Autonomous folding in the membrane proximal HIV peptide gp41659–671: pH tuneability at micelle interfaces†
Craig R.
Gregor
*a,
Eleonora
Cerasoli
b,
Paul R.
Tulip
a,
Maxim G.
Ryadnov
b,
Glenn J.
Martyna
c and
Jason
Crain
ab
aSchool of Physics, The University of Edinburgh, Mayfield Road, Edinburgh, EH9 3JZ, UK. E-mail: c.r.gregor-2@sms.ed.ac.uk
bNational Physical Laboratory, Hampton Road, Teddington, TW11 0WL, UK
cIBM T.J. Watson Research Center, Yorktown Heights, New York, 10598, USA
First published on 8th November 2010
Abstract
The flexibility of the Membrane Proximal Region (MPR) of the HIV-1 gp41 envelope glycoprotein is believed to be relevant to its biological function. Its conformational bias is potentially influenced by the various environmental conditions experienced during viral fusion. Using a combination of Circular Dichroism and Molecular Dynamics simulations, we show that a very short MPR fragment gp41659–671 spanning the 2F5 monoclonal antibody epitope, exhibits autonomous helical folding in the presence of membrane mimicking SDS micelles and the extent of which can be tuned by pH variation: Specifically, the peptide shows no defined fold type at basic pH but is helical at physiological and lower pH environments. By contrast, no such control of helical folding by pH is observed in aqueous solutions in the absence of SDS. Instead, the experimental data imply that unfolded structures persist and that pH has negligible influence on conformational bias. We also explore the pronounced sensitivity to standard empirical potentials and conclude that AMBER-ff03 provides a reasonably accurate description of the solution state structure and is therefore a good choice for future exploration of membrane-induced phenomena.
1. Introduction
A major problem in combating HIV is that the virus has evolved numerous and sophisticated immune-evasion strategies, many of which rely on multiple mutations and sequence variability.1 The Membrane Proximal Region (MPR) of the HIV-1 fusogenic subunit (gp41) is found to be a highly conserved sequence, and therefore an attractive focus for new therapies. Of the few broadly neutralising monoclonal antibodies (mAb) which have been isolated,2 three target gp41 (2F5, Z13 & 4E10)3,4 with epitopes localised in the same continuous MPR; this region encompasses amino acids 660–683 (numbering based on HIV-1 HxB2)5 and also includes the C-terminal residues relevant for the anti HIV-1 fusion inhibitor T-20 (Fig. 1a).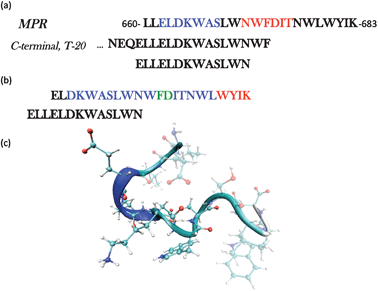 | ||
| Fig. 1 (a) The full sequence of the membrane proximal region (MPR) gp41660–683 highlighting the epitopes for two broadly neutralizing antibodies, 2F5 (blue) and 4E10 (red). The C-terminal sequence is also shown to illustrate the amino acids in common with the peptide ELLELDKWASLWN, the object of this study. (b) The Structure of the peptide ELDKWASLWNWFDITNWLWYIK was solved by NMR in the presence of DPC micelles. The conformation adopted is of a kinked α-helix.6,7 Blue: amino acids in α-helix, Red: 310-helix. (c) Snapshot of the initial structure used in each of the simulations. | ||
Approximately half of the residues in this region are hydrophobic with five of these being tryptophan.8 Mutations in this region (in particular the Trp residues) have been shown to decrease the fusogenicity and inhibit viral entry, suggesting that these residues are critical for the function of this domain.9 However attempts to elicit a neutralising immune response from the membrane-proximal region, and especially the hexapeptide epitope ELDKWA (gp41662–667)10 of the human monoclonal antibody (mAb) 2F5, have not succeeded.11,12 This has prompted several studies to determine the structure of this sequence as well as to establish whether or not additional residues outside of the ‘core’ hexapeptide are required for successful binding to the 2F5 mAb.10 It has also been reported the 2F5 epitope must contain the sequence gp41659–671, where the additional residues enhance the binding affinity by a factor of 103. It is this sequence that we focus on in this study.
Previous solution studies of gp41659–671 have led to conflicting findings regarding conformational preference.10,13 Nuclear Magnetic Resonance data implied that this peptide has a strong conformational bias toward the rare 310-helix in water.13 Conversely, a greater variety of local motifs in this peptide has been revealed by UV resonance Raman scattering14 and independent circular dichroism data10,15 revealed no strong helical bias under aqueous conditions, which was also supported by independent Nuclear Magnetic Resonance data.10 However, parallel tempering molecular dynamics simulations using the CHARMM empirical potential were strongly biased toward helical motifs in water accounting for the experimental data only at unphysically high temperatures.15
Structural data has shown that the sequence gp41662–683 forms a kinked helix in liposomes with a hinge centred on gp41673–674 (Fig. 1b),6,7 which is in contrast to previous results pertaining to the shorter sequence gp41665–683 which revealed a straight α-helix.16 The functional role of these two different conformations has been proposed and linked to mAb neutralising activity; suggesting that the interconversion between the two conformations is an important aspect of the fusion process. Furthermore, it has been proposed that the capping residue Asn671 acts as a switch in this interconversion between the kinked conformation and the longer α-helix.17 These studies have also demonstrated that both the nature of the lipid and the pH were important factors in the structure of the P1 peptide (residues 649–683), located in the conserved MPR of gp41.6 At neutral pH, the N-terminal charged residues of this sequence adopt a disordered structure, while the Trp-rich domain folds into a helix in order to interact with DPC micelles. Under acidic conditions, the Glu-rich region of the P1 peptide also folds into this helical conformation.
We propose that if the hinge gp41673–674 is indeed functionally important, then the sequence gp41659–671 should exhibit an independent capability to interact with a membrane surface. It is also proposed that the two Trp residues are able to fold independently in order to interact with the membrane surface. This autonomous interaction with the surface of a membrane is supported by an investigation into interactions of dipeptides with SDS micelles which probed the effect of neighbouring amino acids on the energetics of the interactions,18 concluding that the presence of either lysine or leucine preceding a Trp residue greatly favoured the Trp-micelle interaction. As both of these amino acids are present in the sequence gp41659–671 (ELLELDKWASLWN), this therefore implies that this peptide should posses a strong affinity for binding. Finally, as these Trp residues may be assigned positions i and i+4 relative to one another, it is logical to assume that these fold into an α-helix.
The effect a membrane has on a proteins structure is dependent partly on the location of the protein relative to the membrane, due to various environmental conditions that it may experience.19 These include diffuse layers where the dielectric constant can be either considered well approximated by aqueous conditions (at far distances) or is considerably reduced (at intermediate regions of closer approach); and the interfacial contact regime where the membrane is in contact or in very close proximity to the protein. Here we examine the secondary structure of gp41659–671 using a combination of computer simulation and experimental measurements under various solvent conditions. These are chosen in order to mimic the distinct physical and chemical environments that this sequence may be exposed to during the viral fusion process. The aim of which is to uncover aspects of structural control and membrane binding of the peptide by chemical and physical processes accessible in the extracellular environment.
2. Results and discussion
2.1 Structure in the diffuse layers surrounding a membrane
Molecular dynamics simulations were performed using three separate force fields in order to determine the secondary structure of gp41659–671 under aqueous conditions and to expose any sensitivity due to the choice in potential. After the initial equilibration, a structural change from the unordered starting configuration (Fig. 1c) had occurred in each of the simulations (SI - Fig. 1), with the two AMBER force fields displaying the greater helical content over the epitope (ELDKWA). This is supported by analysis of the backbone torsional angles over the initial 1ns for each of the simulations (SI - Fig. 2), which revealed that the epitope of the peptide was dominated by a helical structure and only CHARM22 displayed additional secondary structure (attributed to the Asp664 residue). Visualisation of the peptide after 10 ns reveals further structural changes to the peptide in the simulations (Fig. 2). The model using the CHARMM22 force field has folded into an α-helix, spanning the entire length of the peptide chain. The simulation employing the AMBER-ff03 force field however clearly reveals that only the epitope displays an α-helical motif, with residues lying outwith the epitope adopting a more frayed structure. This structure is very similar to that observed after equilibration, indicating that this arrangement of different secondary structures along the peptide chain may be a stable arrangement for the AMBER-ff03 system. Finally, visualisation of the simulation employing the AMBER-ff99SB force field reveals an α-helical structure that extends beyond the epitope region yet not fully encompassing the whole peptide sequence.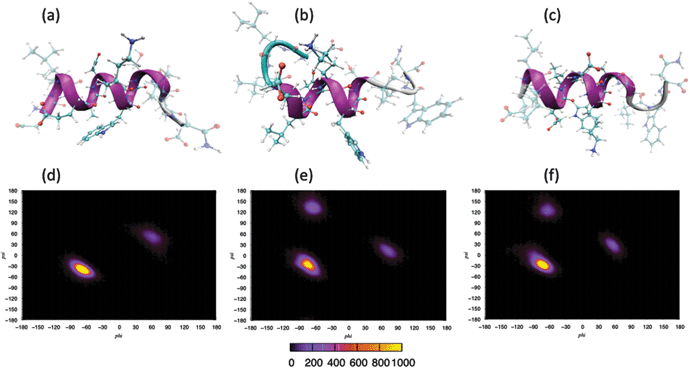 | ||
| Fig. 2 Secondary structure of gp41 after 10 ns: (a) CHARMM, (b) AMBER-ff03, (c) AMBER-ff99SB. Ramachandran plots over last 5 ns of simulation: (d) CHARMM, (e) AMBER-ff03, (f) AMBER-ff99SB. | ||
Analysis of the backbone torsional angles over the last 5 ns of each simulation (Fig. 2) reveals that in the system employing the CHARMM22 force field, the main feature of the Ramachandran plot is a compact peak located within the right-handed α-helical region of dihedral space, with only residue Trp670 presenting a peak outside of this region (located within the left-handed α-helical region). Under the AMBER-ff03 parameterisation, the main feature is also a compact peak located within the right-handed α-helical region, with the peak located within the β-sheet region arising from the torsional angles of the Leu661 and Leu669 residues, and the other peak arising from the Trp670 residue. Finally in the system defined by the AMBER-ff99SB force field, like the other two simulations, the main feature is a compact peak located within the right-handed α-helical region of dihedral space. The other two peaks outwith this α-helical region have been attributed to residue Leu669 and Trp670 for the peaks residing within β-sheet region and left-handed α-helical region respectively.
Bond probability distributions for; i, i+3 (310-helix); i, i+4 (α-helix); and i, i+5 (π-helix) hydrogen bonds reveal evidence of helical structures spanning residues Leu660-Trp670 in the CHARMM22 model (SI - Fig. 3), residues Leu661-Leu669 in the AMBER-ff03 model (SI - Fig. 4), and residues Leu660-Leu669 in the AMBER-ff99SB model (SI -Fig. 5). These are comprised of α-helices and potentially 310-helices, with no π-helical content in any of the three simulations. It is clear that only the CHARMM22 model predicts i, i+4 hydrogen bonding between the two Trp residues (Fig. 3a) as it is the only model to present a peak below an assumed hydrogen bonding threshold of 2.5 Å. In combination with the analysis of the backbone torsional angles (Fig. 2), the length of these helices must be shortened to residues Leu660-Leu669, Glu662-Ser668, and Leu660-Ser668 for the CHARMM22, AMBER-ff03 and AMBER-ff99SB models respectively. This modification is due to aforementioned residues lying out with the right-handed α-helical region of dihedral space. A consequence of this is that all three of the models predict that under aqueous conditions, the two Trp residues do not fold into the α-helical conformation we suspect to be important in the fusion process of gp41659–671.
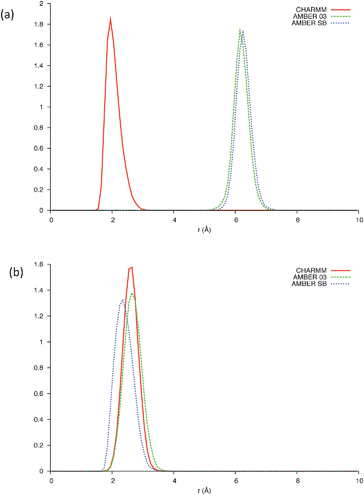 | ||
| Fig. 3 (a) Bond distribution between Trp666 and Trp670 and (b) bond distribution between Glu662 and Lys665, over the last 5 ns of each simulation; CHARMM (Solid line), AMBER-ff03 (Dashed line), AMBER-ff99SB (Dotted line). | ||
The secondary structure of each residue as a function of time was subsequently analysed utilising the STRIDE secondary structure assignment algorithms20 (Fig. 4). The results of which reveal that all models predict the epitope to fold into helical conformation consisting of a mixture of α-helices (major) and the rare 310-helices (minor), with the degree of mixing being dependent on the force field. In each of the three simulations the second Trp residue is not assigned an α-helical conformation, confirming that the two Trp residues do not fold into this co-operatively under aqueous conditions. Of the three force fields used in this study, the CHARMM parameterisation is clearly the most biased towards a helical structure and this finding is certainly consistent with a previous in depth study of classical force fields.21 Additionally it would appear that the simulation utilising the AMBER-ff03 parameterisation gives the best agreement with results obtained previously,10,15 in that the model shows the greatest content of secondary structure over and above that of a helical motif and suggests an ensemble of conformers at the residue level.
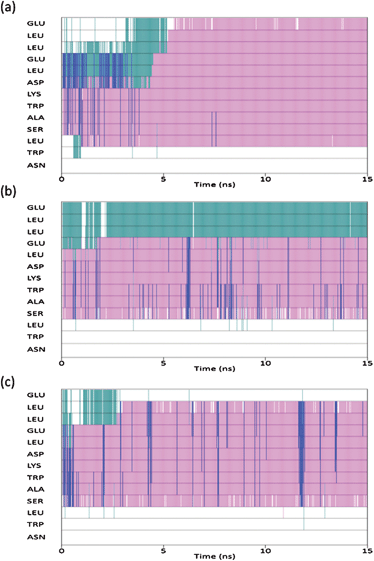 | ||
| Fig. 4 Secondary structure of each residue as a function of time: CHARMM (top), AMBER-ff03 (middle), AMBER-ff99SB (bottom). Pink corresponds to α-helix, blue is 310-helix, green is turn and white corresponds to unordered. | ||
This is supported by far-UV CD spectra of 0.04 mM gp41659–671 in 20 mM Sodium Phosphate (Fig. 5a), which display an aperiodic structure and are also consistent with a rugged energy landscape indicative of a flexible conformational ensemble. The AMBER-ff03 model also demonstrates that regardless of the absence of i, i+4 hydrogen bonding between the two Trp residues, these residues are positioned in order to facilitate simultaneous interaction with a membrane surface (Fig. 6). The lack of an α-helical structure spanning these residues may be attributed to this structure being representative of that under aqueous conditions, and as this peptide is membrane-active then this folding may only transpire in close proximity to a membrane surface.
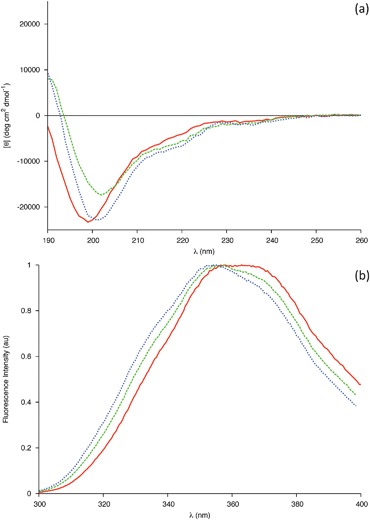 | ||
| Fig. 5 (a) Far-UV CD spectra of 0.04 mM gp41659–671 in 20 mM sodium phosphate buffer pH 6.83 (Solid line), in 20 mM sodium phosphate buffer and 50% Methanol (Dashed line) and in 20 mM sodium phosphate buffer and 50% Acetonitrile (Dotted line). (b) Intrinsic Trp fluorescence of the 0.04 mM gp41659–671 in 20 mM sodium phosphate buffer pH 6.83 (Solid line), in 20 mM sodium phosphate buffer and 50% Methanol (Dashed line) and in 20 mM sodium phosphate buffer and 50% Acetonitrile (Dotted line). | ||
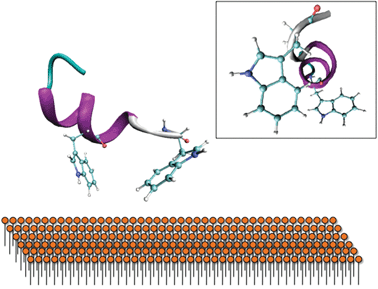 | ||
| Fig. 6 Proposed interaction of the Trp residues with the membrane surface. The secondary structure predicted by AMBER-ff03 reveals that they are orientated in a way to facilitate binding with a membrane surface. Insert: Side-view reveals that these Trp residues are not perfectly aligned, potentially indicating that further folding may occur on a membrane surface. | ||
Mixed water–alcohol solvents have been reported to model a decrease in dielectric constant effectively, with the effect of various alcohols on a peptides' secondary structure reportedly depending entirely on the average dielectric constant of the solution rather than the individual properties of the alcohols.22,23 Previous investigation of this membrane proximal peptide has exploited the fluorinated alcohol trifluoroethanol (TFE);15 therefore it was of interest to expose any differences between fluorinated and non-fluorinated alcohols. Additionally, organic co-solvents are known to promote intramolecular hydrogen bonding through the removal of water molecules in the proximity of the solute (encompassing the solute in a hydrophobic ‘matrix’) and by lowering the dielectric constant. It was therefore also of interest to compare the effect of a solvent with comparable dielectric constant, yet which lacked hydrogen bonding donors. To that end CD spectra of 0.04 mM gp41659–671 in 20 mM Sodium Phosphate were recorded in the presence of either methanol (50% v/v) or acetonitrile (50% v/v); chosen as they had similar dielectric constants to one another and have both been used previously as mimics for membranes.19,24–26
The changes in the CD spectra (Fig. 5a) were qualitatively similar to those previously observed for TFE,15 with the ensemble of peptide conformations shifting towards a higher helical content. This was indicated by a red shift in the position of the band of negative ellipticity from 199 to 202 nm, coupled with an increase in the intensity of a negative shoulder centred around 220 nm and the emergence of a positive band around 190 nm. As Trp residues are known to be capable of contributing to the far-UV CD spectra, Fluorescence spectra were recorded in order to investigate potential changes in the microenvironment of the Trp side chains (Fig. 5b). N-acetyl tryptophan amide (NATA) was used as a model compound for a completely exposed Trp molecule; such a control method is necessary when comparing spectra of different solvent conditions due to such spectra being extremely sensitive to changes in the environment. Upon addition of each solvent, the changes observed for the peptide are identical to those observed for the NATA control (SI - Fig. 6) indicating that the tryptophans are solvent exposed. These results are compatible with an increase in helical population with both Trp residues favourably orientated to interact with a membrane (Fig. 6).
A crude estimate of the helical content was obtained through the value of molar ellipticity at 222 nm, with the % being based on the assumption that a value of −33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deg cm2 dmol−1 is taken for 100% helix content.27 The mixture of 50% methanol (v/v) results in a composition of 13%, which when compared to the previous investigation is found to be half of the value obtained for 30% TFE (SI - Fig. 7).15 This may be due in part to the differences in the dielectric constant values for these two solvents (with the net dielectric constant of the mixed solvent being lower in the case of TFE) and/or to the differences in stabilisation of secondary structure elements by fluorinated solvents.28 A helical content of 15.9% was obtained for the mixture of 50% acetonitrile (v/v). As acetonitrile and methanol have a similar dielectric constant, this small increase in helical fraction may be evidence of the solvents lack of hydrogen bond donors further promoting the formation of intramolecular hydrogen bonds.
000 deg cm2 dmol−1 is taken for 100% helix content.27 The mixture of 50% methanol (v/v) results in a composition of 13%, which when compared to the previous investigation is found to be half of the value obtained for 30% TFE (SI - Fig. 7).15 This may be due in part to the differences in the dielectric constant values for these two solvents (with the net dielectric constant of the mixed solvent being lower in the case of TFE) and/or to the differences in stabilisation of secondary structure elements by fluorinated solvents.28 A helical content of 15.9% was obtained for the mixture of 50% acetonitrile (v/v). As acetonitrile and methanol have a similar dielectric constant, this small increase in helical fraction may be evidence of the solvents lack of hydrogen bond donors further promoting the formation of intramolecular hydrogen bonds.
The solvents used in this mixed-solvent study are all less polar and have a lower dielectric constant than water. As the dielectric constant has a linear relationship to the number of hydrogen bonds in solution, both of these factors reduce the total number of hydrogen bonds between solvent and peptide. This appears to promote the formation of intramolecular hydrogen bonding within the peptide, promoting the helical conformation in each of the solvents studied. As these mixed solvents can mimic the extended regions of a membrane,19 it is therefore natural to conclude that the secondary structure of the membrane proximal peptide gp41659–671 is more ordered in its native state near a membrane surface than under simple aqueous conditions. These results pertaining to the lowering of dielectric constant, appear to be in greater agreement with the results of the MD simulations than those obtained under aqueous conditions. This may be evidence of the force fields over-expressing the helical content in pure water potentially due to an inability to correctly handle the intermolecular and intramolecular competition for hydrogen bonding. The AMBER-ff03 model may in reality represent the secondary structure of this sequence in an environment with a lower dielectric constant than that of water, with the helical conformation spanning residues Glu662-Ser668 being representative of the increase in helical composition observed in the CD spectra (Fig. 5a).
2.2 Structure at the surface of a membrane: mimicked via micelles
Here we replicate the cumulative partial denaturation effects of a membrane surface; that of the negative charges of a membrane29 and the decrease in dielectric constant near the membrane surface.24,25,30 Far-UV CD spectra of 0.04 mM gp41659–671 in 20 mM sodium phosphate buffer were recorded in the presence of varying concentrations of Sodium Dodecyl Sulfate (SDS). Micellar SDS provides an anionic membrane-mimicking environment31 and has been used extensively to study membrane peptides.32–36 The rationale behind using this model system, as opposed to liposomes or vesicles, was because the simplicity of an SDS micelle makes it an ideal starting point to study the secondary structure of the membrane-proximal gp41659–671 and to elucidate the interactions between the peptide and membrane interfaces.18In the CD experiments carried out, the ensemble of the peptide conformation changes upon addition of SDS with a shift in the population towards higher percentages of helical structures (Fig. 7a). This was indicated by a red shift in the band of negative ellipticity, the formation of a positive band around 190 nm and the emergence of a shoulder centred around 227–228 nm. This latter band appears at higher wavelengths when compared to the classical 220 nm position of the α-helix, suggesting a major contribution to the CD spectra from the Trp residues (known to give rise to bands in this region).37 As this band has so far only appeared in the presence of SDS, this may be evidence of the two Trp residues folding into the proposed helical structure in order to directly interact with the micelle surface. Fluorescence spectra of gp41659–671 in the presence of micelles reveal that the microenvironment of the Trp residues is indeed altered (Fig. 7b), with the shift towards lower wavelengths being indicative of the microenvironment of the Trp residues becoming less polar.38 As this difference between the NATA control and the peptide was only found in the presence of micellar SDS (SI - Fig. 8), it implies membrane-mediated folding of the peptide upon the interaction of the tryptophan residues with the micelle.
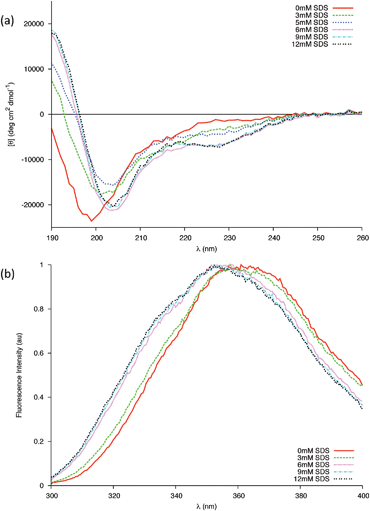 | ||
| Fig. 7 (a) Changes in the far-UV CD spectra of 0.04 mM peptide in 20 mM Phosphate buffer due to increasing concentrations of SDS at 25 °C. Above the cmc all changes are completed. (b) Intrinsic Trp fluorescence of the gp41 peptide in the presence of different concentrations of SDS below (3 mM) and above (6, 9, 12 mM) the cmc. | ||
The first step of the interaction between Trp-rich cationic antimicrobial peptides and membranes is an electrostatic association between these positively charged peptides and the negatively charged micelles.39 As the sequence gp41659–671 carries an overall charge of −2 (localised towards the N-terminus of the peptide) at neutral pH, it is important to investigate the electrostatic interactions between this peptide and the negatively charged head groups of SDS via variations in the pH of the sample. Far-UV CD spectra of gp41659–671 in 20 mM sodium phosphate and 7mM SDS under neutral, acidic and basic conditions were subsequently recorded (Fig. 8).
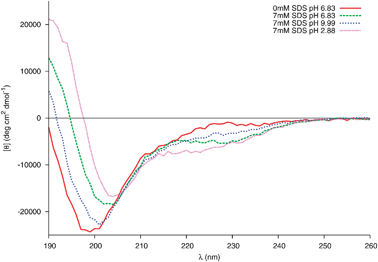 | ||
| Fig. 8 Changes in the far-UV CD spectra of 0.04 mM peptide in 20 mM Phosphate buffer and 7 mM SDS at different pH. | ||
The structure of gp41659–671 at pH 6.83 in the presence of micellar SDS was, as previously mentioned, consistent with the stabilisation of a helical motif. Under acidic conditions, SDS-induced α-helical folding appears to be further enhanced. This is evident through the increased intensity in the band of positive ellipticity and the appearance of an additional negative shoulder near 220 nm; no such increase is detected in the CD spectra of 0.04 mM gp41659–671 in 20 mM sodium phosphate in the absence of SDS (SI - Fig. 9). However under basic conditions this SDS-induced folding is weakened, as the major negative band is blue shifted and accompanied by a decrease in the intensity of the positive band centred around 190 nm.
This structural sensitivity due to pH is only manifested in the presence of SDS micelles; the presence of micelles for all pH values was confirmed using dynamic light scattering (SI - Fig. 10). It is therefore surmised that the molecular mechanism responsible is due to the electrostatic interaction between the negatively charged head groups of SDS and the charged residues of gp41659–671. As previously mentioned, under neutral conditions it appears that the Trp residues (positioned at i and i+4) fold into an α-helix in order to interact with the membrane surface. A strong electrostatic interaction between the Glu662 and Lys665 residues (potentially resulting in a competing i, i+3 electrostatic intrahelical interaction) will also be present; such an interaction is supported by all three of the MD simulations which reveal potential 310hydrogen bonding between these residues (Fig. 3b).
As the two Trp residues bind to the surface of the micelle, the positively charged Lys residue will be attracted to the negatively charged surface and may potentially act as an ‘anchor’ for this helix. There will subsequently be a strong electrostatic repulsion between the anionic head groups of SDS and the Glu residue (Glu662), destabilising this 310-helix. Coupled with the strong electrostatic repulsion between the anionic SDS head groups and Asp664, will result in the N-terminal part of gp41659–671 adopting a more frayed-like structure directed away from the micelle surface. Under acidic conditions, the carboxylic acid groups of the Glu and Asp residues would be protonated, therefore removing the electrostatic repulsion between the negatively charged groups of the peptide and micelle. This protonation subsequently results in the co-operative folding of ELLELD into a helical structure with the sequence spanning the two Trp residues. Finally under extreme basic conditions, deprotonation of the Lys residue also removes the i, i+3 electrostatic interaction and the electrostatic repulsion between the anionic species increases and affects the ability of the two Trp residues to successfully bind to the micelle. This is certainly consistent with the results obtained for the full MPR sequence,6 and suggests that the pH plays an important role in the conformational stability of gp41659–671 at the membrane interface.
Interestingly, an independent study investigating pH dependence on the secondary structure of a peptide (both under aqueous and micellar SDS environments) found that while the aqueous structure was highly dependent on the pH of the system, the secondary structure promoted by the micelle did not vary greatly under the pH conditions tested.40 Such a result was claimed to be expected due to the interaction with SDS potentially imposing constraints on any structural effects caused by variations in the pH. By contrast, we find appreciable pH tuneability suggesting that gp41659–671 is structurally flexible even in close proximity to a membrane surface.
It should be noted that phospholipid vesicles may represent more robust membrane models, due to the small size and high curvature of SDS potentially affecting the nature of the interaction with the peptide.41 Whether utilising such vesicles would give more insight into the membrane interactions of gp41659–671 is the subject of our future studies.
3. Experimental
3.1 Materials
The peptide, ELLELDKWASLWN, was purchased from Cambridge Peptides with a stated final purity of 100%. The peptide stock was prepared by dissolving the peptide powder directly in 20 mM buffer solution (pH 6.83) when applicable. For samples in pure water, the solubilisation of the peptide was helped through additions of 2 μl of 1M NaOH. In order to check for complete dissolution, the absorbance at 350 nm was monitored. The concentration of the stock solutions were determined spectrophotometrically using the calculated extinction coefficient at 280 nm of 14000 M−1 cm−1.13
The peptide stock solutions were used to prepare working solutions of various concentrations of Methanol (50% v/v), Sodium Dodecyl Sulfate (3 mM, 4 mM, 5 mM, 6 mM, 9 mM, 12 mM), and Acetonitrile (50% v/v). Water and 0.2 M buffer solutions were used to ensure the overall buffer concentration remained 20 mM. The concentration of each sample was checked spectrophotometrically prior to any experiments being carried out, using the same extinction coefficient used to determine the concentration of the stock solution.
3.2 Circular dichroism (CD) measurements
Far-UV CD spectra were recorded on a Jasco J-810 spectropolarimeter (Japan Spectroscopic Co., Tokyo) fitted with a Peltier unit for temperature control. The concentration of peptide in solution was 0.04 mM. The spectra are the average of three accumulations in step mode (data pitch of 1 nm, response time of 8 s, bandwidth of 1 nm) acquired in rectangular cuvettes of 0.1 cm path length (unless otherwise stated) at 25 °C. For every sample, an appropriate solvent spectrum was recorded under identical conditions and subtracted from the corresponding sample spectrum. The data collected was expressed in molar ellipticity (deg cm2 dmol−1) in order for a direct comparison with CD spectra reported to date.10,133.3 Dynamic Light Scattering measurements
DLS was used to confirm whether the concentrations of the surfactants were above or below the critical micelle concentration (cmc). The DLS measurements were performed using a Zetasizer Nano (Malvern) and disposable clear plastic cuvettes at 25 °C. For each run, three accumulations were performed to ensure consistent results. Data was subsequently analysed using the manufacturer's Dispersion Technology Software (DTS version 5.10).In order to ensure subsequent interpretation of the CD spectra was handled correctly, it was necessary to confirm which of the concentrations of SDS lay above and below the cmc. DLS was performed on the solutions of various concentrations of SDS in 20 mM sodium phosphate (i.e. without gp41659–671) to determine where they lay relative to the cmc of the surfactant (SI - Fig. 11). Furthermore, in order to verify that the solutions of gp41659–671 and SDS under the different pH conditions tested lay above the cmc, DLS measurements performed on the samples (SI - Fig. 10) revealed that all samples lay above the cmc.
3.4 Fluorescence spectroscopy
Tryptophan fluorescence was measured with a LS55 (Perkin Elmer Instruments, Massachusetts, USA) at 25 °C using a 1cm quartz cuvette (Hellma GmbH, Germany) and 0.0647 mg ml−1protein solution. To selectively excite the tryptophan an excitation wavelength of 295 nm (2.5 nm excitation slit) was used. The spectra were acquired from 300 to 400 nm (emission slit 6nm) and subtracted with the corresponding buffer blank.3.5 Computational methods
In a previous publication conformational equilibria was investigated using classical atomistic molecular dynamics. Three starting configurations based on motifs reported by Barbato et al.10 and Biron et al.13 were employed, which comprised of an α-helix, a type β-turn and an inverse γ-turn. One motif that was reported by Barbato, yet not investigated, was that of the unordered conformation. Furthermore, as the results of the previous study suggested that the CHARMM force field over expressed the helical content in pure water, it was imperative to investigate the effect of employing different force fields for this system. A unit cell was set up, which comprised one gp41 molecule (Fig. 1c), 1853 water molecules and 2 sodium counter ions in order to ensure charge neutrality. The molecules were arrayed on a cubic lattice with random molecular orientations in a cubic box of length 38.2 Å. The water molecules were represented by the rigid, non-polarisable 3-site TIP3P empirical force field. The unit cell was replicated three-fold with the peptide being described by either the CHARMM22,42 AMBER-ff0343,44 or AMBER-ff99SB45 force fields in each of the replicas. The systems were equilibrated at 300 K in the canonical system for 150 ps with a time-step of 0.5 fs to anneal out unphysical contacts. The system was then run for 150 ps with a time step of 0.5 fs in the isothermal-isobaric ensemble to allow for spatial relaxation. The system was then run for a further 150 ps in the canonical ensemble with a time-step of 1 fs being employed, before a production run of 15 ns using a time-step of 2.5 fs from which all data was collected. Periodic boundary conditions were employed and long-range interactions were evaluated via Ewald summation.In order to analyse the structure of the peptide using Molecular Dynamics simulations, we have made use of Ramachandran plots and bond probability distribution functions, as used by Samuelson et al.46 The former allows for the assignment of secondary structure motifs based on dihedral angles, while providing a clear illustration of the conformational space explored during the course of the simulation. The later on the other hand allows for the probability distribution of selected atomic contacts as a function of inter-atomic separation to be assessed. These distribution functions arise from the connectivities between residues i and i+n where n = 3, 4 or 5 for 310−, α- and π-helices respectively, and examination of such functions allows us to assess the conformational and secondary structure preferences for each individual residue at a given temperature.
4. Conclusions
We have studied the structural properties of the HIV peptide gp41659–671 using mixed solvents, anionic micelles and MD simulations in order to mimic the distinct local environments relevant to viral fusion. The results reveal gp41659–671 presents an independent capability to interact with a membrane surface, with different segmental secondary structures being adopted during the fusion process. The bias towards helical folding can be tuned by pH variation to the extent that the membrane proximal peptide shows no defined fold type at basic pH and that it is helical near physiological and lower pH environments. This is in contrast to results obtained in the absence of membrane mimicking SDS, which revealed no such control of helical folding by pH under aqueous conditions. The pronounced sensitivity to standard empirical potentials was also explored and it was concluded that AMBER-ff03 provides a reasonably accurate description of the solution state structure and provides a marked improvement over the previously employed CHARMM force field, which substantially overestimated the helical population. On the basis of this force field comparison study, it would be interesting to determine if the AMBER-ff03 force field transfers to the membrane environment and provides useful input to aide the interpretation of the experimental results.References
- D. C. Montefiori, in Retroviral Immunology: Immune Response and Restoration, Humana Press, Totowa, NJ, 2001, ch. HIV-specific neutralizing antibodies Search PubMed.
- A. Trkola, A. P. Pomales, H. Yuan, B. Korber, P. J. Maddon and G. Allaway, J. Virol., 1995, 69, 6609–6617 CAS.
- T. Muster, F. Steindl, M. Purtscher, A. Trkola, A. Klima and G. Himmler, J. Virol., 1993, 67, 6642–6647 CAS.
- M. B. Zwick, A. K. Labrjin, M. Wang, C. Spenlehauer, E. O. Saphire, J. M. Binley, J. P. Moore and G. Stiegler, J. Virol., 2001, 75, 10892–10905 CrossRef CAS.
- M. Montero, N. E. van Houten, X. Wang and J. K. Scott, Microbiol. Mol. Biol. Rev., 2008, 72, 54–84 CrossRef CAS.
- J. Coutant, H. Yu, M. J. Clement, A. Alfsen, F. Toma, P. A. Curmi and M. Bomsel, FASEB J., 2008, 22, 4338–4351 CrossRef CAS.
- Z. Y. Sun, K. J. Oh, M. Kim, J. Yu, V. Brusic, L. Song, Z. Qiao, J. H. Wang, G. Wagner and E. L. Reinherz, Immunity, 2008, 28, 52–63 CrossRef CAS.
- G. Ofek, M. Tang, A. Sambor, H. Katinger, J. R. Mascola, R. Wyatt and P. D. Kwong, J. Virol., 2004, 78, 10724–10737 CrossRef CAS.
- K. Salzwedel, J. T. West and E. Hunter, J. Virol., 1999, 73, 2469–2480 CAS.
- G. Barbato, E. Bianchi, P. Ingallinella, W. H. Hurni, M. D. Miller, G. Ciliberto, R. Cortese, R. Bazzo, J. W. Shiver and A. Pessi, J. Mol. Biol., 2003, 330, 1101–1115 CrossRef CAS.
- X. Liang, S. Munshi, J. Shendure, G. Mark, M. E. Davies and D. C. Freed, Vaccine, 1999, 17, 2862–2872 CrossRef CAS.
- Y. Lu, Y. Xiao, J. Ding, M. P. Dierich and Y.-H. Chen, Scand. J. Immunol., 2000, 51, 497–501 Search PubMed.
- Z. Biron, S. Khare, A. O. Samson, Y. Hayek, F. Naider and J. Anglister, Biochemistry, 2002, 41, 12687–12696 CrossRef CAS.
- Z. Ahmed and S. Asher, Biochemistry, 2006, 45, 9068–9073 CrossRef CAS.
- P. R. Tulip, C. R. Gregor, R. Z. Troitzsch, G. J. Martyna, E. Cerasoli, G. Tranter and J. Crain, J. Phys. Chem. B, 2010, 114, 7942–7950 CrossRef CAS.
- D. J. Schibli, R. C. Montelaro and H. J. Vogel, Biochemistry, 2001, 40, 9570–9578 CrossRef CAS.
- R. Pejchal, J. S. Gach, F. M. Brunel, R. M. Cardoso, R. L. Stanfield, P. E. Dawson, D. R. Burton, M. B. Zwick and I. A. Wilson, J. Virol., 2009, 83, 8451–8462 CrossRef CAS.
- T. Imamura and K. Konishi, J. Colloid Interface Sci., 1998, 198, 300–307 CrossRef CAS.
- L. A. Munishkina, C. Phelan, V. N. Uversky and A. L. Fink, Biochemistry, 2003, 42, 2720–2730 CrossRef CAS.
- M. Heinig and D. Frishman, Nucleic Acids Res., 2004, 32, W500–W502 CrossRef CAS.
- R. B. Best, N.-V. Buchete and G. Hummer, Biophys. J., 2008, 95, L07–L09 CrossRef CAS.
- K. D. Wilkinson and A. N. Mayer, Arch. Biochem. Biophys., 1986, 250, 390–399 CrossRef.
- E. Dufour, C. Bertrand-Harb and T. Haertlé, Biopolymers, 1993, 33, 589–598 CrossRef CAS.
- V. E. Bychkova, A. E. Dujsekina, S. I. Klenin, E. I. Tiktopulo, V. N. Uversky and O. B. Ptitsyn, Biochemistry, 1996, 35, 6058–6063 CrossRef.
- O. B. Ptitsyn, V. E. Bychkova and V. N. Uversky, Philos. Trans. R. Soc. London, Ser. B, 1995, 348, 35–41 CrossRef CAS.
- J. L. Otvos, G. I. Szendrei, V. M.-Y. Lee and H. H. Mantsch, Eur. J. Biochem., 1993, 211, 249–257 CAS.
- E. K. O’Shea, R. Rutkowski and P. S. Kim, Science, 1989, 243, 538–542 CAS.
- M. Buck, Q. Rev. Biophys., 1998, 31, 297–355 CrossRef CAS.
- T. Endo and G. Schatz, EMBO J., 1988, 7, 1153–1158 CAS.
- V. E. Bychkova and O. B. Ptitsyn, Chemtracts: Biochem. Mol. Biol., 1993, 4, 133–163 CAS.
- C.-S. C. Wu and J. T. Yang, Mol. Cell. Biochem., 1981, 40, 109–122 CAS.
- K. V. Pervushin, V. Y. Orekhov, A. I. Popov, L. Y. Musina and A. S. Arseniev, Eur. J. Biochem., 1994, 219, 571–583 CrossRef CAS.
- D. K. Chang, S. F. Cheng and W. J. Chien, J. Virol., 1997, 71, 6593–6602 CAS.
- C. H. Papavoine, R. N. Konings, C. W. Hilbers and F. J. V. de Ven, Biochemistry, 1994, 33, 12990–12997 CrossRef CAS.
- V. Chupin, J. A. Killian, J. Breg, H. H. de Jongh, R. Boelens, R. Kaptein and B. de Kruijff, Biochemistry, 1995, 34, 11617–11624 CrossRef CAS.
- H. W. van de Hooven, C. C. Doeland, M. V. D. Kamp, R. N. Konings, C. W. Hilbers and F. J. V. D. Ven, Eur. J. Biochem., 1996, 235, 382–393 CAS.
- N. Sreerama and R. W. Woody, Methods Enzymol., 2004, 383, 318–351 CAS.
- E. A. Burstein, N. S. Vedenkina and M. N. Ivkova, Photochem. Photobiol., 1973, 18, 263–279 CAS.
- L. W. Tinoco, J. A. da Silva, A. Leite, A. P. Valente and F. C. L. Almeida, J. Biol. Chem., 2002, 277, 36351–36356 CrossRef CAS.
- G. A. Dykes, R. E. W. Hancock and J. W. Hastings, Biochem. Biophys. Res. Commun., 1998, 247, 723–727 CrossRef CAS.
- M. Magzoub, K. Kilk, L. E. G. Eriksson, U. Langel and A. Gräslund, Biochim. Biophys. Acta, Biomembr., 2001, 1512, 77–89 CrossRef CAS.
- A. MacKerell, D. Bashford, M. Bellott, R. Dunbrack, J. Evanseck, M. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. Lau, C. Mattos, S. Michnick, T. Ngo, D. Nguyen, B. Prodhom, W. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586 CrossRef CAS.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo and T. Lee, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS.
- M. Lee and Y. Duan, Proteins: Struct., Funct., Bioinf., 2004, 55, 620–634 Search PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Bioinf., 2006, 65, 712–725 Search PubMed.
- S. O. Samuelson, D. J. Tobias and G. J. Martyna, J. Phys. Chem. B, 1997, 101, 7592–7603 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional supporting experimental (fluorescene, DLS) and simulation results. See DOI: 10.1039/c0cp01502d |
| This journal is © the Owner Societies 2011 |