On the route to mimic natural movements: synthesis and photophysical properties of a molecular arachnoid†‡
Joceline
Zeitouny
a,
Abdelhalim
Belbakra
b,
Anna
Llanes-Pallas
a,
Andrea
Barbieri
b,
Nicola
Armaroli
*b and
Davide
Bonifazi
*ac
aDipartimento di Scienze Farmaceutiche, Università di Trieste, Piazzale Europa 1, 34127 Trieste, Italy
bIstituto per la Sintesi Organica e la Fotoreattività del CNR, Via Gobetti 101, 40129 Bologna, Italy. E-mail: nicola.armaroli@isof.cnr.it; Fax: +39 051 6399844
cDepartment of Chemistry, University of Namur (FUNDP), Rue de Bruxelles 61, B-5000, Namur, Belgium. E-mail: davide.bonifazi@fundp.ac.be; Fax: +32 81 725433
First published on 4th October 2010
Abstract
The synthesis, photoswitchability and NIR emitting properties of a novel π-extended pyrene derivative, peripherally decorated with four azobenzenyl-ethynyl legs, are reported.
One of the most intriguing aspects in contemporary supramolecular chemistry is the engineering of architectures that bridge the gap between molecules and applications, taking advantage of molecular motion to perform programmed functions in devices or materials.1 Proposed strategies often find inspiration from components of natural (e.g. physiological and biochemical processes of living systems, such as muscle contraction/extension)2 or manmade (e.g. cogwheels and shuttles)3 machineries from the macroscopic world. In this respect, one of the first examples is the nanoscale elevator reported by Stoddart, Balzani, Credi and co-workers,4 where movement occurs via pH stimulus and develops an exceptional force of up to 200 pN. Landmark results have been also reported by Feringa and co-workers, who showed that macroscopic movements of a liquid crystal, induced by unidirectional changes in the molecular chirality, reversibly rotate clockwise or anticlockwise microscale objects placed on top of its surface.5 Among others, light-induced switchable azobenzenes6 are good candidates for triggering movements at the molecular scale, as they can reversibly switch from the thermodynamically stable trans configuration to the cis one, by irradiation with UV or VIS light.7 The back cis →trans reaction can be accomplished either by light or heat. The versatility of azobenzenes has been demonstrated in several supramolecular structures8 and at the interfaces9 such as in self-assembled monolayers (SAMs). Recently, the group of Tour has employed the azobenzene group as the skeleton of a molecule equipped with fullerene appendages, that, upon light irradiation, should exhibit a worm-like movement if deposited on a surface.10 As part of our research program aiming at the preparation of multicomponent architectures exhibiting, at the same time, light switching properties, robust chromophoric character and luminescence properties, herein we describe the synthesis and the photophysical studies in solution of a novel π-conjugated compound in which a 1,3,6,8-tetrasubstituted pyrene derivative (molecule 1, Fig. 1) has been peripherally connected to four azobenzenyl moieties (the legs) through ethynyl linkers. Our goal is to construct a system that may show molecular motion of its azobenzenyl legs upon light stimulation. The syntheses of the azobenzenyl legs and of the tetrasubstituted pyrene conjugate 1 are outlined in Scheme 1 (see also ESI†). 4,4′-diiodoazobenzene 4 was prepared by oxidative coupling reaction of 4-iodo-aniline in the presence of KMNO4 and CuSO4·5H2O in CH2Cl2. In parallel, 4-aminophenylethynyl 3 was synthesized by reacting trimethylsilylacetylene (TMSA) with 4-iodo-N,N-dihexylaniline 2 under Sonogashira-type11 cross-coupling conditions ([Pd(PPh3)4], CuI and iPr2NH), followed by cleavage of the TMS protecting groups upon addition of a 2 M aq. solution of KOH. Subsequent Sonogashira-type reaction between an equimolar mixture of aminophenylacetylene 3 and diiodoazobenzene 4 afforded unsymmetrical azobenzene 6 as an orange solid. Doubly-substituted azobenzene 5 was also obtained as side product. Addition of the terminal ethynyl units (molecule 8) was performed in 79% of yield by Pd-catalysed cross-coupling reaction between 6 and TMSA followed by a cleavage of the protecting group using TBAF. At last, cross-coupling reaction between 1,3,6,8-tetrabromopyrene 912 at rt with five equivalents of azobenzene 8 afforded tetra(4,4′-azobenzenyl)-pyrene 1 (12%) and diazobenzene derivative 12 as darkish red and orange solids, respectively. Separation of compounds 1 and 12 was achieved by repeated column chromatography. In order to study the electronic effects of the azobenzenyl legs on the pyrene emitting properties, tetrasubstituted pyrene derivative 11 bearing four non-switchable aminophenylethynyl legs has been also synthesized starting from tetra(ethynyl)pyrene 10 and 4-iodo-N,N-dihexylaniline 2 (see ESI†). All the structural assignments were supported by 1H-NMR and UV-VIS-NIR spectroscopies (see ESI†) and mass spectrometry. In particular, the molecular mass of each compound was unambiguously established by HR-MALDI-TOF-MS (matrix: DCTB), that displayed for 1 the molecular ion as prominent peak at m/z 2151.2788 (1, M+, C152H158N12+, calc. 2151.2732).
![Reagents and conditions: i, TMSA, [Pd(PPh3)4], CuI, iPr2NH, 50 °C, 20 h (82%); ii, 1 M aq. KOH, MeOH/CH2Cl2, 2 h (98%); iii, 4, [Pd(PPh3)4], CuI, Et3N/THF, rt, 20 h; iv, TMSA, [PdCl2(PPh3)2], CuI, iPr2NH/THF, rt, 20 h; v, TBAF/THF, rt, 20 h; vi, 8, [PdCl2(PPh3)2], CuI, PPh3, iPr2NH/THF, rt, 20 h; vii, TMSA, [PdCl2(PPh3)2], CuI, PPh3, iPr2NH/THF, 80 °C, 12 h; viii, 1 M aq. KOH, MeOH/CH2Cl2, 12 h; ix, 2, [PdCl2(PPh3)2], CuI, Et3N, 45 °C, 12 h. Right bottom: electronic absorption spectra of molecules tttt-1, t-8, 11 and tt-12 in toluene solution at 298 K.](/image/article/2011/CC/c0cc03045g/c0cc03045g-s1.gif) | ||
| Scheme 1 Reagents and conditions: i, TMSA, [Pd(PPh3)4], CuI, iPr2NH, 50 °C, 20 h (82%); ii, 1 M aq. KOH, MeOH/CH2Cl2, 2 h (98%); iii, 4, [Pd(PPh3)4], CuI, Et3N/THF, rt, 20 h; iv, TMSA, [PdCl2(PPh3)2], CuI, iPr2NH/THF, rt, 20 h; v, TBAF/THF, rt, 20 h; vi, 8, [PdCl2(PPh3)2], CuI, PPh3, iPr2NH/THF, rt, 20 h; vii, TMSA, [PdCl2(PPh3)2], CuI, PPh3, iPr2NH/THF, 80 °C, 12 h; viii, 1 M aq. KOH, MeOH/CH2Cl2, 12 h; ix, 2, [PdCl2(PPh3)2], CuI, Et3N, 45 °C, 12 h. Right bottom: electronic absorption spectra of molecules tttt-1, t-8, 11 and tt-12 in toluene solution at 298 K. | ||
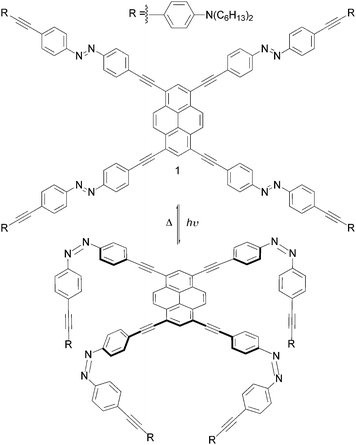 | ||
| Fig. 1 Schematic representation of the full switching of the molecular arachnoid 1 from the 1-tttt to the 1-cccc configuration. | ||
UV irradiation of azobenzene derivatives 1, 8 and 12 afforded the all-trans isomers in toluene solutions, their electronic absorption spectra are depicted in Scheme 1 (bottom right) along with that of tetrasubstituted pyrene reference 11. The spectral onset is progressively red-shifted from 8 to 12 to 1 with enhanced π-delocalization. Upon irradiation of tttt-1 at 470 nm (see exp. section in ESI†), spectral changes are observed, which are stopped after about 2 min. An isosbestic point is detected at 372 nm, suggesting the occurrence of a clean photoreaction (Fig. 2a). By keeping the solution in the dark, the initial spectrum is recovered after 2 hours. The same result is obtained upon light excitation at 313 nm but in a much shorter time, i.e. about 90 seconds (Fig. 2b). Taken together, the above observations suggest the occurrence of a reversible trans–cis interconversion. Photoisomerization experiments have been carried out also with the linear derivative t-8. Due to the lack of the strongly-absorbing pyrenyl unit, this molecule showed more pronounced spectral changes upon illumination (Fig. S1, ESI†). The intense absorption features of 1 and 8, which are attributable to the pyrene core and/or the extended π-delocalization, do not allow us to individuate the weak characteristic absorption fingerprints of the azobenzene moieties around 450 nm, thus hampering the determination of the quantum yield of photoisomerisation.13 In order to get further insight on the control of the molecular movements, semi-empirical (PM3) quantum mechanical calculations on the minimised geometrical structures of the nine possible configurational isomers of molecule 1 have been performed and their relative stability has been assessed (Table S2 and Fig. S3, ESI†). Results show that the all-trans (tttt) is the most stable isomer and that each trans → cisisomerization destabilizes the structure by roughly 2.6 kcal mol−1.14 Furthermore, it has been found that the isomers with the same number of cis and transazobenzene moieties show similar enthalpies of formation (Fig. S2 and S3, ESI†), thereby supporting the idea that the four legs behave like independent units in the gas phase, as the central pyrenyl core is large enough to keep them far apart, avoiding steric congestion and repulsion.
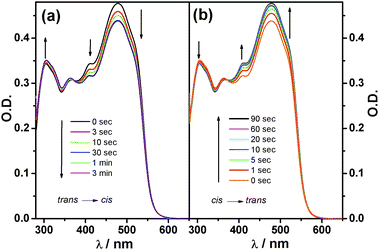 | ||
| Fig. 2 (a) Time evolution of the absorption spectra of a 4.0 × 10−6 M solution of tttt-1 in toluene under 460 nm irradiation, until the photostationary state is reached. (b) Back reaction observed over time under 313 nm irradiation. The initial spectrum of tttt-1 is fully recovered showing complete reversibility. | ||
Despite a few scattered reports claiming weak emission,15azobenzene systems are known to be virtually non-luminescent. Indeed, a detailed analysis of azobenzene 8 does not reveal any reliable luminescence signals in a variety of experimental conditions. On the contrary, pyrene and phenylacetylene derivatives are typically strong emitters16 and in fact reference molecule 11 exhibits an intense luminescence band (λmax = 578 nm, Φfl = 0.70, τ = 3.1 ns, CH2Cl2), comparable to that of unsubstituted pyrene. Molecule 1 unexpectedly shows a weak emission band centred at 740 nm, only observable with NIR sensitive photodetectors (quantum yield ≤ 0.001, Fig. 3). Very interestingly, also bisazobenzene derivative 12 exhibits an almost identical NIR-centred emission, with perfect matching of absorption and excitation spectra (Fig. 3). This proves that the population of the electronic excited states of molecules 1 and 12 yields sensitization of the NIR emitting state. Structurally, both molecules 1 and 12 are, respectively, linked to four and two fragments of 8 through a π-conjugated spacer (a bisethynyl and pyrenyl, respectively). In both cases, the central spacer allows extensive electronic delocalization between the 8-type branches, generating a low-lying NIR emitting level. This shows that both bisethynyl and pyrene π-spacers have a similar effect on the electronic delocalization between the azobenzene units.
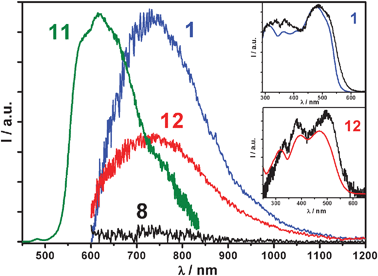 | ||
| Fig. 3 Emission spectra of tttt-1, t-8, 11 and tt-12 (λexc = 330 nm) in toluene at 298 K. Inset: absorption (colored lines) vs.excitation spectra (black lines) of tttt-1 and tt-12. The emission spectrum of 11 is recorded with a conventional UV-Vis photodetector, the others with a NIR sensitive one. Luminescence spectra are corrected for the detector responses. | ||
In summary, we have prepared an arachnoid-like molecular system made of a chromophoric pyrene core and azobenzenyl-ethynyl photoswitchable legs, which exhibits NIR emission, a quite uncommon feature for organic switchable chromophores. We are now carrying out more detailed photophysical investigations to exploit the photoswitchable character of this complex system also on surfaces and to rationalize and tune its intriguing luminescence properties.
This work was supported by EU (MC-RTN “PRAIRIES” MRTN-CT-2006-035810 and MC-ITN FINELUMEN PITN-GA-2008-215399), the CNR (PM.P04.010, MACOL), the FRS-FNRS (2.4.625.08.F & F.4.505.10.F), the “Loterie Nationale”, the ‘TINTIN’ ARC project (09/14-023), the University of Namur and the University of Trieste.
Notes and references
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS; S. Saha and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 77–92 RSC; J. Michl and E. C. H. Sykes, ACS Nano, 2009, 3, 1042–1048 CrossRef CAS.
- T. Mirkovic, N. S. Zacharia, G. D. Scholes and G. A. Ozin, Small, 2010, 6, 159–167 CrossRef CAS; J. Wang, ACS Nano, 2009, 3, 4–9 CrossRef CAS; S. Hiyama, R. Gojo, T. Shima, S. Takeuchi and K. Sutoh, Nano Lett., 2009, 9, 2407–2413 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC; E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS; V. Balzani, G. Bergamini and P. Ceroni, Coord. Chem. Rev., 2008, 252, 2456–2469 CrossRef CAS.
- J. D. Badji, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2006, 303, 1845–1849.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef.
- J. Griffiths, Chem. Soc. Rev., 1972, 1, 481–493 RSC; P. Bortolus and S. Monti, J. Phys. Chem., 1979, 83, 648–652 CrossRef CAS; H. Rau and E. Luddecke, J. Am. Chem. Soc., 1982, 104, 1616–1620 CrossRef CAS.
- N. Takamiya, M. Tada, T. Kishimoto, T. Takano and Y. Sato, Bull. Sci. Eng. Res. Lab., Waseda University, 1986, 114, 12–16 Search PubMed.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC; R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS; M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS.
- G. Pace, V. Ferri, C. Grave, M. Elbing, M. Zharnikov, M. Mayor, M. A. Rampi and P. Samorì, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9937–9942 CrossRef CAS; V. Ferri, M. Elbing, G. Pace, M. Zharnikov, P. Samorì, M. Mayor and M. A. Rampi, Angew. Chem., Int. Ed., 2008, 47, 3407–3409 CrossRef CAS; J. Zeitouny, C. Aurisicchio, D. Bonifazi, R. De Zorzi, S. Geremia, M. Bonini, C.-A. Palma, P. Samori, A. Listorti, A. Belbakra and N. Armaroli, J. Mater. Chem., 2009, 19, 4715–4724 RSC.
- T. Sasaki and J. M. Tour, Org. Lett., 2008, 10, 897–900 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467–4470 CrossRef CAS.
- A. Llanes-Pallas, C.-A. Palma, L. Piot, A. Belbakra, A. Listorti, M. Prato, P. Samori, N. Armaroli and D. Bonifazi, J. Am. Chem. Soc., 2009, 131, 509–520 CrossRef CAS.
- Handbook of Photochemistry, ed. M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, CRC Press, Boca Raton, FL, 3rd edn., 2006 Search PubMed.
- S. Kucharski, R. Janik, H. Motschmann and C. Radüge, New J. Chem., 1999, 23, 765–771 RSC.
- H. Satzger, S. Sporlein, C. Root, J. Wachtveitl, W. Zinth and P. Gilch, Chem. Phys. Lett., 2003, 372, 216–223 CrossRef CAS.
- C. Aurisicchio, B. Ventura, D. Bonifazi and A. Barbieri, J. Phys. Chem. C, 2009, 113, 17927–17935 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details for the experimental procedures, computational studies and photophysical characterization. See DOI: 10.1039/c0cc03045g |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |