Hybrid cyclic peptide–thiourea cryptands for anion recognition†‡
Philip G.
Young
a,
Jack K.
Clegg§
a,
Mohan
Bhadbhade
b and
Katrina A.
Jolliffe
*a
aSchool of Chemistry, The University of Sydney, 2006 NSW, Australia. E-mail: kate.jolliffe@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 2297
bSolid State and Elemental Analysis Unit, Analytical Centre, The University of New South Wales, Kensington, 2052 NSW, Australia
First published on 1st October 2010
Abstract
Two cyclic peptide derived cryptands incorporating thioureas in the framework were synthesised as neutral anion receptors and bind acetate ions with high affinity and, in one case, good selectivity.
The development of molecular receptors capable of selective anion recognition using non-covalent interactions is of current interest because anions play significant roles in areas as diverse as biology, medicine, catalysis and the environment.1,2 In biological systems (e.g. the sulfate-binding and phosphate-binding proteins), selective and strong binding of anions in aqueous media is frequently achieved using a precise arrangement of hydrogen-bonding residues.3 Therefore, the design and synthesis of synthetic receptors with convergent hydrogen bonding sites that match a target anionic species is of particular interest.
We4 and others5 have previously described the use of analogues of the Lissoclinum family of naturally occurring cyclic peptides as scaffolds for the construction of molecular receptors. These cyclic peptides feature oxazole or thiazole heterocycles (derived from serine, threonine or cysteine residues) alternating with amino acid residues to provide macrocycles that are rigidified by a network of bifurcated hydrogen bonds. When all of the amino acid side chains are of the same configuration, they are presented on one face of the macrocycle in a convergent manner.
Herein we report the synthesis of two cryptands, 1 and 2 (Scheme 1), based on such azole-modified cyclic peptide scaffolds. While several cages based on this class of cyclic peptide have been previously reported,6 none have been investigated for their ability to bind guest molecules. We envisaged that the incorporation of thioureas into the cyclic peptide side chains would provide hydrogen bond donor sites for anion binding that could potentially be complemented by the amide bonds in the macrocycle itself.7 In addition, to provide a highly preorganised framework with convergent hydrogen bond donors necessary for strong and selective binding, we chose to constrain the cyclic peptide side chains to provide cryptand-type structures. Surprisingly, despite the utility of thioureas in anion binding there are relatively few examples in the literature of cryptand-type structures incorporating them.8
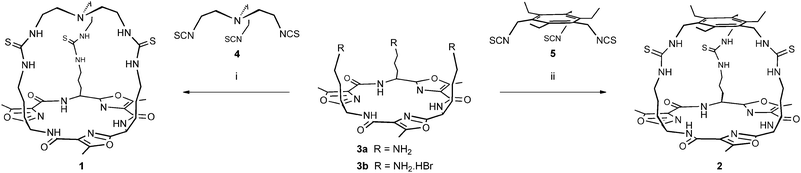 | ||
| Scheme 1 Synthesis of tren-capped cryptand 1 and 1,3,5-triethylbenzene-capped cryptand 2. Reagents and conditions: (i) 3a, CHCl3, 54% or3b, triethylamine, CHCl3, DMF, 62%. (ii) 3a, CHCl3, 69% or3b, triethylamine, CHCl3, DMF, 50%. | ||
We chose tris(aminoethyl)amine (tren) and a 1,3,5-trisubstituted-2,4,6-triethylbenzene as trivalent capping units with complementary geometries and size for a Lissoclinum-type cyclic peptide containing three oxazole units. Both capping units have been widely demonstrated to be effective scaffolds for the construction of molecular receptors9,10 and CPK models indicated that both would provide cryptands in which the thioureas could adopt a conformation with all NHs directed towards the interior of the cage in a convergent manner.
To synthesise 1 and 2 we used the known cyclic peptide tris-amine derivative 311 as one of the trivalent components and condensed this with tris-isothiocyanates 412 and 5,13 respectively (Scheme 1). Compounds 4 and 5 were readily prepared from the corresponding triamines upon treatment with carbon disulfide and N,N′-dicyclohexylcarbodiimide (DCC) in 64% and 76% yields, respectively. Under optimised conditions (high dilution with dropwise addition of both the free triamine 3a and either 4 or 5 to CHCl3 at 35–40 °C), the desired tren-capped cryptand 1 and 1,3,5-triethylbenzene-capped cryptand 2 were obtained in 54% and 69% yields, respectively, following purification by flash column chromatography. Trisamine 3 is most readily obtained as the hydrobromide salt 3b (by acid catalysed removal of side chain protecting groups)11 and when this species was used directly as the source of 3 in the condensation reaction in the presence of triethylamine, both 1 and 2 were obtained in increased crude yields. However, it was extremely difficult to separate these compounds from the triethylammonium bromide byproduct, suggesting that the cryptands were binding to the bromide ions present. Eventually, we found that purification of material obtained in this manner by reverse phase HPLC provided 1 in an improved yield of 62%, while cryptand 2 was obtained in a reduced yield of 50% using this procedure. The improvement in the yield obtained for 1 from 3b suggested that bromide ions might be templating this condensation reaction. However, all further attempts to investigate templation of the formation of 1 using the free amine 3a and added bromide or chloride salts (e.g.tetrabutylammonium chloride) were hindered by difficulties in isolation and purification of the products. Notably, the lower yield obtained for 2 from condensation of the tris-hydrobromide salt 3b compared to that obtained from the free amine 3a may be attributed to these difficulties.
Single crystals of 1 and 2 suitable for X-ray diffraction analysis were obtained from slow crystallisation in MeOH/CHCl3 and acetonitrile/water/trifluoroacetic acid solutions, respectively (Fig. 1).¶ The crystal structure of [MeOH⊂1]·TFA·0.875MeOH·0.625H2O indicates that 1 crystallises with two molecules in the asymmetric unit. In each case as illustrated in Fig. 1a the thiourea moieties are orientated such that the thiourea hydrogen donor atoms point outside of the cryptate cavity; hydrogen bonding to solvent molecules forming a one-dimensional chain that propagates parallel to the crystallographic a-axis. Adjacent chains sit closely to one another with strong π–π stacking present between oxazole rings (centroid–centroid distances = 3.36 Å). Located within the cryptate cavity is a methanol guest molecule that strongly hydrogen bonds (DA 2.743(13) Å, DHA 153.9°) to the capping tertiary amine which has adopted an endo-arrangement.
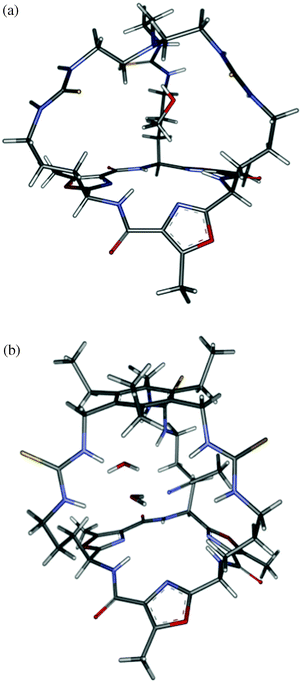 | ||
| Fig. 1 Schematic representations of the X-ray crystal structures of (a) cryptand 1 with a molecule of methanol located inside the cavity and (b) cryptand 2 with two water and one acetonitrile molecules inside the cavity. Numerous other solvent molecules and regions of disorder have been removed for clarity. | ||
2 also crystallises with two molecules per asymmetric unit. While each of the molecules is chemically equivalent they are host to different guest molecules. The first (Fig. 1b) encapsulates an acetonitrile and two water molecules while the second encapsulates three water molecules. Hence the species is best described as {[(H2O)2MeCN⊂2][(H2O)3⊂2]}·14.5H2O. The encapsulation of the larger number of guests is perhaps a reflection of the larger cavity size formed by the use of the phenylene cap compared to the tren in 1. In contrast to the previous structure, the thiourea groups are arranged such that their H-atoms are directed into the cavity and hydrogen bond to the solvent guest molecules. There is also hydrogen bonding directly between the solvent guests and with the solvents that are outside the cavity. There is π–π stacking between the oxazole units in adjacent capsules in a similar fashion to that observed in the structure of 1.
Anion binding studies with 1 and 2 were performed by titrating them with the tetrabutylammonium salts of a range of monovalent anions in 0.5% v/v H2O/DMSO-d6. The results are shown in Table 1.14 In this solvent, the signals observed in the 1H NMR spectrum of 1 were broad at 300 K, presumably due to slow conformational change on the NMR timescale resulting from the relatively rigid three dimensional structure and intramolecular hydrogen bonding interactions. However, at 330 K the signals were sharp enough to allow exact chemical shifts to be determined, so apparent stability constants for 1 were determined at this temperature. The 1H NMR spectrum of 2 at 300 K was much sharper than that of 1. This can be attributed to the greater structural rigidity imposed by 1,3,5-triethylbenzene cap, which prevents intramolecular hydrogen bonding interactions.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 binding model. Errors < 15% except where noted
∶1 binding model. Errors < 15% except where noted
| Anion guest | 1 a | 2 b |
|---|---|---|
| a Determined at 330 K. b Determined at 300 K. c Titration displayed slow exchange on the NMR timescale. d Titration resulted in deprotonation of the thiourea binding sites as evidenced by the appearance of a signal attributable to HF2− at 16 ppm. e Peak broadening after addition of 2 equiv. H2PO4− prevented a stability constant from being determined. | ||
| F− | >104c |
d |
| Cl − | >104 | 87 |
| Br − | 2.7 × 103 | 11 (±6) |
| I− | <10 | <10 |
| AcO − | >104 | >104 |
| NO3− | <10 | <10 |
| HSO4− | <10 | <10 |
| H2PO4− | e | e |
Apparent stability constants for 1 and 2 with a variety of anions were determined by following the downfield shifts of both of the thiourea NH proton signals and fitting the titration data to a 1∶1 binding model. In all cases except for fluoride, fast exchange processes were observed. There are some notable differences in binding behaviour for 1 and 2 towards the halides, with 1 binding chloride and bromide at least three orders of magnitude more strongly than 2, and both compounds showing some selectivity for the smaller halides. Neither 1 nor 2 had any observable affinity for iodide, nitrate or hydrogensulfate, but both compounds showed strong affinity for acetate ions. Indeed, 2 exhibits good selectivity towards acetate, with an apparent stability constant for this anion over three orders of magnitude higher than those of any other anions investigated. An explanation for both this selectivity and the role of the amide protons in binding requires further investigation,15 but we postulate that the ability of the tren-capped cryptand 1 to bind a greater range of anions is a result of the increased flexibility of this molecule compared to that of the rigid 1,3,5-triethylbenzene capped 2, allowing the accommodation of a wider range of anions.
In conclusion, two novel cryptands based on a cyclic peptide scaffold and featuring three conformationally constrained thiourea groups for anion binding have been synthesised in good yields. Anion binding studies indicated that 1 binds fluoride, chloride, bromide and acetate but not iodide, nitrate or hydrogensulfate in 0.5% H2O/DMSO-d6. In contrast, cryptand 2 shows selectivity towards acetate ions. We are currently extending our investigations of the anion binding affinities and selectivities of these receptors.
We thank Dr Kelvin Picker (USyd) and Dr Ian Luck (USyd) for technical assistance with HPLC and NMR, respectively, and the Australian Research Council for financial support. Part of this research was undertaken on the macromolecular crystallography beamline at the Australian Synchrotron, Victoria, Australia. We thank Dr Tom Caradoc-Davies (Australian Synchrotron) for assistance.
Notes and references
- For reviews see: C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520 Search PubMed; J. L. Sessler, P. A. Gale, and W.-S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, 2006 RSC; in Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman James, and E. García-España, Wiley-VCH, New York, 1997 RSC; P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 RSC.
- For representative recent articles on anion recognition see: N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis and W. A. Harrell, Jr., Chem. Commun., 2010, 46, 6252 Search PubMed; P. A. Gale, J. R. Hiscock, S. J. Moore, C. Caltagirone, M. B. Hursthouse and M. E. Light, Chem.–Asian J., 2010, 5, 555 RSC; A. N. Swinburne, M. J. Pateron, A. Beeby and J. W. Steed, Org. Biomol. Chem., 2010, 8, 1010 CrossRef CAS; A. J. Lowe and F. M. Pfeffer, Org. Biomol. Chem., 2009, 7, 4233 RSC; K. J. Winstanley, S. J. Allen and D. K. Smith, Chem. Commun., 2009, 4299 RSC; J. N. Babu, V. Bhalla, M. Kumar, R. K. Puri and R. K. Mahajan, New J. Chem., 2009, 33, 675 RSC.
- J. W. Pflugrath and F. A. Quiocho, Nature, 1985, 314, 257 CrossRef CAS; J. W. Pflugrath and F. A. Quiocho, J. Mol. Biol., 1988, 200, 163 CAS; J. J. He and F. A. Quiocho, Science, 1991, 251, 1479 CrossRef CAS.
- K. A. Jolliffe, Supramol. Chem., 2005, 17, 81 CrossRef CAS; M. J. McDonough, A. J. Reynolds, W. Y. G. Lee and K. A. Jolliffe, Chem. Commun., 2006, 2971 RSC.
- D. Mink, S. Mecozzi and J. Rebek, Jr., Tetrahedron Lett., 1998, 39, 5709 CrossRef CAS; A. J. Lucke, J. D. A. Tyndall, Y. Singh and D. P. Fairlie, J. Mol. Graphics Modell., 2003, 21, 341 CrossRef CAS; Y. Singh, M. J. Stoermer, A. J. Lucke, T. Guthrie and D. P. Fairlie, J. Am. Chem. Soc., 2005, 127, 6563 CrossRef CAS; A. Pintér, G. Haberhauer, I. Hyla-Kryspin and S. Grimme, Chem. Commun., 2007, 3711 RSC; A. Pintér and G. Haberhauer, Synlett, 2009, 3082 CAS.
- G. Pattenden and T. Thompson, Chem. Commun., 2001, 717 RSC; G. Pattenden and T. Thompson, Tetrahedron Lett., 2002, 43, 2459 CrossRef CAS; G. Haberhauer, T. Oeser and F. Romiger, Chem.–Eur. J., 2005, 11, 6718 CrossRef CAS; Y. Singh, N. Sokolenko, M. J. Kelso, L. R. Gahan, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2001, 123, 333 CrossRef CAS.
- M. Schnopp, S. Ernst and G. Haberhauer, Eur. J. Org. Chem., 2009, 213 CrossRef CAS.
- I. Hisaki, S. Sasaki, K. Hirose and Y. Tobe, Eur. J. Org. Chem., 2007, 607 CrossRef CAS; N. G. Lukyaneko, T. I. Kirichencko and S. V. Scherbakov, J. Chem. Soc., Perkin Trans. 1, 2002, 2347 RSC; N. G. Lukyanenko, A. V. Bogatskii, T. I. Kirichenko, S. V. Scherbakov and N. Y. Nazarova, Synthesis, 1984, 137 CrossRef CAS.
- G. Hennrich and E. V. Anslyn, Chem.–Eur. J., 2002, 8, 2218 CrossRef CAS.
- I. Ravikumar, P. S. Lakshminarayanan, M. Arunachalam, E. Suresh and P. Ghosh, Dalton Trans., 2009, 4160 RSC , and references therein.
- R. J. G. Black, V. J. Dungan, R. Y. T. Li, P. G. Young and K. A. Jolliffe, Synlett, 2010, 551 CAS.
- X. Zhang and W.-D. Woggon, J. Am. Chem. Soc., 2005, 127, 14138 CrossRef CAS.
- Q. P. B. Nguyen, T. N. Le and T. H. Kim, Bull. Korean Chem. Soc., 2009, 30, 1743 CrossRef CAS.
- Initial binding studies with Bu4NBr in the less polar 10% DMSO-d6/CDCl3 indicated that in this solvent, stability constants for both 1 and 2 were too high to be determined by 1H NMR (>104).
- The signal for the amide NH protons of 1 shifts upfield upon addition of anions, while that for the same protons in 2 shifts downfield. In both cases the data can be fit to a 1∶1 binding model to give identical apparent stability constants (within experimental error) to those obtained from the urea NH protons.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and spectroscopic data for new compounds; CIFs for 1 and 2. CCDC 783199 and 783200. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02223c |
| § Current address: University of Cambridge, Department of Chemistry, Lensfield Road, Cambridge CB2 1EW, UK. |
| ¶ Crystal data for [MeOH⊂1]·TFA·0.875MeOH·0.625H2O: formula C79.75H121.50F6N26O21S6, M 2086.89, monoclinic, space groupP21(#4), a 17.302(4), b 15.233(3), c 21.662(4) Å, β 113.54(3)°, V 5234.3(22) Å3, Dc 1.324 g cm−3, Z 2, crystal size 0.02 by 0.01 by 0.01 mm, light yellow, habit Plate, temperature 120(2) K, λ(Synchrotron) 0.65256 Å, μ(Synchrotron) 0.217 mm−1, 2θmax 42.50, hkl range −19 19, −16 16, −24 24, N 56007, Nind 14942(Rmerge 0.1995), Nobs 7088(I > 2σ(I)), Nvar 1258, residuals R1(F) 0.0978, wR2(F2) 0.3028, GoF(all) 0.773, Δρmin,max −0.439, 0.998 e− Å−3. Crystal data for {[(H2O)2MeCN⊂2][(H2O)3⊂2]}·14.5H2O: formula C92H162N25O31.50S6, M 2314.83, triclinic, space group P1(#1), a 13.8640(12), b 14.5750(10), c 16.8430(12) Å, α 69.705(5)°, β 77.952(3)°, γ 75.179(3)°, V 3058.8(4) Å3, Dc 1.261 g cm−3, 0.20 by 0.050 by 0.01 mm, colourless, habit needle, temperature 150(2) K, λ(MoKα) 0.71073 Å, μ(MoKα) 0.192 mm−1, 2θmax 41.82, hkl range −13 13, −14 14, −16 16, N 62257, Nind 12759(Rmerge 0.0640), Nobs 10214(I > 2σ(I)), Nvar 1531, residuals R1(F) 0.0492, wR2(F2) 0.1272, GoF(all) 1.049, Δρmin,max −0.308, 0.628 e− Å−3. |
| This journal is © The Royal Society of Chemistry 2011 |