complexes.24–28 Yang et al. have succeeded in encapsulating the anticancer drugs cisplatin and carboplatin within the apo-Fr cage, and found that the composites exert a cytotoxic effect on tumor cells.27 It has been found that a composite of apo-Fr with Rh(nbd) complexes can serve as a nano-reactor for a polymerization reaction and effectively restrict the molecular weight distribution of the polymer products.24 While apo-Frs containing metal complexes have been found to be applicable as catalytic reagents, drug delivery systems, and for the development of biotechnological applications, there have been few reports on the detailed mechanism of coordination of metal complexes within the ferritin cage.13 In this communication, we report that the coordination arrangement of Pd(allyl) complexes on the interior surface of the apo-Fr cage can be precisely controlled as a result of adjustment of the positions of cysteine and histidine residues. This work is expected to contribute to the establishment of guidelines for the design of artificial metalloproteins adapted to contain synthetic metal complexes in well defined environments.
We have reported on the incorporation of [Pd(allyl)Cl]2 (allyl = η3-C3H5) into the cage of wild-type apo-rHLFr (recombinant L-chain apo-Fr from horse liver).29 The crystal structure of Pd(allyl)·apo-rHLFr reveals that one subunit of apo-rHLFr has two binding domains for Pd(allyl) complexes, on the interior surface of the apo-rHLFr cage and centered at Cys48 in the accumulation center and at Cys126 in the three-fold channel (Fig. 1). At each binding site, a dinuclear Pd center is formed through the thiol-bridging ligation of the cysteine residues. The nearby histidines, allyl ligands and water molecules work together with the cysteines to bind Pd ions with square-planar coordination geometry (Fig. 1b and 2a). Moreover, we found that replacing either cysteine or histidine residues in these two binding sites with alanines dramatically changes the metal binding structures within the cages of the Pd(allyl)·apo-rHLFr mutants.13 These results indicate that cysteine and histidine are essential residues which restrain the accumulation of Pd complexes, and that it is possible to construct new binding sites for Pd complexes within the apo-rHLFr cage by inserting or removing cysteine and histidine residues. On the basis of this hypothesis, new Pd(allyl)·apo-rHLFr composites were prepared using the rationally-designed mutants apo-E45C/C48A, E45C/R52H, and E45C/H49A/R52C-rHLFrs.
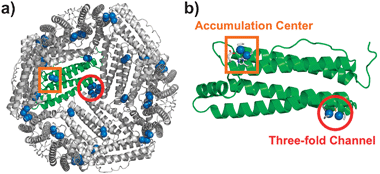 | ||
| Fig. 1 Crystal structure of Pd(allyl)·apo-rHLFr: (a) interior surface of 24-mer and (b) the subunit structure. The Pd atoms and the ligands are shown as sphere and tube models, respectively. | ||
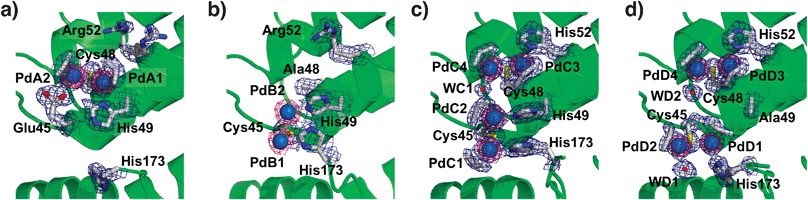 | ||
| Fig. 2 The accumulation centers of Pd(allyl)·apo-rHLFr (a), Pd(allyl)·apo-E45C/C48A-rHLFr (b), Pd(allyl)·apo-E45C/R52H-rHLFr (c), and Pd(allyl)·apo-E45C/H49A/R52H-rHLFr (d). The Pd atoms are indicated as sphere models colored with cyan. The O atoms of water molecules are shown as red spheres. The anomalous difference Fourier maps at 4.0σ as shown in magenta indicate the positions of palladium atoms. The selected 2|Fo| − |Fc| electron density maps at 1.0σ are shown in blue. | ||
The apo-rHLFr mutants were prepared by site-directed mutagenesis using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene Co., USA). Pd(allyl)·apo-rHLFr mutants were prepared according to procedures described in our previous report.29 The exact number of encapsulated Pd ions per apo-rHLFr cage was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES) and the bicinchoninate (BCA) method. These composites were crystallized by the hanging drop vapor diffusion method using precipitant solutions containing 0.5–1.0 M (NH4)2SO4 and 15–20 M CdSO4 for X-ray crystal structure analysis. The crystal structures of Pd(allyl)·apo-E45C/C48A-rHLFr, Pd(allyl)·apo-E45C/R52H-rHLFr, and Pd(allyl)·apo-E45C/H49A/R52C-rHLFr were refined at 1.86, 1.52, and 1.48 Å resolutions, respectively (see ESI‡). The selected bond distances of the Pd dinuclear structures of each composite are listed in Table S2 (ESI‡). The dinuclear centers at the three-fold channels in each mutant retain the same structures as that of Pd(allyl)·apo-rHLFr (Fig. S1, ESI‡).
Comparison of the accumulation centers of Pd(allyl)·apo-Fr and Pd(allyl)·apo-E45C/C48A-rHLFr indicates that PdA1–PdA2 disappears upon substitution of Cys48 with alanine. The replacement of Glu45 with cysteine produces a new dinuclear center, PdB1–PdB2, which has typical Pd–Pd and Pd–S bond lengths (Fig. 2a and b, and see ESI‡).18 Both His49 and His173 can serve as supporting ligands for the PdB1–PdB2 center because they are located at appropriate distances from the PdB1–PdB2 center. The number of Pd atoms in the cage of Pd(allyl)·apo-E45C/C48A-Fr determined by the ICP-OES/BCA method (98 ± 1 Pd atoms per apo-rHLFr) is almost identical to the number estimated from the crystal structure (96 Pd atoms per apo-rHLFr). These results indicate that the PdB1 and PdB2 atoms are rigidly bound to Cys45, although their allyl ligands have weak electron density maps below 1σ. This weak density might be caused by flexible conformations of the coordinated residues (Fig. 2b).
The crystal structure of Pd(allyl)·apo-E45C/R52H-rHLFr shows that two dinuclear sites are formed at the accumulation center (Fig. 2c). The binding center has two cysteine residues, Cys45 and Cys48, as well as two original histidines, His49 and His173, and a new histidine (His52) introduced by the replacement of Arg52 which cannot coordinate to Pd atoms of Pd(allyl)·apo-rHLFr (Fig. 2a). The PdC1–PdC2 structure retains a structure similar to that of PdB1–PdB2 (Fig. 2b). The other dinuclear site, PdC3–PdC4 (Fig. 2c), is formed by the ligation of Cys48, His52 and a molecule of H2O (WC1) to maintain a structure similar to that of PdA1–PdA2 (Fig. 2a). All ligand structures which include a water molecule and allyl molecules have electron density maps which are much clearer than those of Pd(allyl)·apo-E45C/C48A-rHLFr (Fig. 2b). It is worth mentioning that all of the histidine residues participating in the coordination of Pd are fixed by hydrogen bonding interactions with surrounding nearby amino acid residues (Fig. S2, ESI‡). The number of Pd atoms assessed by X-ray crystallography (144 Pd per Fr) is close to the number determined by the ICP/BCA method (154 ± 10 Pd per Fr). Therefore, it is expected that the newly introduced PdC3–PdC4 core suppresses the flexibility of His49 by providing steric hindrance, which in turn stabilizes the two dinuclear centers at the accumulation center, because the B-factor values of the imidazole ring are lower than that of Pd(allyl)·apo-E45C/R52H-rHLFr.
Pd(allyl)·apo-E45C/
H49A
/
R52H-Fr also has two dinuclear sites at the accumulation center with typical coordination geometry (Table S2, ESI‡) although this mutation remarkably alters the position of the dinuclear Pd complex bound to Cys45 by the replacement of His49 (Fig. 2d). The orientation of the PdD1–PdD2 pair is nearly perpendicular to the PdC1–PdC2 pair in Pd(allyl)·apo-E45C/R52H-rHLFr (Fig. 2c). The coordination of His173 to PdD1 is stabilized by a hydrogen bond interaction between Nε of His173 and the main chain C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Glu167 (Fig. S2, ESI‡). The composite contains 144 Pd atoms in the 24-mer cage of the mutant, according to ICP/BSA data (154 ± 3 Pd per Fr) which assume that these four Pd ions have rigidly settled coordination. These observations indicate the significance of the histidine residues with respect to modulation of the coordination structure of Pd(allyl) complexes. Previous studies on the accumulation of Pd2+ ions in apo-rHLFr have indicated that the conformational changes of His49 and Arg52 promote an increase in the number of Pd atoms bound to the accumulation center.14 Furthermore, Cys48 is a key residue for the binding of Pd ions in the initial stage of the accumulation.14 Thus, the arrangement of Pd(allyl) complexes is cooperatively modulated by appropriately placed cysteine residues, which capture the Pd complexes, and histidine residues, which control the direction of the center by supplementary ligation.
O of Glu167 (Fig. S2, ESI‡). The composite contains 144 Pd atoms in the 24-mer cage of the mutant, according to ICP/BSA data (154 ± 3 Pd per Fr) which assume that these four Pd ions have rigidly settled coordination. These observations indicate the significance of the histidine residues with respect to modulation of the coordination structure of Pd(allyl) complexes. Previous studies on the accumulation of Pd2+ ions in apo-rHLFr have indicated that the conformational changes of His49 and Arg52 promote an increase in the number of Pd atoms bound to the accumulation center.14 Furthermore, Cys48 is a key residue for the binding of Pd ions in the initial stage of the accumulation.14 Thus, the arrangement of Pd(allyl) complexes is cooperatively modulated by appropriately placed cysteine residues, which capture the Pd complexes, and histidine residues, which control the direction of the center by supplementary ligation.
We evaluated the catalytic reactivity of the Pd(allyl)·apo-rHLFr composites for the Suzuki coupling reaction by assessing the reaction of p-I–PhNH2 and PhB(OH)2 in an aqueous solvent.29 The turnover frequencies ([product (mol)] per apo-rHLFr (mol) per hour) of the coupling reactions were calculated based on the consumption of p-I–PhNH2 and the yield of the product. The turnover frequency of Pd(allyl)·apo-E45C/C48A-rHLFr (4200 ± 100 h−1) is slightly higher than that of Pd(allyl)·apo-rHLFr (3500 ± 400 h−1).29 Although Pd(allyl)·apo-E45C/R52H-rHLFr and Pd(allyl)·apo-E45C/H49A/R52H-rHLFr have 1.5-fold more Pd complexes than Pd(allyl)·apo-E45C/C48A-rHLFr, their turnover frequencies (4300 ± 300 and 4200 ± 200 h−1, respectively) do not represent a significant improvement relative to that of Pd(allyl)·apo-E45C/C48A-rHLFr. Our previous work proves that (1) the substrates penetrate the Fr cage through the 3-fold channel before they react with the Pd complexes on the interior surface of apo-rHLFr, and (2) the replacement of His with Glu to coordinate to the Pd(allyl) moiety has little effect on the catalytic reactivity.29 Thus, the process of the penetration is thought to be a rate-limiting step. This may explain why the addition of Pd(allyl) complexes into the apo-rHLFr mutant cavity does not effectively accelerate the catalytic reaction.
In summary, rearrangements of coordination structures of Pd(allyl) complexes in the apo-rHLFr cage can be obtained by appending or removing cysteine and histidine residues. To design such structures, two factors should be primarily considered: (1) Cys residues are essential components for capturing two Pd complexes for the formation of the dinuclear structures at the accumulation center, and (2) His residues, which have flexible side chains, are needed in the vicinity of the Cys residues in order to control the diverse direction of the dinuclear Pd(allyl) moieties at the center. They are also supported by fundamental investigations as recently reported by us.13 Such arrangements of metal complexes are too complicated to be constructed only by using synthetic multimeric ligands. Thus, we believe that this work should be amenable to other proteins and synthetic metal complexes and will provide insights into the rational design of proteins containing metal complexes to produce candidates for the development of useful materials such as biocatalysts, biosensors, and metal-containing drugs.
We thank the members of BL38B1 of SPring-8 for assistance during the collection of diffraction data (No. 2009A1185 and 2009B1065). This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas (“Coordination Programming” Area 2107, No. 22108513 for T.U.) from MEXT, Japan, and PRESTO and JST for T.U.
Notes and references
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS.
- J. Steinreiber and T. R. Ward, Coord. Chem. Rev., 2008, 252, 751–766 CrossRef CAS.
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- T. Ueno, S. Abe, N. Yokoi and Y. Watanabe, Coord. Chem. Rev., 2007, 251, 2717–2731 CrossRef CAS.
- D. E. Benson, M. S. Wisz and H. W. Hellinga, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6292–6297 CrossRef CAS.
- V. A. Roberts, B. L. Iverson, S. A. Iverson, S. J. Benkovic, R. A. Lerner, E. D. Getzoff and J. A. Tainer, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 6654–6658 CrossRef CAS.
- J. E. Debreczeni, A. N. Bullock, G. E. Atilla, D. S. Williams, H. Bregman, S. Knapp and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 1580–1585 CrossRef CAS.
- L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2007, 251, 1633–1648 CrossRef CAS.
- A. Datta, J. M. Hooker, M. Botta, M. B. Francis, S. Aime and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 2546–2552 CrossRef CAS.
- X. Huang, L. M. Bronstein, J. Retrum, C. Dufort, I. Tsvetkova, S. Aniagyei, B. Stein, G. Stucky, B. McKenna, N. Remmes, D. Baxter, C. C. Kao and B. Dragnea, Nano Lett., 2007, 7, 2407–2416 CrossRef CAS.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS.
- A. Pordea and T. R. Ward, Chem. Commun., 2008, 4239–4249 RSC.
- S. Abe, T. Hikage, Y. Watanabe, S. Kitagawa and T. Ueno, Inorg. Chem., 2010, 49, 6967–6973 CrossRef CAS.
- T. Ueno, M. Abe, K. Hirata, S. Abe, M. Suzuki, N. Shimizu, M. Yamamoto, M. Takata and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 5094–5100 CrossRef CAS.
- S. Kang, C. C. Jolley, L. O. Liepold, M. Young and T. Douglas, Angew. Chem., Int. Ed., 2009, 48, 4772–4776 CrossRef CAS.
- E. C. Theil, Annu. Rev. Biochem., 1987, 56, 289–315 CrossRef CAS.
- N. D. Chasteen, Iron Transport and Storage in Microorganisms, Plants, and Animals, Marcel Dekker, New York, 1998 Search PubMed.
- P. Santambrogio, S. Levi, P. Arosio, L. Palagi, G. Vecchio, D. M. Lawson, S. J. Yewdall, P. J. Artymiuk, P. M. Harrison, R. Jappelli and G. Cesareni, J. Biol. Chem., 1992, 267, 14077–14083 CAS.
- L. Zhang, J. Swift, C. A. Butts, V. Yerubandi and I. J. Dmochowski, J. Inorg. Biochem., 2007, 101, 1719–1729 CrossRef CAS.
- C. A. Butts, J. Swift, S. G. Kang, L. Di Costanzo, D. W. Christianson, J. G. Saven and I. J. Dmochowski, Biochemistry, 2008, 47, 12729–12739 CrossRef CAS.
- T. Ueno, M. Suzuki, T. Goto, T. Matsumoto, K. Nagayama and Y. Watanabe, Angew. Chem., Int. Ed., 2004, 43, 2527–2530 CrossRef CAS.
- K. K. W. Wong and S. Mann, Adv. Mater., 1996, 8, 928–932 CrossRef CAS.
- I. Yamashita, J. Hayashi and M. Hara, Chem. Lett., 2004, 33, 1158–1159 CrossRef CAS.
- S. Abe, K. Hirata, T. Ueno, K. Morino, N. Shimizu, M. Yamamoto, M. Takata, E. Yashima and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 6958–6960 CrossRef CAS.
- S. Aime, L. Frullano and S. G. Crich, Angew. Chem., Int. Ed., 2002, 41, 1017–1019 CrossRef CAS.
- J. M. Dominguez-Vera and E. Colacio, Inorg. Chem., 2003, 42, 6983–6985 CrossRef.
- Z. Yang, X. Y. Wang, H. J. Diao, J. F. Zhang, H. Y. Li, H. Z. Sun and Z. J. Guo, Chem. Commun., 2007, 3453–3455 RSC.
- J. Lucon, M. J. Abedin, M. Uchida, L. Liepold, C. C. Jolley, M. Young and T. Douglas, Chem. Commun., 2010, 46, 264–266 RSC.
- S. Abe, J. Niemeyer, M. Abe, Y. Takezawa, T. Ueno, T. Hikage, G. Erker and Y. Watanabe, J. Am. Chem. Soc., 2008, 130, 10512–10514 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, tables, and figures. See DOI: 10.1039/c0cc02221g |
| § Atomic coordinates are deposited in the Protein Data Bank under accession numbers 3NP2, 3NOZ, and 3NP0 for Pd(allyl)·apo-E45C/C48A-rHLFr, Pd(allyl)·apo-E45C/R52H-rHLFr and Pd(allyl)·apo-E45C/H49A/R52H-rHLFr, respectively. |
| This journal is © The Royal Society of Chemistry 2011 |