A tetraalkylated pyrene building block for the synthesis of pyrene-fused azaacenes with enhanced solubility†‡
Niksa
Kulisic
ab,
Sandeep
More
ab and
Aurelio
Mateo-Alonso
*ab
aFreiburg Institute for Advanced Studies (FRIAS), School of Soft Matter Research, Albert-Ludwigs-Universität Freiburg, Albertstrasse 19, D-79104 Freiburg i. Brsg., Germany. E-mail: amateo@frias.uni-freiburg.de
bInstitut für Organische Chemie und Biochemie (IOCB), Albert-Ludwigs-Universität Freiburg, Albertstrasse 21, D-79104 Freiburg i. Brsg., Germany
First published on 25th October 2010
Abstract
The synthesis and characterisation of a soluble pyrene-fused tetraazaoctacene derivative has been achieved by developing a key pyrene-based building block with four solubilising groups.
Semiconducting organic molecules and polymers are expected to open up possibilities for new electronic products that will not compete with the existing technology, but instead will create a new market that could reach the size of silicon. n-Acenes are polyaromatic hydrocarbons composed of linearly fused benzene rings (where n refers to the number fused rings), which are among the most promising organic semiconductors. Although higher acenes (n > 5) are known to be unstable, a lot of effort has been dedicated to study their properties. Several stabilisation strategies have allowed the preparation of higher acenes1–5 (5 ≤ n ≤ 9), which in some cases are stable for several days. The optoelectronic properties observed on the larger acenes have motivated scientists to explore other members of the acene family.
Azaacenes have re-emerged as an alternative platform to acenes since in some cases they can be more stable and easier to synthesise. Azaacenes show the same degree of aromaticity than acenes but structurally present N instead of C atoms in some positions of the aromatic framework.6 By expanding laterally the conjugation of the linear aromatic backbone with additional aromatic rings, sextets of π-electrons are localised on such external rings, which results in the enhancement of the stability. This stabilisation strategy has allowed scientists to prepare not only higher pyrene-fused azaacenes7,8,10–23 (6 ≤ n ≤ 16) but also polyazaacenes7–9 to study their length/property characteristics. One of the common features of pyrene-fused azaacenes relies on their synthesis, which proceeds through the cyclocondensation of 1,2-diamines and diketones to fuse different polyaromatic residues through pyrazine rings. A key building block in the preparation of such oligoazaacenes is 4,5,9,10-tetraketopyrene24 (the substitution numbering of pyrene is shown on Scheme 1). However the azaacenes that evolve from 4,5,9,10-tetraketopyrene are highly insoluble,21 which hampers their deposition through solution-based techniques. Even if the introduction of alkyl groups on pyrene has been reported,24 the current solubilisation strategies are based either on the use of soluble terminal building blocks or on the postfunctionalisation of the azaacene core with solubilising groups.11,13,14,16,22,23 Ideally, for facilitating the preparation and characterisation of higher pyrene-fused azaacenes, it would be desirable to use pyrene building blocks displaying suitable solubilising groups that yield directly soluble azaacenes during the cyclocondensation step, without the need of functionalisation at a later stage.
 | ||
| Scheme 1 Synthetic route for the preparation of tetraazaoctacenes 4 and 10. (a) tBuCl, AlCl3, rt, 3 h; (b) NaIO4, RuCl3, CH2Cl2, CH3CN, water, 35 °C, 24 h, 36%; (c) pyridine, reflux, 3 days, 32%; (d) Br2, PhNO2, reflux, 4 h; (e) CuI, Pd(PPh3)2Cl2, iPr2NH, THF, 80 °C, overnight, 71%; (f) H2, Pd/C (10%), 1 atm, rt, overnight, 89%; (g) NaIO4, RuCl3, CH2Cl2, CH3CN, water, 45 °C, 3 days, 20%; (h) AcOH, rt overnight, then reflux 3 days, 17%. | ||
With all this in mind, we have developed a convenient synthetic route (Scheme 1) for preparing 1,3,6,8-tetraoctyl-4,5,9,10-tetraketopyrene (9). By using this building block, we have conveniently synthesised a tetraazaoctacene derivative (10) with four n-octyl solubilising chains in the pyrene core that displays enhanced solubility in neutral organic solvents.
In a first attempt to synthesise soluble azaacenes, we focused on the synthesis of tetraazaoctacene derivatives with two solubilizing groups on the pyrene core (Scheme 1). tert-Butyl groups can be introduced in azaacenes by using 2,7-di-tert-butyl-4,5,9,10-tetraketopyrene (2), which is currently the most commonly used building block since it can be synthesised in only two steps.24 Accordingly, pyrene was functionalised with two tert-butyl groups through Friedel–Crafts alkylation to give 2,7-di-tert-butylpyrene (1). Subsequently, 1 was oxidised with NaIO4 in the presence of RuCl3 catalyst to tetraketone 2. The reaction of 2 with 2,3-diaminonaphthalene (3) in a degassed pyridine solution yielded the desired pyrene-fused tetraazaoctacene 4 that was collected in pure form by filtration. Azaacene 4 is sparingly soluble in neutral organic solvents. The best solvents for solubilising 4 are strong acids, among which, sulfuric and trifluoroacetic acid (TFA) are the most accessible. The presence of the tert-butyl groups enhanced the solubility in TFA and no dilution was necessary to obtain a resolved NMR spectrum, in contrast to previous observations on an analogous pyrene-fused tetraazaoctacene with no solubilizing groups.21 The structure of 4 was indeed confirmed by NMR (TFA-d1, see ESI‡) and MS.
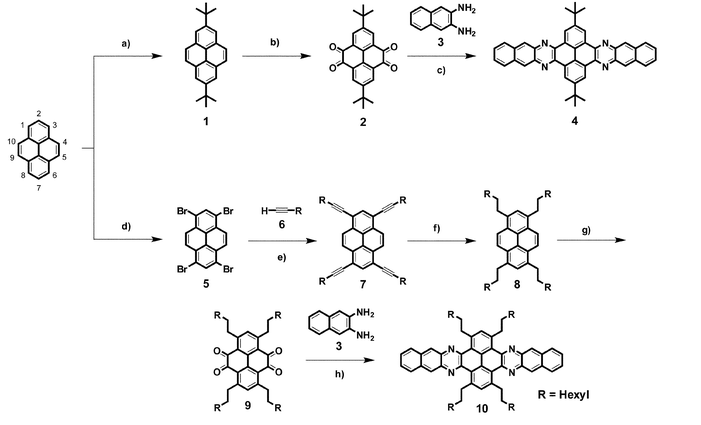
To overcome the lack of solubility of tert-butyl functionalised azaacenes in neutral solvents, more efficient solubilising groups must be incorporated in the pyrene core. A route to introduce n-alkyl groups on positions 2,7 of 4,5,9,10-tetraketopyrene derivatives has been reported.24 Nevertheless, this route is not straightforward as it proceeds through several reduction and oxidation steps that we found difficult to reproduce. Our alternative approach is conveniently based on the reaction of pyrene with Br2 to give 1,3,6,8-tetrabromopyrene 5.25 This reaction is straightforward and 5 can be obtained in a multigram scale without the need of other purification than filtration, as 5 is insoluble in organic solvents. Tetrabromopyrene 5 was functionalised with 1-octyne (6) under Sonogashira conditions. Eventhough a wide variety of alkynes functionalised with alkyl chains are commercially available, 6 was chosen because of the high solubilising power of octyl chains and also because of its reasonable price. The Sonogashira C–C coupling proceeded smoothly by using CuI and Pd(PPh3)2Cl2 at 80 °C. Filtration and chromatography yielded compound 7 as a pure material, which was characterised by NMR and MS. Crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of a solution of petrol ether (40–60 °C), which confirmed the structure (Fig. 1).
 | ||
| Fig. 1 X-Ray structure of compound 7. | ||
Pyrene 7 presents already a very high solubility in the most common organic solvents. However the acetylene groups may interfere in the oxidation of pyrene to tetraketopyrene. For this reason, the acetylene residues of 7 were reduced to methylenes by catalytic hydrogenation to yield 1,3,6,8-tetraoctylpyrene (8). The reaction also proceeded smoothly and 8 was obtained in pure form by filtrating off the catalyst and evaporating the solvent. However it should be noted that when the reaction times were exceeded 4,5-dihydropyrene derivatives were detected by NMR.
The resulting tetraoctylpyrene 8 was oxidised to 1,3,6,8-tetraoctyl-4,5,9,10-tetraketopyrene (9) by oxidation with NaIO4 catalysed by RuCl3. Tetraketone 9 was obtained in pure form as an orange solid after chromatography. Reaction of 2,3-diaminonaphthalene (3) with tetraketone 9 in a degassed and refluxing solution of AcOH yielded, after chromatography, the desired pyrene-fused tetraazaoctacene 10 displaying four n-octyl solubilising groups. Compound 10 presents enhanced solubility in organic solvents (≫20 mg mL−1 in CHCl3), which facilitated the confirmation of its structure by NMR (see ESI‡).
Taking advantage of the enhanced solubility of 10, its electronic properties were investigated by steady-state UV-Vis and fluorescence spectroscopy (Fig. 2). In non-polar solvents such as toluene and CHCl3, well-resolved absorption features of 10 along the UV and the visible were observed, with no sign of aggregation upon dilution. The optical HOMO–LUMO gap of 10 (2.5 eV) was calculated from the absorption spectrum onset of a CHCl3 solution (Table 1). The emission spectra of 10 were obtained by excitation with UV or visible light. The spectra showed a peak centered at 478 nm with a shoulder at 454 nm. Excitation spectra mirrored the features observed on emission and Stokes shifts of 93 nm were observed (see ESI‡).
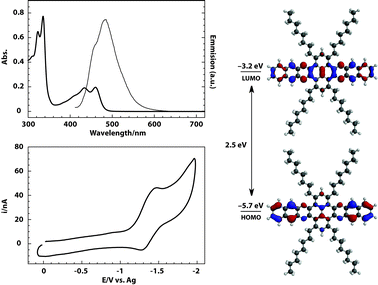 | ||
| Fig. 2 Top left: absorption (thick line) and emission (thin line) spectra of 10. Bottom left: cyclic voltammogram of 10 in 0.1 M TBAP/ODCB. Right: experimental HOMO–LUMO gaps and LUMO level. Calculated shapes of HOMO and LUMO levels. | ||
| λ onset a | λ em a | E gap(opt) b | E gap(calcd) c | E 1/2 d | E onset d | E LUMO(CV) e | E LUMO(calcd) c |
|---|---|---|---|---|---|---|---|
| a Measured in CHCl3. b Estimated from absorption onset. c Calculated B3LYP/6-31G* using Spartan 08. d Measured from 0.1 M ODCB/TBAPvs. Ag wire, potential window (+1.1 to −2.0 V). e Estimated from EONSET according to ELUMO = −4.8 − e (EONSET − E1/2 Fc) eV where E1/2 Fc = 0.48 V. | |||||||
| 488 nm | 478 nm | 2.5 eV | 2.5 eV | −1.37 V | −1.13 V | −3.2 eV | −2.7 eV |
Electrochemical properties of 10 were investigated by cyclic voltammetry in a degassed solution of 1,2-dichlorobenzene and tetrabutyl ammonium perchlorate (TBAP/ODCB) (Fig. 2). On the reduction scan, one reversible reduction wave was observed with half-wave potential of E1/2 = −1.37 V. On the oxidative scan, no waves were observed. The LUMO energy (−3.2 eV) was estimated from the reduction onset of the first reduction wave (Table 1).
A theoretical estimation of the HOMO–LUMO gaps in vacuo (2.5 eV, B3LYP/6-31G*) is in good agreement with the measured optical bandgaps (Table 1). The overestimation by 0.5 eV of the calculated LUMO level (−2.7 eV) is reasonable due to the lack of the solvent in the simulations. As expected both HOMO and LUMO are delocalized along the azaacene core of 10 (Fig. 2).
In summary we have developed a synthetic route to prepare 1,3,6,8-tetraoctyl-4,5,9,10-tetraketopyrene, a new and key building block for the synthesis of soluble pyrene-fused azaacenes. Indeed, by using this building block, we have conveniently synthesised a pyrene-fused tetraazaoctacene derivative with four n-octyl solubilising chains in the pyrene core that displays enhanced solubility in neutral organic solvents (≫20 mg ml−1). Overall, this methodology opens the door for preparing pyrene fused azaacenes with enhanced solubility, facilitating their synthesis, characterisation, and the establishment of their properties. The use of 1,3,6,8-tetraoctyl-4,5,9,10-tetraketopyrene to prepare higher azaacenes and polymers is currently underway and the results will be reported in due course.
This work was carried out with support of the Freiburg Institute for Advanced Studies (FRIAS Junior Research Fellowship) and the Verband der Chemischen Industrie (SK 185/13). We thank Dr M. Keller, Dr J. Wörth and Dr J. Geier (U. Freiburg) for assistance on NMR, MS and X-ray characterisation, respectively.
Notes and references
- M. M. Payne, S. R. Parkin and J. E. Anthony, J. Am. Chem. Soc., 2005, 127, 8028–8029 CrossRef CAS.
- D. Chun, Y. Cheng and F. Wudl, Angew. Chem., Int. Ed., 2008, 47, 8380–8385 CrossRef CAS.
- I. Kaur, N. N. Stein, R. P. Kopreski and G. P. Miller, J. Am. Chem. Soc., 2009, 131, 3424–3425 CrossRef CAS.
- C. Tönshoff and Holger F. Bettinger, Angew. Chem., Int. Ed., 2010, 49, 4125–4128.
- I. Kaur, M. Jazdzyk, N. N. Stein, P. Prusevich and G. P. Miller, J. Am. Chem. Soc., 2010, 132, 1261–1263 CrossRef CAS.
- U. H. F. Bunz, Chem.–Eur. J., 2009, 15, 6780–6789 CrossRef CAS.
- J. K. Stille and E. L. Mainen, J. Polym. Sci., Part B, 1966, 4, 665–667 Search PubMed.
- J. K. Stille and E. L. Mainen, Macromolecules, 1968, 1, 36–42 CrossRef CAS.
- K. Imai, M. Kuhihara, L. Mathias, J. Wittman, W. B. Alston and J. K. Stille, Macromolecules, 1973, 6, 158–162 CrossRef CAS.
- F. E. Arnold, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 749–753 CrossRef CAS.
- D. C. Lee, K. Jang, K. K. McGrath, R. Uy, K. A. Robins and D. W. Hatchett, Chem. Mater., 2008, 20, 3688–3695 CrossRef CAS.
- A. Mateo-Alonso, C. Ehli, K. H. Chen, D. M. Guldi and M. Prato, J. Phys. Chem. A, 2007, 111, 12669–12673 CrossRef CAS.
- J. Hu, D. Zhang, S. Jin, S. Z. D. Cheng and F. W. Harris, Chem. Mater., 2004, 16, 4912–4915 CrossRef CAS.
- B. R. Kaafarani, L. A. Lucas, B. Wex and G. E. Jabbour, Tetrahedron Lett., 2007, 48, 5995–5998 CrossRef CAS.
- F. M. Jradi, M. H. Ai-Sayah and B. R. Kaafarani, Tetrahedron Lett., 2008, 49, 238–242 CrossRef CAS.
- D. C. Lee, K. K. McGrath and K. Jang, Chem. Commun., 2008, 3636–3638 RSC.
- L. A. Lucas, D. M. DeLongchamp, L. J. Richter, R. J. Kline, D. A. Fischer, B. R. Kaafarani and G. E. Jabbour, Chem. Mater., 2008, 20, 5743–5749 CrossRef CAS.
- K. K. McGrath, K. Jang, K. A. Robins and D.-C. Lee, Chem.–Eur. J., 2009, 15, 4070–4077 CrossRef CAS.
- K. Jang, J. M. Kinyanjui, D. W. Hatchett and D.-C. Lee, Chem. Mater., 2009, 21, 2070–2076 CrossRef CAS.
- M. Luo, H. Shadnia, G. Qian, X. Du, D. Yu, D. Ma, James S. Wright and Zhi Y. Wang, Chem.–Eur. J., 2009, 15, 8902–8908 CrossRef CAS.
- A. Mateo-Alonso, N. Kulisic, G. Valenti, M. Marcaccio, F. Paolucci and M. Prato, Chem.–Asian J., 2010, 5, 482–485 CrossRef CAS.
- S. Leng, L. H. Chan, J. Jing, J. Hu, R. M. Moustafa, R. M. V. Horn, M. J. Graham, B. Sun, M. Zhu, K.-U. Jeong, B. R. Kaafarani, W. Zhang, F. W. Harris and S. Z. D. Cheng, Soft Matter, 2010, 6, 100–112 RSC.
- B. X. Gao, M. Wang, Y. X. Cheng, L. X. Wang, X. B. Jing and F. S. Wang, J. Am. Chem. Soc., 2008, 130, 8297–8306 CrossRef CAS.
- J. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS.
- G. Venkataramana and S. Sankararaman, Eur. J. Org. Chem., 2005, 4162–4166 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Full experimental procedures and characterisation of compounds 4, 7, 8, 9, and 10. Structure of 7 in CIF format. CCDC 783204. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02249g |
| This journal is © The Royal Society of Chemistry 2011 |