Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuel cells
Frédéric
Jaouen
*a,
Eric
Proietti
a,
Michel
Lefèvre
a,
Régis
Chenitz
a,
Jean-Pol
Dodelet
*a,
Gang
Wu
b,
Hoon Taek
Chung
b,
Christina Marie
Johnston
b and
Piotr
Zelenay
*b
aInstitut National de la Recherche Scientifique, Énergie, Matériaux & Télécommunications, 1650 Bd Lionel Boulet, Varennes, Québec J3X 1S2, Canada. E-mail: jaouen@emt.inrs.ca; Fax: +450 929 8102; Tel: +450 929 8176; dodelet@emt.inrs.ca; Fax: +450 929 8198; Tel: +450 929 8142
bMaterials Physics and Applications Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: zelenay@lanl.gov; Fax: +505 662 4292; Tel: +505 667 0197
First published on 18th October 2010
Abstract
Hydrogen produced from water and renewable energy could fuel a large fleet of proton-exchange-fuel-cell vehicles in the future. However, the dependence on expensive Pt-based electrocatalysts in such fuel cells remains a major obstacle for a widespread deployment of this technology. One solution to overcome this predicament is to reduce the Pt content by a factor of ten by replacing the Pt-based catalysts with non-precious metal catalysts at the oxygen-reducing cathode. Fe- and Co-based electrocatalysts for this reaction have been studied for over 50 years, but they were insufficiently active for the high efficiency and power density needed for transportation fuel cells. Recently, several breakthroughs occurred that have increased the activity and durability of non-precious metal catalysts (NPMCs), which can now be regarded as potential competitors to Pt-based catalysts. This review focuses on the new synthesis methods that have led to these breakthroughs. A modeling analysis is also conducted to analyze the improvements required from NPMC-based cathodes to match the performance of Pt-based cathodes, even at high current density. While no further breakthrough in volume-specific activity of NPMCs is required, incremental improvements of the volume-specific activity and effective protonic conductivity within the fuel-cell cathode are necessary. Regarding durability, NPMCs with the best combination of durability and activity result in ca. 3 times lower fuel cell performance than the most active NPMCs at 0.80 V. Thus, major tasks will be to combine durability with higher activity, and also improve durability at cell voltages greater than 0.60 V.
 Frédéric Jaouen | Frédéric Jaouen is a research associate in Jean-Pol Dodelet's group at the Institut National de la Recherche Scientifique since 2004. He obtained his PhD at the Royal Institute of Technology of Stockholm in 2003. His PhD focused on the modeling and experimental characterization of Pt-based cathodes. He was then a postdoctoral researcher in the same group and developed an efficient air-breathing PEM fuel cell. His interests include the synthesis and characterization of noble or non-noble metal catalysts for the oxygen reduction and modeling of kinetics and transport phenomena in porous electrodes. |
 Eric Proietti | Eric Proietti is a Mechanical Engineering graduate from McGill University and is currently completing his PhD in Energy and Materials Science in Professor Jean-Pol Dodelet's research group at the Institut national de la recherche scientifique. His studies focus on the development of non-precious metal catalysts for oxygen reduction in polymer electrolyte membrane fuel cells. Eric is also a co-founder of Canetique Electrocatalysis, Inc., a spin-off company focused on the development and commercialization of non-precious metal catalysts for fuel cells and metal-air batteries. |
 Michel Lefèvre | Michel Lefèvre is currently a research associate in Jean-Pol Dodelet's group at the Institut National de la Recherche Scientifique. He completed his PhD in 2003 at INRS on the synthesis and surface characterization, using ToF-SIMS and XPS, of non-precious metal catalysts for oxygen reduction in PEM fuel cell. He was then a postdoctoral researcher at NRC Industrial Materials Institute and worked on bipolar plate made by graphite compaction for PEM fuel cells. Michel is also a co-founder of Canetique Electrocatalysis, Inc., a spin-off company focused on the development and commercialization of non-precious metal catalysts for fuel cells and metal-air batteries. |
 Gang Wu | Gang Wu completed his Ph.D. studies in 2004 at the Harbin Institute of Technology. His Ph.D. research was focused on electrodeposition and electrocatalysis. After postdoctoral appointments at Tsinghua University, the University of South Carolina, and Los Alamos National Laboratory (LANL), he became a research scientist at LANL in 2010. His current research focuses on nanostructured materials for electrochemical energy conversion and storage. He has published over 50 papers in scientific journals and received, to date, 700 citations. Recently he was awarded 2011 LANL Early Career LDRD funding to develop non-precious metal cathodes for lithium-air batteries. |
 Hoon Taek Chung | Hoon Taek Chung received his Ph.D. in Materials Science and Engineering from the Korea Advanced Institute of Science and Technology (KAIST), South Korea, in 1989. He did his post-doctoral research at the Tokyo Institute of Technology, Japan, in 1993–1994, where he performed crystal structure analysis of lithium ionic conducting materials using the X-ray diffraction Rietveld method. He has published over 40 papers in peer-reviewed journals in the field of lithium batteries. In 2007, he joined Los Alamos National Laboratory (LANL) as a visiting scientist to pursue the development of novel non-precious metal catalysts for the oxygen reduction reaction. |
 Christina Marie Johnston | Christina M. Johnston completed her Ph.D. studies in Chemistry at the University of Illinois at Urbana-Champaign in 2005 as an NSF graduate fellow. Her dissertation work involved fundamental studies of methanol electro-oxidation catalysts. She was awarded a Director's Postdoctoral Fellowship at the Los Alamos National Laboratory (LANL) in 2005 to evaluate alternative support materials for platinum catalysts in PEFCs. She became a research scientist at LANL in 2008. Her current work focuses on non-precious metal catalysts and novel electrode structures for PEFCs. |
Broader contextThe use of catalysts in the chemical and pharmaceutical industry is widespread. Their use in electrochemical applications is of paramount importance, especially in power sources such as batteries and fuel cells. Today, hydrogen/air polymer electrolyte fuel cells (PEFCs) are considered a promising technology to replace internal combustion engines for automotive propulsion. However, a major drawback of current PEFC technology is their high cost, in large part due to the use of platinum-based catalysts at both the anode (10%) and cathode (90%). Recently, two paths have been considered to reduce the cost of PEFCs cathode catalysts: (a) Improve the activity for oxygen reduction of Pt-based catalysts by nano-structuring or alloying, or (ii) replace Pt-based catalysts altogether with lower-cost, non-precious metal catalysts (NPMCs). This review focuses on NPMCs obtained from the heat treatment, at temperatures above 600 °C, of carbon, nitrogen and Fe or Co precursors. The recent activity and stability breakthroughs as well as the synthesis procedures and understanding that led to these breakthroughs are summarized. The remaining improvements required for NPMCs to replace Pt-based catalysts are also highlighted. |
1. Introduction
1.1. Future energy sources and fuels for automotive transportation
Transportation accounts for about one third of the 140,000 TWh of primary energy (or 45,000 TWh of useful energy†) consumed annually.1 The primary energy demand is, all sectors included, met at 80–85% by fossil fuels and, in the transportation sector, this figure reaches 95%. In the long-term future, hydrogen produced from renewable energies could supplant fossil fuels.Both the internal combustion engine (ICE) and the fuel cell can be fuelled with hydrogen. Today, the most adequate fuel cell for automotive propulsion is the hydrogen/air polymer electrolyte fuel cell (PEFC), but the cost and performance targets for this application are challenging.2,3 Since the production cost of clean H2 from water will be non-negligible, its efficient conversion into mechanical energy is required to achieve an acceptable fuel cost per km. Efficiency is also important for the driving range. Storing enough H2 for a driving range greater than 400 km is challenging because of the low energy density of compressed H2 (Table 1, Row 3). This drawback, however, is partly compensated by the higher efficiency of the PEFC/electric motor system compared to that of an ICE/conventional power train (Table 1, Row 4).4,5 A driving range of 480 km has been demonstrated by PEFC cars carrying compressed H2.6,7 Alternatively, and without any fuel, plug-in battery cars with 100–150 km driving range could be interesting for urban use. Limitations are the charging time and the battery cost and weight (Table 1, Row 5).
| row | Gasoline | Biodiesel | Ethanol | Compressed H2 (690 bars) | Li-ion battery | Ni-MH battery | |
|---|---|---|---|---|---|---|---|
| a Based on higher heating value. b With an ICE. c With a PEFC. d 12.9 kWh of mechanical energy per person and per 100 km was assumed.101 The weight of fuel per person and per 100 km is calculated from 12.9 kWh × (100/Efficiency)/(kWh kg−1). e Electrode materials only. f With packaging. g H2 only. h Assuming a storage system with gravimetric and volumetric capacities of 4.5 wt % H2 and 22 gH2/L, respectively,98 corresponding to state-of-art storage systems for compressed H2 (690 bars). | |||||||
| 1 | Energy densitya/kWh kg−1 | 13.497 | 11.197 | 8.397 | 39.3g,97 | 0.4–1e,99 | 0.24e,99 |
| 1.8h,98 | <0.2f | <0.1f | |||||
| 2 | Density/kg of fuel L−1 | 0.72 | 0.88 | 0.79100 | 0.0615g | — | — |
| 0.022h,98 | |||||||
| 3 | Energy Densitya/kWh L−1 | 9.7 | 9.8 | 6.6 | 2.4g | — | — |
| 0.9h | <0.4f | <0.25f | |||||
| 4 | Efficiency/% | 20.0b | 25.0b | 20.0b | 50.0c | 85.0 | 85.0 |
| 5 | Weight per person per 100 kmd/kg | 4.8 | 4.6 | 7.8 | 0.8g | 75.9f | 151.8f |
| 17.9h | |||||||
| 6 | Volume per person per 100 kmd/L | 6.6 | 5.3 | 9.8 | 13.4g | 37.9f | 60.7f |
| 35.8h |
1.2. Importance of catalysts for high efficiency and power density of PEFC
While ICEs generate power without any catalysts, needing precious metals only for catalytic converters in order to eliminate noxious gases, the catalysts found in electrodes are key to electric power generation by PEFCs. Therefore, R&D efforts in the catalysis of the PEFC reactions (1) and (2) have been continuously pursued since the 1990's.| 2H2 → 4H+ + 4e Anode reaction | (1) |
| O2 + 4H+ + 4e → 2H2O Cathode reaction | (2) |
The only viable catalyst used today in large-scale PEFCs is nano-structured platinum, dispersed or not on a support, and found either as a pure Pt-phase or as an alloy (Pt3Co, Pt3Ni, etc). Even though platinum is a rare and precious metal, its replacement in high power-density PEFCs has, to date, been prohibited by insufficient activity for reactions (1) and (2) of more abundant catalysts. Due to the five-orders-of-magnitude slower kinetics (Ref. 8, pages 51–52) of the oxygen reduction reaction on Pt (ORR, Reaction 2) compared to that of the hydrogen oxidation reaction (Reaction 1), most platinum is located at the cathode side. The latter usually contains ten times more Pt than the anode.9 Nonetheless, the cathode still remains the major source of losses in efficiency and power density. Several paths have been identified to reduce the Pt loading in cathodes without decreasing their performance:10 (1) enhancing Pt mass activity for ORR via alloying or core-shell nano-structuring, (2) improving mass-transport properties of Pt-based cathodes, and (3) developing well-performing non-precious metal catalysts (NPMCs) for ORR. This review focuses on option (3) and, in particular, on metal/nitrogen/carbon (Me/N/C) catalysts obtained via the heat-treatment of Fe and/or Co precursors, nitrogen- and carbon precursors. However, before delving into this topic, the implications of using Pt-based catalysts in PEFCs for large-scale production are discussed.
1.3. Platinum demand under a scenario of massive PEFC production
The Pt-utilization target for 2015, as defined by the U.S. Department of Energy (U.S. DOE), is 0.2 g of Pt per kW at 55% efficiency for a transportation PEFC stack.11 Based on this target and under the assumption that all cars in the future would be powered by PEFCs, a global annual production of 100 million PEFC cars (today, 70 million new ICE vehicles a year) rated at 50 kW each would require a steady Pt demand of 1000 tons a year. In recent years, the global Pt production has only been ca. 200 tons a year. An eventual global fleet of 1 billion PEFC cars would commit 10,000 tons of Pt. The total world Pt reserves, proven and inferred, are estimated at 40,000 tons, if mining down to 2 km is assumed.12 Based on these numbers, it is clear that the Pt recycling rate must be very high for such a fleet to be sustainable. Under such market pressure, the Pt cost would likely rise significantly. James et al. forecast that the Pt catalyst alone would account for 38–56% of the stack cost,13 assuming a low Pt price of $1,100 per troy ounce and a relatively low production volume of 500,000 PEFC stacks a year. Therefore, replacing platinum-based catalysts with a more abundant material would greatly improve the outlook for the widespread development of automotive fuel cells.2. Activity and performance of NPMCs for the ORR in PEFCs
2.1. A summary of the knowledge in 200614
The ORR is a ubiquitous reaction, occurring not only in fuel cells but also in life on Earth. To catalyze the ORR, Nature has devised complex cytochrome enzymes, in which the active center is an Fe-ion ligated to four nitrogen atoms, FeN4.15 In 1964, Jasinski discovered that a simple metal-N4 molecule, cobalt phthalocyanine, catalyzes the ORR.16 Other Co- and FeN4 macrocycles were later found to catalyze the ORR in alkaline or acid media.17 Although unstable in acid solution, a heat-treatment of such macrocycles between 500 and 900 °C in inert atmosphere greatly improved their stability and activity.18,19 Progress in the ORR electrocatalysis using macrocycle-based NPMCs is still ongoing.20–24 A second route was opened in 1989 when a NPMC was synthesized without resorting to macrocycles.25 An iron salt and polyacrylonitrile (–CH2CHCN–) were heat-treated at 800 °C under Ar. Later, the metal ion, nitrogen and carbon precursors were all separately introduced as a metal salt, NH3 and a carbon support, respectively.26From these studies, it followed that NPMCs are obtained after a heat-treatment at 500–1000 °C if elemental metal ions (Fe, Co, etc) a source of carbon (carbon support, molecule, polymer), and a source of nitrogen (MeN4 macrocycle, N-bearing molecule or polymer, N-containing gas) are simultaneously present during the heat-treatment.
Several reviews on such NPMCs have been published, discussing results obtained up to and including 2006.14,27–34 A generic notation of the active sites created at high temperature is MeNxCy.35–38 The actual presence of a metallic ion in the active sites is still questioned by some research groups claiming that, while Fe or Co is needed to create active sites during the heat treatment, the metal species only acts as a catalyst for site formation but is not necessarily found in the formed catalytic site.39–41 The latter would be composed only of carbon and nitrogen; an idea first proposed in the 1980's.29,42 This hypothesis is difficult to prove or disprove since it is not possible to completely remove Fe or Co from a heat-treated catalyst, even with strong acids. Claims that active NPMCs were free of metal have hitherto been based on the absence of XPS signal for metals.41 However the sensitivity of XPS for Fe or Co (ca 0.1 wt %) is too low to draw such a conclusion since it has been reported that Fe or Co contents as low as 0.02 wt% significantly increase the ORR activity vs. a quasi metal-free catalyst.43 In conclusion, while the ORR activity of truly metal-free CNx sites is non-zero, its value has been hitherto too low to be of practical interest for high power density applications.39,44 To obtain more active catalysts, there is a consensus that a metal species must be present during at least one heat treatment step.
The activities that have been reported for various NPMCs broadly differ. The volumetric activity of an NPMC under given experimental conditions may formally be regarded as the product of the site density with the average single-site activity.10,43
| Iv(E) = SD·TOF(E)·e | (1) |
Despite extensive R&D efforts, by 2005, NPMCs had failed to show ORR activity that would make them viable for transportation application.10,14,33 In 2005, the highest ORR activity in PEFC reported for an NPMC was not even 1/100th of U.S. DOE's 2010 catalyst activity target. No good durability lasting longer than 100 h in PEFC had been shown either. Only recently have major advances in the activity and durability of NPMCs in PEFC been reported.48–55 Today, the most active NPMC for ORR has already reached 75% of U.S. DOE's 2010 target for ORR activity54 and durability up to 450–600 h in PEFC has been demonstrated for other active NPMCs.51,55 The present review focuses on these recent achievements.
2.2. ORR activity of non-precious metal catalysts reported in 2006–2009
Reviewed here are catalysts with ORR activity measured using either the rotating disk electrode (RDE) at room temperature or a fully-humidified PEFC at 80 °C. Activity is defined as either the mass- or volume-specific current (A g−1 or A cm−3) obtained at 0.80 V vs. RHE and with the cathode under kinetic control. The activity values are obtained from Tafel plots of oxygen reduction with slopes of, typically, 55–70 mV/dec. As gas pressures used in various PEFC measurements sometimes differed, all ORR activity values reported here were normalized to 1 bar O2 and H2 partial pressures using eqn (2).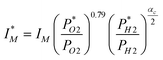 | (2) |
| I*V = ρeffI*M | (3) |
Table 2 gives an overview of the synthesis procedures and activities at 0.80 V vs. RHE under reference conditions (columns 10–11). Prior to 2008, the highest volumetric activity was only 1.4 A cm−3 (Row 10);57 an activity of 2.9 A cm−3 was reported in 2008 (Row 16),50 followed by a major breakthrough in 2009. The most recent achievements include the following: a volumetric ORR activity of (A) 30 and 98 A cm−3 reported by the INRS group (Rows 12–13);54 (B) 23 and 50 A cm−3 reported by the Los Alamos National Laboratory (LANL) group [H. Chung, C. M. Johnston, and P. Zelenay, in preparation] (Rows 17–18), and (C) 5–6 A cm−3 reported by the groups of Bogdanoff and Dahn (Rows 4,14).53 The highest activity reported in 2009 came close to the U.S. DOE's 2010 activity target of 130 A cm−3.
| Column → | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Row↓ | Precursors | Impregnation or dry mix (wt% metal) | Ball-mill | 1st Heat-treatment | Acid-wash | Impregnation or dry mix (wt % metal) | 2nd Heat-treatment | 3rd Heat-treatment | Mass-activitya (RDE) | Mass-activityb (PEFC) | Volume-activityc (PEFC) | Label and/or reference | |
| a At 20 °C, pH 1, 0.80 V vs. RHE (equivalent to 0.50 V vs. SCE at pH 1); in A g−1. b At 80 °C, 0.80 V iR-free cell voltage, corrected to 1 bar O2 and H2 pressures, 100% RH; in A g−1. c At 80 °C, 0.80 V iR-free cell voltage, corrected to 1 bar O2 and H2 pressures, 100% RH; in A cm−3. d Derived from the volumetric activity targets assuming the apparent carbon density of 0.4 g cm−3 in porous cathode. e Acid-washing with 1 M HCL was performed after the second heat treatment. f Ballmilling was performed between the 2nd and 3rd heat treatment. g FeOx means Fe oxalate. h Activity extrapolated from Tafel kinetics observed at higher voltage. i CB means carbon black. j (non)-μp means (non)-microporous. k PF means pore-filler molecule. l Cannot be applied similarly to Pt/C catalysts. | |||||||||||||
| Type T1 | 1 | FeTMPP + CBi | X (0.2 Fe) | — | X (NH3:H2:Ar) | — | — | — | — | — | 0.5 | 0.2 | Ref. 58 |
| 2 | FeTMPP + CB | — | — | X (NH3:H2:Ar) | — | X (0.2 Fe) | X (NH3:H2:Ar) | — | — | 0.2 | 0.1 | Ref. 58 | |
| 3 | FeTMPP + CB | X (4.5 Fe) | — | X (Ar) | — | — | — | — | — | 0.7 | 0.3 | 66%FeTMPP59 | |
| 4 | FeTMPP + FeOxg + S | X (25.0 Fe) | — | X (N2) | X (HCl) | — | X (N2:H2)e | X (CO2) | 18.0 | 13.0h | 5.3h | UK6353 | |
| 5 | CoTMPP + SiO2 | X (7.4 Co) | — | X (N2) | X (KOH) | — | — | — | 1.0 | 2.0 | 0.8 | CoTMPP70053 | |
| Type T2 | 6 | FeAc + non-μp CBj | X (0.2 Fe) | — | X (NH3) | — | — | — | — | 4.5 | 3.3 | 1.3 | FC28053 |
| 7 | FeAc + non-μp CB | X (0.8 Fe) | — | X (NH3) | — | — | — | — | 3.1 | — | — | Ref. 43 | |
| 8 | FeAc + non-μp CB | X (0.2 Fe) | — | X (NH3) | — | — | — | — | 2.4 | 1.7 | 0.7 | Ref. 43 | |
| 9 | FeAc + non-μp CB | — | — | X (Air) | — | X (0.2 Fe) | X (NH3) | — | — | 0.9 | 0.4 | Ref. 61 | |
| 10 | FeAc + non-μp CB | X (0.2 Fe) | — | X (NH3) | — | — | — | — | 5.0 | 3.6 | 1.4 | Ref. 57 | |
| 11 | FeAc + μp CB | X (0.2 Fe) | — | X (NH3) | — | — | — | — | 0.5 | 0.6 | 0.2 | Ref. 52 | |
| 12 | FeAc + PFk + μp CB | X (1.0 Fe) | X | X (NH3) | — | — | — | — | — | 76.0h | 30.2h | PFM(1)54 | |
| 13 | FeAc + PF + μpCB | X (1.0 Fe) | X | X (Ar) | — | — | X (NH3) | — | — | 246.0h | 98.3h | PFM(2)54 | |
| Type T3 | 14 | FeCl2 + SiO2 + Pyrrole | X (0.56 Fe) | — | X (Ar) | X (HF) | X (0.2 Fe) | X (NH3) | — | 1.2 | 14.0h | 5.6h | DAL900C53 |
| 15 | Fe & Cu salts + Adenine + Glucose | X (0.47Fe + 0.53Cu) | — | X (air) | — | — | X (Ar) | — | 0.6 | 3.2 | 1.3 | GadFeCu53 | |
| 16 | FeCl2 + Nitroaniline + non-μp CB | X (Fe) | — | X (N2) | — | — | X (NH3:N2)f | X (NH3:N2) | — | 7.3h | 2.9h | Ref. 50 | |
| 17 | FeII sulfate + | X (6.6 Fe) | — | X (N2) | X (H2SO4) | — | X (N2) | — | — | 58.0h | 23.0h | CM-Fe-C | |
| 18 | Cyanamide + μp CB | X (4.9 Fe) | — | X (N2) | X (H2SO4) | — | X (N2) | — | — | 124.0h | 50.0h | CM-Fe-C(2) | |
| REFS. | 19 | Target for 2010 | — | 325.0d | 130.0 | Ref. 10 & Ref. 11 | |||||||
| 20 | Target for 2015 | — | 750.0d | 300.0 | |||||||||
| 21 | 47 wt % Pt/C | —l | 1300.0 |
2.3. General comments about the synthesis of NPMCs found in Table 2
Metal and nitrogen precursors, together with the type of carbon used, are given in Column 1. If NH3 was used as a gaseous N precursor, this is indicated in Columns 4, 7–8. The precursors (Column 1) were first mixed, either as dry powders with a mortar or by planetary ball-milling, or by wet-impregnation. If the support (carbon or silica) was first subjected to a heat-treatment without a metal precursor, the addition of the metal precursor occurred before the second treatment (Columns 6–7). Some catalysts were subject to acid washing, after either an intermediate heat-treatment or after the final one, in order to leach inactive metal species (e.g. Row 4) or to remove a non-carbonaceous template material (e.g. Row 5). All these NPMCs can be classified into three types: (T1) NPMCs obtained from MeN4-macrocycles acting as a precursor for both the metal ion and nitrogen (Rows 1–5,20,53,58,59) (T2) NPMCs derived from metal salts and NH3 gas as metal and nitrogen precursors, respectively (Rows 6–13,43,54,57,60–62) and (T3) NPMCs obtained from metal salts and a N-containing molecule as metal and nitrogen precursors, respectively (Rows 14–18,50,53).Some catalysts escape this simple classification. For example, catalysts in Rows 1–2 use both a MeN4-molecule and NH3 as nitrogen precursors; catalysts in Rows 13–14 and 16 use both a nitrogen-containing molecule and NH3 as nitrogen precursors. Moreover, T2-catalysts (Rows 6–13) can be further subdivided into three types depending on the carbon support used: (T2a) non-microporous carbon (Rows 6–10), (T2b) microporous carbon (Row 11) and (T2c) microporous carbon filled with a pore-filler molecule prior to heat-treatment (Rows 12–13).
Another distinct feature in the catalyst synthesis is the use of either inert or reactive gases during the heat-treatment(s). The presence of a reactive gas implies that the carbon found either in the support or in the solid nitrogen precursor is continuously gasified during the heat-treatment. The extent of gasification depends on the heat-treatment temperature and duration. Gasification creates micropores during the heat-treatment, which appears to be of great importance for obtaining high activity with certain NPMCs (Rows 6–13).54,60
2.4. NPMCs prepared from metal salt, NH3 gas and a carbon support (T2 - Rows 6–13 in Table 2)
Optimizing this type of NPMC led to the activity breakthrough reported in 2009 (Ref. 54 and Rows 12–13 in Table 2). In the optimum synthesis to date for this type of NPMCs, a microporous carbon black (Black Pearls 2000) was filled with a pore filler (phenanthroline) and iron acetate (FeAc).54 It should be noted that with a similar synthesis procedure, but without pore filler, the use of Black Pearls 2000 resulted in mediocre activity (Row 11 in Table 2,62). The four important steps that led to this breakthrough are detailed below.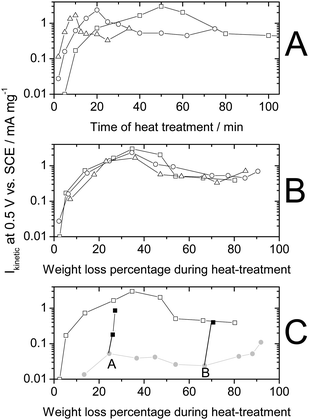 | ||
| Fig. 1 Effect of heat-treatment time and temperature on the activity measured with RDE at 20 °C in a sulfuric-acid solution of pH 1. Reprinted from Jaouen et al., J. Phys. Chem. B, 110, 5553 (2006). Kinetic current of oxygen reduction at 0.50 V vs. SCE (0.80 V vs. RHE) per mass of catalyst against. (A) Heat-treatment time and (B) weight loss percentage of carbon during heat-treatment in pure NH3 at 900 °C (open square), 950 °C (open circle) or 1000 °C (open triangle). All catalysts prepared with 0.2 wt % Fe initial loading on a non-microporous carbon black. (C) Two-step heat-treatment: first step is heat-treating the carbon (950 °C in NH3) without iron (grey circles), thereafter adsorbing iron acetate on the heat-treated carbon (point A or B) and re-heat-treating such compounds at 900 °C in NH3 (filled squares, 2–5 min). | ||
It was also found that the NPMC synthesis could be sequenced in two separate heat-treatments, the first involving the carbon alone in NH3 followed by impregnation with FeAc and a second, shorter, heat-treatment in NH3 (Fig. 1C). While the first heat-treatment slowly produces micropores but no active sites (no Fe precursor yet present), the second heat-treatment generates active ORR sites. Maximum ORR-activity was reached for a weight loss of ca. 35 wt% in all cases involving this non-microporous carbon (Fig. 1). N2-adsorption isotherms were carried out to determine the pore-size distribution. Only the contribution of micropores (pore size ≤2 nm65) showed a maximum at ca. 35 wt%, i.e. the weight-loss corresponding to the highest ORR activity of the catalysts. Moreover, plotting ORR activity against the micropore specific area in log-log coordinates produced a linear relationship (Fig. 6 in Ref. 60), clearly indicating that ORR active sites exist in micropores. This correlation was later, notwithstanding some scattering, convincingly verified for other NPMCs synthesized at INRS66 or at other laboratories53 (Rows 4–6, 14–15 in Table 2).
While microporous carbons as such cannot be efficiently used due to lack of disordered carbon,62 their micropores can, however, be filled with small hydrocarbon molecules (pore-filler) before the heat-treatment in NH3. In one approach taken at INRS, a planetary ball-milling technique was used to fill the micropores. Following optimization of the iron and pore-filler contents, heat-treatment number, sequence, duration and gas used, the ORR activity of the resulting catalyst was enhanced by more than a factor 100.54 The highest activity (98 A cm−3, Row 13) was obtained by mixing Black Pearls 2000 (BP-2000) with 1,10-phenanthroline (50/50 weight ratio) and 1 wt% Fe (from FeAc) with planetary ball milling, then heat-treating the resulting catalyst precursor first in argon at 1050 °C and then at 950 °C in NH3. The optimization of the heat-treatment duration in NH3 turned out to be paramount to reaching such high ORR activities. Fig. 2 shows the change in mass activity, micropore area and surface nitrogen content as a function of the weight loss during the heat-treatment in NH3 for (i) an FeAc-impregnated non-microporous carbon support (hollow circles),60 (ii) FeAc-impregnated BP-2000 without a pore-filler (hollow squares),62 and (iii) BP-2000 filled with a pore-filler and FeAc (filled squares).54 The arrow in Fig. 2B highlights the efficient filling of pores by the pore-filler during planetary ball milling.
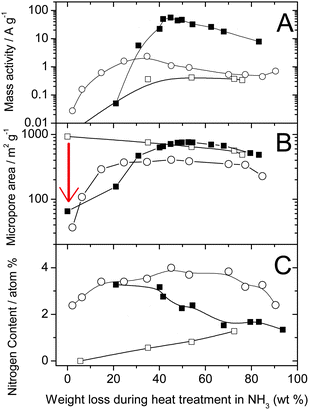 | ||
| Fig. 2 Comparison of catalysts made from a non-microporous carbon black (circle), a microporous carbon black (square) and the same microporous carbon black but filled with a pore filler (filled square). (A) Mass activity at 0.80 V vs. RHE, (B) micropore surface area and (C) nitrogen surface concentration as a function of the weight loss during the heat-treatment in NH3. For a same series, different weight loss corresponds to different heat-treatment times. Circles: 0.2 wt % FeAc on 86977 (BET area 71 m2g−1), heat-treated at 950 °C in NH3 for various times (same data as shown as circles in Fig. 1A). Squares: 0.2 wt % FeAc on Black Pearls 2000 (BET area 1379 m2g−1), heat-treated at 1050 °C in NH3 for various times. Filled squares: 0.2 wt % FeAc in (BlackPearls 2000 + PTCDA, 50/50 wt%), planetary ballmilled at 400 rpm for 3h, then heat-treated at 950 °C in NH3 for various times. NB: activities measured in PEFC (open and filled squares) were normalized to 1 bar O2 and H2 using eqn (1). | ||
2.5. NPMCs prepared from metal salt, N-containing molecule, with or without a carbon support (T3 – Rows 14–18 in Table 2)
The catalysts in Rows 14 and 15 were prepared without carbon support. In Row 14, the catalyst was templated with silica, which was later removed by leaching in HF after a first heat-treatment.53 Pyrrole was used as a precursor for both nitrogen and carbon. A second heat-treatment in NH3 increased the ORR activity significantly, to reach 5.6 A cm−3 (Column 11). In Row 15, no carbon support or template was used. The nitrogen precursor was adenine and the carbon precursors were adenine and glucose.53 After a first heat-treatment in air at 150 °C to dehydrate glucose, the precursors were heat-treated in argon at 1000 °C. The final ORR activity was 1.3 A cm−3 (Column 11). The catalyst in Row 16 used a non-microporous carbon, iron chloride and nitroaniline.50 The precursors were subject to: (i) a first heat-treatment at 300 °C in N2, (ii) a second heat-treatment at 800–1000 °C in NH3, (iii) ball-milling to break agglomerates, (iv) a third heat-treatment at 800–1000 °C in NH3 and (v) acid-washing. The final ORR activity (converted to correspond to U.S. DOE reference conditions) was 2.9 A cm−3 (Column 11).The remainder of this section focuses on the catalysts in Rows 17 and 18, prepared by the LANL group and using relatively simple N-precursors, in contrast to the N-bearing polymers used for the synthesis of more durable, but less active, catalysts discussed in Section 3. The precursors used were (i) FeII sulfate (FeSO4·7H2O), (ii) cyanamide (CN2H2, labeled CM) and (iii) a commercial microporous carbon support.69 Cyanamide plays a dual role as a source of both nitrogen and carbon. The first formulation of this catalyst was developed in 2007–2008 and achieved good ORR activity (Row 17, 23 A cm−3) compared to previous NPMCs.70 For this catalyst, Ketjenblack EC-300J (labeled KJ-300J) was pretreated in nitric acid before addition to a slurry of cyanamide and FeII sulfate. After drying, the material was heat-treated at 1050 °C for 1 h in flowing N2, acid-leached, and finally heat-treated again at 1050 °C for 1 h in N2 (Table 2).
The effect of using alternate precursors on the final ORR activity was then investigated. Replacing cyanamide with ethylenediamine generated a 40% less active catalyst, demonstrating the importance of the nitrogen/carbon source (unpublished work). The use of an iron precursor free of sulfur, FeAc, resulted in a catalyst with 15–25% lower activity (unpublished work). Recent results by Hermann et al. showing that sulfur addition improved the activity of a Co-porphyrin/Fe-oxalate-derived NPMC may be relevant.71
Yet another variable to consider is the final carbon structure. Notably, the hydrophilicity of the catalyst greatly increases after the heat-treatment at 1050 °C, suggesting that functionalities present on the surface were modified (e.g., edge plane exposure72). While micropores generated during heat-treatment have been shown to be important for NPMCs of type T2 (Section 2.4), this has not yet been verified for the CM-Fe-C catalysts (Rows 17–18 in Table 2) but porosimetry measurements are ongoing.
A more active catalyst prepared by the LANL group (Row 18 of Table 2, 50 A cm−3) was prepared in a similar manner as that in Row 17, except that (i) a different carbon support was chosen and (ii) the ratio of cyanamide to Fe- and C-precursors was increased. BP-2000 was selected for its higher total and microporous areas compared to KJ-300J. The increase in cyanamide content was likely needed to compensate for higher surface area. These two changes resulted in an increase in the ORR activity by a factor of 2 (Rows 17–18 in Table 2). When the cyanamide mass fraction was kept the same as that used in row 17, the activity still increased but only by 50%. Further improvements to CM-Fe-C catalysts should be possible since no parameter has been fully optimized.
2.6. NPMCs prepared from MeN4 macrocycles, with or without carbon support (type T1 - Rows 1–5 in Table 2)
Catalysts in Rows 1–2 were prepared by mixing Cl-FeTMPP with a non-microporous carbon black and heat-treating them in NH3. These catalysts were prepared at INRS in 2006 using the older method involving a temperature-ramp and 1-hour heat-treatment, a non-optimized duration.58 Achieved ORR activities were low, 0.1–0.2 A cm−3 (Table 2). Next, the catalyst in Row 3 was the most active of a series of catalysts prepared by loading Cl-FeTMPP (various loadings) on a non-microporous carbon black, and heat-treating in argon at 900 °C for 1h. The highest ORR activity (0.3 A cm−3) was obtained by loading 66 wt% Cl-FeTMPP (i.e. 4.5 wt% Fe) on the carbon support. Although this ORR activity is mediocre, this catalyst is interesting because it was stable in PEFC.59 The reason for its stability is discussed in Section 3.2.The remainder of this section details the synthesis of catalysts that do not involve a carbon support (Rows 4–5 in Table 2). Three salient features of the catalyst in Row 4 are (a) the absence of carbon support or template material, (b) utilization of iron oxalate, and (c) the use of sulfur. In the absence of a support, heat-treating an MeN4-molecule usually results in a low BET area of 200–300 m2g−1 and low activity (Row 3 in Table 2).59,73 This is due to the graphitization during the heat-treatment. In order to promote the formation of meso- and micropores, a foaming agent such as Fe oxalate can be added. The foaming agent technique was reported by Bogdanoff et al. in a study that involved mixing Co-tetramethoxy-phenylporphyrin (CoTMPP) with Fe oxalate microparticles.23 Oxalates other than the Fe oxalate were also shown to work as foaming agents.74
Certain inactive iron and cobalt compounds present in the catalyst cannot be removed by acid-leaching in 1 M HCl, but can be eliminated via addition of elemental sulfur to the catalyst precursor;73 the approach results in increased ORR activity.75 Complete removal of inactive Fe and Co species is also important for the fundamental study of the nature of active ORR sites with spectroscopic techniques.76 The synthesis of the catalyst in row 4 of Table 2 involved an additional heat-treatment in CO2, which enhanced the activity by a factor of ca. 5, due to the creation of new pores, to reach an ORR activity of 5.3 A cm−3.76
Finally, the catalyst in Row 5 was prepared by impregnating a silica template with CoTMPP dissolved in tetrahydrofuran, evaporating the solvent, heat-treating in N2, and finally removing the silica template with KOH. The corresponding ORR activity was 0.8 A cm−3 (Row 5).
2.7. Performance of NPMC-based cathodes reported in 2006–2010
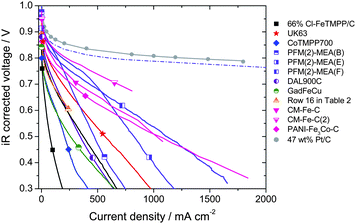
A clear distinction must be made between the ORR kinetic activity of a cathode and its performance. The ORR activity is the current obtained, per mass of catalyst or per volume of electrode, when the cathode is limited only by electro-catalytic processes. The ORR activity is therefore usually measurable only at low or medium current densities (high voltage). In contrast, the cathode performance refers to the entire polarization curve of the cathode (iR-corrected), which, at high current density, is set not only by electro-catalytic processes but also by transport processes of ORR-related species (H+, e−, O2, H2O). In the absence of transport limitations, a cathode would only show a ca. 70 mV/decade performance loss (cf. grey circles, Fig. 3). In reality, E-log I plots almost always show deviation from Tafel behavior at high current density (Fig. 3). An ensuing difference between the activity and performance is evidenced by the dotted line in Fig. 3 that reflects kinetic behavior (activity) of a cathode labeled PFM(2)-MEA(B). The dotted line is the extrapolation of the Tafel slope (kinetic behavior), observable only at low current density, to higher current densities.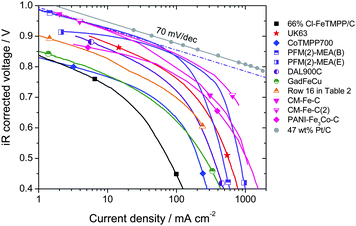 | ||
| Fig. 3 Tafel plots of the PEFC iR-corrected polarization curves with O2/H2. PEFC at 80 °C, gases at 100% RH, Pt/C at anode. Membranes, geometric area, backpressures and NPMC cathode loadings vary (Table 3). The dashed line represents an extrapolation to high current densities of the kinetic behavior observed at low current density for the cathode PFM(2)-MEA(B), assuming a 70 mV-per-decade Tafel slope. | ||
The ultimate goal of NMPC-research is to develop cathodes capable of delivering at any fuel cell voltage the same current density as that delivered by state-of-art Pt-based cathodes. The grey curve in Fig. 3 represents the performance of an approximately 10 μm-thick PEFC cathode and containing 0.4 mg cm−2 of platinum (as well as 0.45 mg cm−2 of carbon). In an attempt to reach this performance, it is acceptable for NPMC-based cathodes to use higher loadings due to their lower cost. At present, a typical NPMC cathode loading ranges from 1 to 6 mg cm−2 (Table 3), which corresponds to a cathode thickness of 25–150 μm. Such high NPMC loadings are still required because the volumetric activity for ORR of NPMCs is up to now, at best, ca. 1/13th that of a 47 wt% Pt/C catalyst (Table 2, Rows 13 & 21).
| Catalyst label | Corresponding Row in Table 2 | Loading/mg cm−2 | Geometric area/cm2 | Membrane | Nafion-to-catalyst ratio | Anode and cathode gauge pressures/bar | HFR/ohm cm2 |
|---|---|---|---|---|---|---|---|
| a Measurements were done close to sea level except for CM-Fe-C, CM-Fe-C(2) and PANI-Fe3Co-C (measurement at 2,200 m altitude; i.e. atmospheric pressure 0.76 atm). b Value estimated from membrane thickness. c Cathode has two layers with different Nafion content, one painted on the membrane, and the other on the GDL. | |||||||
| 66%Cl-FeTMPP/C | 3 | 2.0 | 1.14 | N117 | 1.0 | 1.0/1.0 | 0.20 |
| UK63 | 4 | 4.0 | 1.14 | N117 | 2.0 | 1.0/1.0 | 0.18 |
| CoTMPP700 | 5 | 1.0 | 1.14 | N117 | 2.0 | 1.0/1.0 | 0.24 |
| PFM(2)-MEA(B) | 13 | 5.3 | 1.14 | N117 | 1.5 | 1.0/1.0 | 0.23 |
| PFM(2)-MEA(E) | 13 | 4.2 | 1.14 | NRE-211 | 1.5 | 1.0/1.0 | 0.21 |
| PFM(2)-MEA(F) | 13 | 3.5 | 5.0 | N117 | 1.5 | 1.0/1.0 | 0.19 |
| DAL900C | 14 | 4.0 | 1.14 | N117 | 1.0 | 1.0/1.0 | 0.27 |
| GadFeCu | 15 | 1.0 | 1.14 | N117 | 2.0 | 1.0/1.0 | 0.20 |
| 16 | 1.0 | 50 | 2 × 35 μm | ? | 2.0/3.4 | 0.10b | |
| CM-Fe-C | 17 | 6.0 | 5.0 | N117 | 0.6 | 2.1/2.1 | 0.16 |
| CM-Fe-C(2) | 18 | 3.2 | 5.0 | N117 | 0.6 | 2.1/2.1 | 0.18 |
| PANI-Fe3Co-C | — | 4.0 | 5.0 | N1135 | 0.7/0.5c | 2.1/2.1 | 0.08 |
| 47 wt% Pt/C | 21 | 0.4 (Pt) | 50 | 25 μm | ? | 0.5/0.5 | ? |
Fuel cell polarization plots with NPMC cathodes synthesized in seven different laboratories are shown in Fig. 3. All polarization plots in Fig. 3 were recorded at 80 °C using fully-humidified H2 and O2. The main differences in the experimental conditions were (Table 3): cathode loading, geometric area of the membrane-electrode assembly, type of membrane used, and cathode and anode operating pressures. As stated earlier, eqn (2)–(3) can be used to normalize activities measured at different H2 and O2 and pressures to U.S. DOE reference conditions (Table 2) for as long as the cathode performance is under kinetic control. However, such normalization is not possible for the cathode under mixed kinetic and mass- or charge-transport control (at high current densities). Therefore, the data in Fig. 3 are as-measured, uncorrected for the differences in gas pressures. In practice, the plots in Fig. 3 can be viewed as cathode polarization curves since the anode overpotential can be neglected when pure H2 and anode Pt loadings greater than 0.2 mg cm−2 are used.
At voltages higher than 0.85 V, recent NPMC-based cathodes, with a loading of 3–6 mgNPMC cm−2 (Ref. 54 and: H. Chung, C. M. Johnston, and P. Zelenay, in preparation) virtually match the state-of-the-art fuel cell polarization plots obtained with 0.4 mgPt cm−2. These NPMCs come close to meeting U.S. DOE's 2010 target of 1/10th the volumetric activity of state-of-the art Pt catalysts (Table 2). Recently, a polymer-derived catalyst, obtained by heat treating in inert atmosphere Fe-phthalocyanine and a phenolic resin, showed a performance close to the best ones reported in Fig.3–4.77 However, fuel cell polarization plots obtained with NPMC-based cathodes enter a regime of mixed-control by kinetics and transport at a relatively low current density of 0.1 A cm−2 (Fig. 3). By comparison, the reference Pt/C-based cathode remains under kinetic control up to 2 A cm−2 (grey circles in Fig. 3). Susceptibility of NMPC-cathodes to transport limitations can be linked to (i) their greater thickness (e.g. those based on PFM(2), CM-Fe-C and CM-Fe-C(2) in Fig. 3 are 6–12 times thicker than the reference Pt-based cathode) or to (ii) local mass- and/or charge-transport within the micropores, which are believed to host the active sites of NPMCs.53,60,66
 | ||
| Fig. 4 PEFC iR-corrected polarization curves of the data presented in Fig. 3 but with the x-axis as a linear scale. The meaning of the dashed line is explained in Fig. 3 caption. | ||
Fig. 4 is equivalent to Fig. 3 except for the use of a linear x-axis scale, chosen to emphasize the performance at high current density, a regime that is important for generating high power output from a fuel cell. However, generating high current densities at very low voltages is of limited utility because the efficiency is inherently low. The efficiency of converting the chemical energy (H2) into electrical energy is proportional to the fuel cell voltage. For example, a voltage of 0.59 V corresponds to ca. 50% efficiency. Under these conditions, ca. half of the chemical energy is lost as heat. While some NPMC-based cathodes show ORR-kinetics similar to that of Pt-based cathodes (Fig. 3, cell voltages greater than 0.80 V) their performance at e.g. 1 A cm−2 is much lower (Fig. 4). This is the reason why a clear distinction must be made between activity and performance.
In order for NPMC-based cathodes to reach 1 A cm−2 at the same voltage as a Pt-based cathode, both the ORR activity of NPMC-based cathodes and the transport of ORR-relevant species within the latter must be high. While today's thick NPMC-based cathodes meet the activity target, they fail to allow fast transport of ORR-relevant species. The transport problem can be tackled in two ways: (i) further increase in the NPMC activity (U.S. DOE's 2015 target of 300 A cm−3) ultimately resulting in thinner cathodes, and/or (ii) improvements in transport characteristics of the NPMC-based cathodes.
The latter goal can be achieved through innovative electrode design and fabrication, better control of the micropore volume and orientation, and the use of new proton-conducting materials specifically developed for such catalysts. In the present review, a modeling approach is used to define target values for the electronic and protonic conductivities within the porous cathode, which would result in charge-transport efficient cathode up to a thickness of 100 μm (Section 4).
3. Durability of NPMCs in PEFC
Besides the requirement for high activity, NPMCs must also demonstrate sufficient durability to be used in a practical device. This has been a major challenge for NPMCs for which high activity is often coupled with a fast degradation rate.78 Until 2005, active NPMCs (HT-FeTMPP79 or HT-CoTPP80) typically showed a performance loss in fuel cells of no less than 40% after 100 h operation at 0.40–0.50 V.78 In 2006, an NPMC having both reasonably high activity and good durability in H2/air PEFC tests was reported, generating (at 2.7 bar) 0.13 A cm−2 at 0.40 V, and 0.2 A cm−2 at 0.50 V, for over 100 h without any decrease in current density.48 Testing at 0.70 V without performance loss was also possible for over 100 h, but the current density was low.48 The key difference between the preparation of this catalyst and all others discussed in this review was the absence of any heat-treatment in its synthesis.Although much remains to be investigated in the area of polymer-based, non-heat-treated catalysts, attempts to follow a similar synthesis path as described in Ref. 48 with different metal precursors, polymer types, or support types did not result in sufficiently improved ORR activity.78,80,81 Thus, a new strategy combining polymer precursors and heat-treatment was pursued starting in 2008.78 The guiding concept was that the more ordered chemical structure of the polymer, as compared to small molecule precursors, could possibly template the formation of a more ordered and thus more stable carbon-based active layer during heat-treatment. Polypyrrole (PPy) was used initially, but it was soon discovered that polyaniline (PANI) derived catalysts (PANI-Me-C) were more active and durable.51,55 Indeed, the PANI-Fe3Co-C-catalyst has shown good performance and durability for up to 700 h at 0.40 V (Fig. 5).
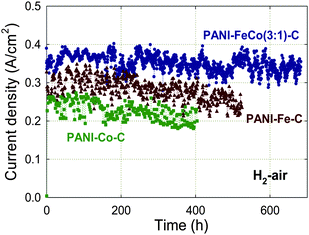 | ||
| Fig. 5 Effect of metals used in the synthesis of PANI-derived non-precious catalysts on fuel cell performance at 0.40 V for 400–600 h. PEFC at 80 °C, H2 and air at 100% RH with a backpressure of 2.1 bar (absolute pressure 2.8 bar), Pt/C with 0.25 mgPt cm−2 at anode, catalyst loading 4 mg cm−2 at cathode, Nafion membrane 1135. The catalysts' synthesis is described in Section 3. Reproduced with permission from ECS Trans.25, 1299 (2009). Copyright 2009, The Electrochemical Society. | ||
The synthesis of PANI-Me-C catalysts involves several steps, beginning with mixing aniline and the metal salt of choice in 0.5 M HCl (procedure slightly changed from Ref. 51). The oxidative polymerization of aniline by ammonium persulfate, (NH4)2S2O2, follows, which is notable also because the oxidant contains sulfur (see Sections 2.5–2.6 regarding role of sulfur). During this step, a carbon support is introduced, usually KJ-300J. After the reaction is finished, the resulting powder is heat-treated in N2, acid-leached in 0.5 M H2SO4 (also sulfur-containing), and heat-treated again in N2, at the same temperature as used for the first heat-treatment, to result in the final catalyst. The second heat-treatment results in a 15–30 mV positive shift in the half-wave potential measured in RDE. Whether the improvements in activity obtained so far by changes in heat-treatment conditions or precursors relate to micropore generation (section 2.4) is currently under study.
When analyzing and discussing durability data it must be kept in mind that several factors may play a role. Note that while some PANI-Me-C catalysts show a good combination of activity and durability, other PANI-Me-C catalysts are less durable. Also, all PANI-Me-C catalysts are less durable when operated at 0.60 V versus 0.40 V (Section 3.3). Whether the active site structure undergoes chemical changes (e.g., irreversible oxidation) during operation is currently under investigation. Spectroscopic studies detailing the fate of detectable Fe- and nitrogen-species will be forthcoming. The stability of NPMCs has been frequently correlated with the amount of peroxide they release during the ORR.14 Such a trend is also observed for PANI-Me-C catalysts, although it is not perfectly obeyed (see below). Peroxide can either directly oxidize active sites, or indirectly attack them by first reacting with Fe to generate HO· radicals, a much more oxidizing species.14 The stability of the carbon layer formed by heat-treating PANI, a polymer that already contains benzene moieties, could also increase the stability of active sites embedded within either graphene planes or pores. If the average coordination number of carbon atoms near active sites is increased, implying a higher degree of graphitization, then active sites may be more stable and durable. Less defective carbon is also likely to form only a small amount of hydrophilic groups. The latter may induce local flooding of the active sites. The puzzle is further complicated by the presence of multiple types of carbon at the end of the synthesis, as observed by TEM: polymer-derived carbon, modified carbon support, and, in certain cases, large graphene-like structures.82 A number of insights regarding catalyst stability, however, have been gained already from changing the metal precursor and the carbon support type, as discussed in Sections 3.1–3.2.
3.1 Effect of the transition metal
The metal precursor used to prepare PANI-derived catalysts was varied to generate PANI-Fe-C, PANI-Co-C, and PANI-Fe3Co-C;82 the durability tests of these catalysts are shown in Fig. 5. The best-performing catalyst of this series, PANI-Fe3Co-C, loses only 3–8% of performance during ca. 650 h. The PANI-Fe-C catalyst shows a performance drop of 13–14% after 500 h, while the PANI-Co-C catalyst is the least durable, losing 18–19% performance after 400 h. The lower durability in PEFC tests of PANI-Co-C vs. PANI-Fe-C is in line with RRDE results showing a higher H2O2 generation for PANI-Co-C (5% at 0.40 V). As for the higher durability of PANI-Fe3Co-C compared to PANI-Fe-C, there is no complete explanation yet. RRDE tests showed a slightly higher level of peroxide generation for PANI-Fe3Co-C compared to PANI-Fe-C, but the value was still less than 1%. SEM and TEM data show a higher occurrence of large graphene-like structures in PANI-Fe3Co-C than in PANI-Fe-C,82 which could prevent active site flooding or increase the resistance of active sites to oxidative attack (see discussion above). The addition of Co may also increase the number of peroxide electro-reduction/chemical decomposition sites, such as Co oxides.83 Further microscopy and spectroscopy studies are required to discriminate between these possibilities.3.2 Effect of carbon support
Next, the carbon support was found to influence the durability of the catalysts to an even greater degree than the choice of metal. Fuel cell durability tests obtained with PANI-Fe catalysts created on various carbon blacks (Vulcan XC-72, KJ-300J and BP-2000) and multi-walled carbon nanotubes (MWNTs) are given in Fig. 6.55 The MWNT-based catalyst shows virtually no performance degradation for more than 500 h at a constant cell voltage of 0.40 V. This represents an improvement over all three carbon black-based catalysts, including the KJ-300J used in Fig. 5. Interestingly, the amount of peroxide detected by RRDE is higher for the MWNT-based catalyst (2% at 0.40 V) than for the Ketjenblack-based catalyst (<1%). The XC-72-supported catalyst, which is durable but has lower activity, generates an unexpectedly high level of H2O2 of 3% at 0.40 V. On the other hand, the BP-2000-based catalyst is the least durable and generates significantly more peroxide than any other catalyst (6%).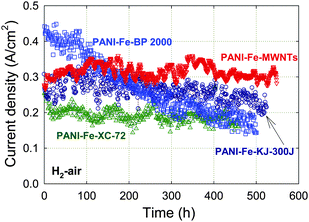 | ||
| Fig. 6 Fuel cell performance of PANI-Fe catalysts obtained using MWNTs or several carbon blacks as supports at 0.40 V for over 400 h. PEFC at 80 °C, H2 and air at 100% RH with a backpressure of 2.1 bar (absolute pressure 2.8 bar), Pt/C with 0.25 mgPt cm−2 at anode, catalyst loading 4 mg cm−2 at cathode, Nafion membrane 1135. The catalysts' synthesis is described in Section 3. Reproduced with permission from ECS Trans.25, 1299 (2009). Copyright 2009, The Electrochemical Society. | ||
The somewhat mixed results concerning the correlation, or lack of, between low H2O2 yield and durability suggest that other factors may contribute to the stability, as discussed above. A point to consider is the higher degree of graphitization of MWNTs compared to other supports, which may template a more ordered catalyst from the PANI decomposition during heat-treatment. If true, then the result could be improved stability of the ORR active site(s) as well as less local flooding because fewer hydrophilic groups are present. The effect of catalyst morphology on water transport in electrode layers and on H2O2 reduction and/or decomposition may also affect the catalyst stability during operation. It is notable that the durability of the PANI-Fe3Co-C catalyst was exceeded by the MWNT-based PANI-Fe catalyst without the addition of Co, although its activity was slightly lower. Graphene-like sheets similar to those seen in PANI-Fe3Co-C were also observed in PANI-Fe-MWNT by TEM, which provides an important clue for future work.
A correlation between the graphitic nature of less active NPMCs and their stability in PEFC has indeed been reported recently.59 A series of NPMCs was obtained by wet-impregnating various amounts of Cl-FeTMPP onto a non-microporous carbon black (BET area 74 m2g−1) and heat-treating the resulting catalyst precursors at 950 °C for 10 min in argon. The stability in PEFC increased with increased wt % of Cl-FeTMPP onto the carbon support. For PEFC tests of ca. 20 h, stability was observed when the wt% of Cl-FeTMPP impregnated on the carbon was equal to, or greater than, 66%. The evolution of the graphitic structure with wt% Cl-FeTMPP was investigated with XRD (Fig. 6 in Ref. 59). With increased wt% Cl-FeTMPP, the average size of graphitic platelets in resulting catalysts increased. Moreover, the loss in kinetic current observed over 20 h in PEFC decreased with increased average height of these graphitic platelets. Therefore, the stability of this type of NPMC could be associated with its highly graphitic structure. This structure emerged from the organization during the heat-treatment in argon of the carbon atoms initially present in FeTMPP molecules. Surprisingly, stable catalysts in this series released a large amount of H2O2 during the ORR (25–35%), as measured by RRDE. In contrast, a more active but instable catalyst was obtained if Cl-FeTMPP was impregnated on the carbon and heat-treated in NH3, instead of argon. The resulting catalyst was poorly graphitic (as the carbon support) and released only 5% H2O2, yet was unstable. While the PEFC performance obtained with the stable catalyst resulting from the heat-treatment in argon of 66 wt% Cl-FeTMPP/C was too low to be of practical interest (see Fig.3–4 and row 3 in Table 2), the extent of graphitization of NPMCs seems to be an important parameter for their stability and durability.
3.3 Effect of the operating cell voltage
Variations in the fuel cell voltage (cathode potential) are likely to affect several key properties of the cathode catalyst, including the oxidation state of the active species (Fe-based and/or carbon-based) as well as the hydrophilicity/hydrophobicity of the carbon in the catalytic layer.84 Cathode performance was tested at LANL at different operating fuel cell voltages, including the open cell voltage (OCV), corresponding to the highest potential experienced by the cathode catalysts under regular fuel cell operation.A 100-hour recording of OCV for an H2-air PEFC operating with PANI-Fe3Co-C at the cathode is shown in Fig. 7. The OCV value remains unchanged at 0.90 V in air, indicating good stability of the catalyst. It should be noted that, unlike the durability test at 0.40 V where stable performance is maintained (Fig. 5), about 50% performance loss was observed when testing at 0.60 V for 100 h. Approximately 90% of the loss could be recovered, however, after flowing dry gases in the fuel cell for several hours. This observation implies that flooding of active sites or of the electrode was responsible for much of the performance loss in this instance. The existence of such a recoverable performance decay has also been reported by Popov et al.85 Future work will need to address the tolerance to water of the catalytic layer, and maybe even of catalytic particles themselves. Because the PEFC efficiency is proportionally dependent on the operating voltage (see Section 2.7), the performance durability at high voltages (greater than 0.60 V) now becomes the biggest challenge facing the use of NPMCs in a fuel cell to power cars.84
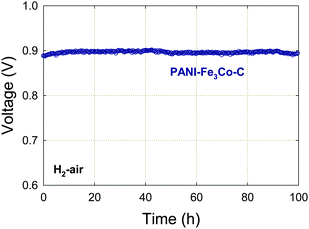 | ||
| Fig. 7 Long-term stability test of a PANI-Fe3Co-C catalyst at OCV. PEFC at 80 °C, H2 and air at 100% RH with a backpressure of 2.1 bar, Pt/C with 0.25 mgPt cm−2 at anode, catalyst loading 4 mg cm−2 at cathode, Nafion membrane 1135. The catalyst synthesis is described in Section 3. Reproduced with permission from ECS Trans.25, 1299 (2009). Copyright 2009, The Electrochemical Society. | ||
3.4 Effect of a heat-treatment in NH3
While it was shown in section 2.4 how a heat-treatment in NH3 results in active NPMCs, these NMCs have hitherto not shown good stability. Typically, the performance loss observed for such catalysts (Rows 12–13 in Table 2) at 0.4–0.5 V is 40–50% over 100 h.54Recently, the INRS group investigated a series of catalysts made from the impregnation of 66 wt % Cl-FeTMPP on a non microporous carbon black, and heat-treating this catalyst precursor in pure Ar, or in a mixture of Ar and NH3.86 The percentage of NH3 in Ar was varied from 1.3 to 100%, in volume. The activity increased drastically from 0 to 1.3 vol. % NH3, but at the same time the catalyst behavior changed from stable to slightly unstable. Another interesting aspect is that the catalysts prepared using 1.3–10 vol. % NH3 displayed interesting activity at high voltage but had poor mass transport properties. Better performing catalysts were obtained when the volumetric content of NH3 was larger than 20%. However, the catalysts became simultaneously much more unstable (70% loss of activity at high voltage over 4h). It shows that the beneficial effect of a heat-treatment in NH3 usually results in unstable catalysts, either because the micropores formed under NH3 are prone to flooding or because activity-enhancing N-functionalities formed on the carbon surface during the heat-treatment in NH3 undergo undesired chemical changes in the acidic medium. Recently, Popov and coworkers have proposed that protonation of pyridinic nitrogen atoms explains the deactivation of their catalysts in acidic medium.87,88
Micropores, generally speaking, do not necessarily lead to unstable catalysts since micropores are also created during the fabrication of the comparatively durable PANI-Me-C catalysts with an overall positive correlation with activity.89 The nitrogen content of the PANI-derived catalysts is relatively high (4 at% by XPS) but it is necessarily more homogeneously distributed because it derives from the polymer rather than from the reaction between NH3 and disordered carbon. Thus, the micropores created in PANI-derived catalysts without NH3 gas could have a locally lower nitrogen content, resulting in less hydrophilic properties and hence less likelihood to flooding.90
Alternatively, the reaction of NH3 with disordered carbon may generate inherently less stable carbon-nitrogen structures. Such hypotheses are difficult to verify given the heterogeneous nature of the samples and the small length scales that must be probed. Further studies are required to draw conclusions.
4. Modeling of charge transport in porous cathode
NPMCs do not necessarily need to reach the same volumetric activity as that of state-of-art 40–50 wt% Pt on carbon. In fact, one-tenth to one-third of the latter activity is acceptable (U.S. DOE targets of 2010 and 2015). The remaining difference can be offset by using higher loadings, i.e. thicker cathodes, for NPMCs than for Pt/C. This is economically acceptable due to the low cost of NPMCs. The object of this section is (i) to estimate the voltage loss due to transport of electrons and protons within a 100-μm thick cathode and (ii) to estimate the desired values of electronic and protonic conductivities within a cathode in order to expect small voltage losses due to these charge transports within a 100-μm thick electrode.The steady-state model described in this section is the same as the one presented previously,91 except that here not only the effect of limited proton conduction inside the porous cathode is taken into account, but also that of limited electron conduction. The full model including the latter effect has been presented in Ref. 92. For the present analysis, O2 diffusion through the cathode was assumed to be sufficiently fast. Calculations were carried out by assuming Tafel kinetics for the ORR and by using effective protonic (σeff,L) and electronic (σeff,S) conductivity values within the porous cathode. The electric potentials in the solid (ϕS) and electrolyte phases (ϕL) across the cathode were assumed to follow Ohm's law
| dϕS/dy = (j − jtot)/σeff,S | (4) |
| dϕL/dy = −j/σeff,L | (5) |
| dη/dy = j·(1/σeff,S + 1/σeff,L) − jtot/σeff,S | (6) |
Owing to charge balance, we obtain the second differential equation for the system of variables η and j, which relates the gradient of the proton flow to the rate of the electrochemical reaction per unit volume, iv (in A m−3, defined <0). The latter is related to the overpotential by the Tafel law (right handside of eqn (7)).
| dj/dy = iv = iv,0·exp (−αr·F·η/(RT)) | (7) |
The system of differential equations for η and j (eqn (6)–(7)) was solved numerically. Last, the total (or measurable) cathode potential, Etot, is obtained from Etot = ϕS(y = 0) − ϕL(y = L). The positions y = 0 and y = L correspond to the current collector and membrane sides, respectively (Fig.8A). Fig. 8A is an example of calculated electric potentials and local volumetric current of oxygen reduction across the cathode, as calculated for one set of parameters and one given current density jtot. The curves of Fig.8A correspond in fact to one single point (0.6V, 1.84 A cm−2) of curve (b) in Fig.8B. Fig. 8A shows that the cathode reaction is, on top of kinetic limitations (Tafel law), also limited by proton and electron conduction since both the solid phase and electrolyte phase potentials do change across the cathode. The stronger limitation by proton conduction (230 mV loss) drifts most of the reaction toward the membrane side. This however implies that a major part of the electron flow entering the cathode must be conducted across the whole cathode thickness before they can react with O2 and protons, resulting in an additional 80 mV loss.
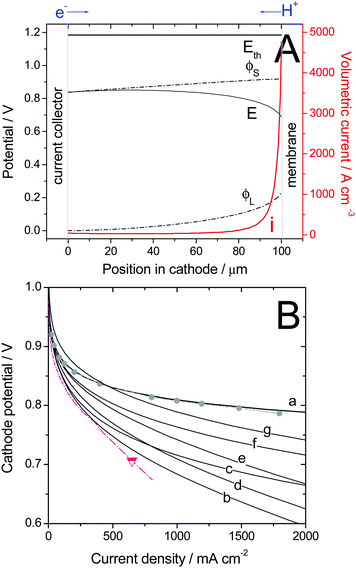 | ||
| Fig. 8 Calculated polarization curves for a 100-μm thick cathode based on a hypothetical NPMC having the activity defined by U.S. DOE's 2010 activity target (curves a–f) or 2015 activity target (curve g) and with a Tafel slope of 70 mV/dec. Calculations were performed under the following assumptions: 100% RH, 80 °C, 1 bar O2 and H2 (i.e. 1.5 bar total gas pressure including 0.5 bar of water vapour), and using various values of the electronic- and protonic effective conductivities in the cathode, σeff,S and σeff,L, respectively. (A) Example of profiles of electric potentials across a cathode at 0.6 V (1.84 A cm−2) for σeff,S = 20 S m−1 & σeff,L = 1 S m−1 (corresponding to highest current of curve (b) in (B)). The overall cathode potential used for the polarization curve is Etot = ϕS(0 μm) − ϕL(100 μm) = 0.6V. (B) Polarization curves computed for different sets of conductivities. (a) σeff,S & σeff,L infinitely high (b) σeff,S = 20 S m−1 & σeff,L = 1 S m−1 (c) σeff,S = 100 S m−1 & σeff,L = 1 S m−1 (d) σeff,S = 20 S m−1 & σeff,L = 2 S m−1 (e) σeff,S = 20 S m−1 & σeff,L = 5 S m−1 (f–g) σeff,S = 100 S m−1 & σeff,L = 5 S m−1. Experimental data: Cathode with 0.4 mgPt cm−2 using 47 wt% Pt/C (grey circles), ca. 10 μm-thick cathode, or with 3.2 mg cm−2 of CM-Fe-C(2) (triangle), ca. 80 μm-thick cathode. | ||
Fig. 8B shows complete polarization curves. The choice of the parameter sets is now described. The values for σeff,L assumed for the model range between 1 and 5 S m−1 and those for σeff,S range between 20 and 100 S m−1.
Typical experimental values for the effective protonic conductivity, σeff,L, at 80 °C and 75–100% relative humidity are 1–2 S m−1 (equivalent to a resistivity of 50–100 Ω cm).93–95 This effective protonic conductivity inside the composite cathode (carbon, polymer, pores) is about 1/10th the value of the bulk protonic conductivity in Nafion membranes, which is 10–17 S m−1. This difference is due to (i) the composite nature of a PEFC cathode, whose volume typically consists of about 50% pores, 25% catalyst (a volume of carbon, mostly) and only 25% Nafion ionomer and (ii) tortuosity and/or connectivity factors. The latter factors depend on the carbon substrate onto which the ionomer is deposited.95
Typical experimental values for the effective electronic conductivity in a PEFC cathode are 100–400 S m−1 as measured ex-situ on dry Pt/Vulcan-based porous electrodes.96 The swelling of Nafion ionomer contained in the cathode may significantly decrease the values of σeff,S under PEFC operation (high humidity). Also, the carbon microstructure of some NPMCs obtained from the pyrolysis of organic molecules may be inappropriate and may result in lower effective electronic conductivity in the corresponding cathode, compared to a Pt/carbon black-based cathode.
For the model, the ORR activity of the NPMC was first assumed to match the U.S. DOE's 2010 volumetric activity target of 130 A cm−3 at 0.80 V (iR-free). Several plots were generated for different values of effective electronic and protonic conductivities (Fig. 8B, Plots a–f). Plot g in Fig. 8B corresponds to an NPMC with an assumed volumetric activity of 300 A cm−3 (U.S. DOE's 2015 target). Fig. 8B also shows two experimental plots recorded with (i) a reference Pt/C-catalyst (circles) or (ii) with the best-performing NPMC in Fig. 3 (triangle). The volumetric activity of the Pt/C-catalyst is ca. 1300 A cm−3 at 0.80 V, assuming a ca. 10 μm-thick cathode.
Plot b, generated using σeff,S = 20 S m−1 and σeff,L = 1 S m−1, matches the experimental data of the best-performing NPMC-based cathode quite well. An increase in the value of effective electronic conductivity from 20 to 100 S m−1 has a positive effect on the electrode performance (Plot c). However, in order to attain the performance of the Pt/C-based cathode reference (circles), the assumed value of effective protonic conductivity must be increased significantly. Plot f (σeff,S = 100 S m−1 and σeff,L = 5 S m−1) is only 46 mV below the Pt reference curve at 1 A cm−2. Finally, Plot g was computed with the same electronic and protonic conductivities as those used to generate Plot f, but the assumed volumetric activity of the NPMC was 300 A cm−3. The NPMC-based cathode performance shown by Plot g is, at 400 mA cm−2, better than that of the Pt/C reference (circles) and, at 1 A cm−2, trails the reference by only 22 mV.
The modeling data described above predicts that, in order to perform well enough at high current density, a 100 μm-thick NPMC cathode needs to have effective electronic and protonic conductivities of ca. 100 S m−1 and 5 S m−1, respectively. These are challenging numbers, especially for the protonic conductivity as it represents 50% of the bulk protonic conductivity of a Nafion membrane at 80 °C. Cathodes with well-controlled architecture and a diminished tortuosity of protonic pathways would help in reaching such a value. Ionomers with higher bulk protonic conductivity than Nafion could also be of interest in that regard.
5. Conclusions
Metal/nitrogen/carbon (Me/N/C) catalysts obtained via the heat-treatment of Fe and/or Co precursors, nitrogen- and carbon precursors were reviewed with respect to the electrocatalysis of the oxygen reduction reaction. Catalytic activity, performance and durability of these catalysts in PEFC were discussed.Concerning the activity, the most active Me/N/C-catalysts reported to date were prepared using a metal salt, phenanthroline or NH3 gas or cyanamide, and a microporous carbon black as Fe-, N- and C-precursors, respectively. The use of planetary ball-milling for filling the micropores of the carbon support was shown to be crucial when using phenanthroline as N-precursor. A common factor is that, in order to obtain these very active Me/N/C-catalysts, at least one heat-treatment at a temperature above 600 °C was necessary. The heat-treatment was performed either in an inert atmosphere (nitrogen, argon) or in NH3. Further improvements in activity are still possible and expected since these new approaches in the synthesis of Me/N/C-catalysts are multi-parametric, including the choice of each of the three precursors, the weight ratio of each of them, the process used to mix them, the heat-treatment temperature and duration and gas used, the number of heat-treatments, etc. The U.S. DOE's target for 2010 for NPMCs has been almost reached (equivalent to 1/10th the volumetric activity of state-of-art Pt-based catalyst) but it is desirable to further increase the activity by a factor of 2–3 to reach the U.S. DOE's target for 2015.
From a performance standpoint, it was noted that even if cathodes based on today's most active Me/N/C-catalysts reach the activity (i.e. current at 0.90 V) of state-of-art Pt-based cathodes, their performance at lower potential (hence, higher current density) is still notably lower. This is partly due to the greater thickness of NPMC-based cathodes, i.e. usually 50–100 vs. 10 μm for Pt-based cathodes. This problem can be tackled in two ways: (i) further increase the volume-specific activity to make thinner NPMC-based cathodes and (ii) improve the transport characteristics of NPMC-based cathodes. The latter goal can be achieved through innovative electrode fabrication and the use of new proton-conducting materials. With the help of a model, the desired effective protonic conductivity for efficient use of a 100 μm-thick cathode was here predicted to be 5 S m−1, significantly higher than the present typical value of 1 S m−1. The effective electronic conductivity, not currently known for NPMC-based cathodes, should at the same time be 100 S m−1.
As for the durability, arguably the toughest challenge facing Me/N/C-catalysts today, progress has been made but combining high activity, high performance and good durability has yet to be achieved. Non-heat-treated catalysts based on cobalt and polypyrrole were shown in 2006 to have much improved durability but they showed low activity. However, great improvements in ORR activity while retaining durability were achieved by preparing heat-treated polymer-based catalysts. The best-performing catalysts to date in terms of both activity and durability are based on polyaniline (PANI), Fe and Co in a weight ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and either Ketjenblack or multiwall-nanotubes as a carbon support. Unlike activity, the fundamental characteristics determining the stability/durability of Me/N/C-catalysts are not yet clearly identified. Peroxide generation during the ORR and the graphitic character of the Me/N/C-catalysts appear to be two important factors. Other, yet unrevealed characteristics may be at play and more fundamental investigations are needed. Recent reports showing durability for 650 h with reasonably high catalytic activity provide an encouraging basis for future work.
:1, and either Ketjenblack or multiwall-nanotubes as a carbon support. Unlike activity, the fundamental characteristics determining the stability/durability of Me/N/C-catalysts are not yet clearly identified. Peroxide generation during the ORR and the graphitic character of the Me/N/C-catalysts appear to be two important factors. Other, yet unrevealed characteristics may be at play and more fundamental investigations are needed. Recent reports showing durability for 650 h with reasonably high catalytic activity provide an encouraging basis for future work.
Acknowledgements
Financial support of NSERC and General Motors of Canada for an industrial chair in electrocatalysts for PEFCs at INRS and financial support of the DOE-EERE Fuel Cell Technologies Program for non-precious metal catalysis research at Los Alamos National Laboratory are gratefully acknowledged.References
- International Energy Agency, Key World Energy Statistics, http://www.iea.org Search PubMed.
- M. Mathias, H. Gasteiger, R. Makharia, S. Kocha, T. Fuller, T. Xie and J. Pisco, ACS Division of Fuel Chemistry, Preprints, 2004, 49, 471 Search PubMed.
- M. F. Mathias, R. Makharia, H. A. Gasteiger, J. J. Conley, T. J. Fuller, C. J. Gittleman, S. S. Kocha, D. P. Miller, C. K. Mittelsteadt, T. Xie, S. G. Yan and P. T. Yu, The Electrochem. Soc. Interf., 2005, 14, 24 Search PubMed.
- U. Wagner, R. Eckl and P. Tzscheutschler, Energy, 2006, 31, 3062 CrossRef CAS.
- F. Villatico and F. Zuccari, Int. J. Hydrogen Energy, 2008, 33, 3235 CrossRef CAS.
- A. Baker, Opening Doors to Fuel Cell Commercialization ( 2005) http://www.fuelcelltoday.com/media/pdf/archive/Article_940_GM%20Sequel.pdf Search PubMed.
- R. F. Service, Science, 2009, 324, 1257 CrossRef CAS.
- J. Larminie, A. L. Dicks, Fuel Cell Systems Explained, 2nd Edition, John Wiley and Sons Inc., Hoboken, NJ, 2003 Search PubMed.
- H. A. Gasteiger, J. E. Panels and S. G. Yan, J. Power Sources, 2004, 127, 162 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9 CrossRef CAS.
- U.S. Department of Energy - Energy Efficiency and Renewable Energy, Multi-Year Research, Development and Demonstration Plan: Planned Program Activities for 2005–2015, http://www1.eere.energy.gov/hydrogenandfuelcells/mypp/pdfs/fuel_cells.pdf Search PubMed.
- R. G. Cawthorn, South African Journal of Science, 1999, 95, 481 Search PubMed.
- B. D. James, J. A. Kalinoski, in U.S. Department of Energy-Hydrogen Program, 2008 Annual Progress Report, http://www.hydrogen.energy.gov/pdfs/progress08/v_a_2_james.pdf Search PubMed.
- J. P. Dodelet, in N4-Macrocyclic Metal Complexes, ed. J. H. Zagal, F. Bedioui and J. P. Dodelet, Springer Science + Business Media Inc., New-York, 2006, p. 83 Search PubMed.
- R. Boulatov, in N4-Macrocyclic Metal Complexes, ed. H. Zagal, F. Bedioui and J. P. Dodelet, Springer Science + Business Media, New-York, 2006, p. 1 Search PubMed.
- R. Jasinski, Nature, 1964, 201, 1212 CAS.
- H. Jahnke, M. Schönborn and G. Zimmermann, Top. Curr. Chem., 1976, 61, 133 CAS.
- J. A. R. Van Veen and H. A. Colijn, Ber. Bunsenges. Phys. Chem., 1981, 85, 700 CAS.
- V. S. Bagotzky, M. R. Tarasevich, K. A. Radyushkina, O. E. Levina and S. I. Andrusyova, J. Power Sources, 1978, 2, 233 CrossRef.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356 CrossRef CAS.
- J. M. Ziegelbauer, T. S. Olson, S. Pylypenko, F. Alamgir, C. Jaye, P. Atanassov and S. Mukerjee, J. Phys. Chem. C, 2008, 112, 8839 CrossRef CAS.
- J. Maruyama, J. Okamura, K. Miyazaki, Y. Uchimoto and I. Abe, J. Phys. Chem. C, 2008, 112, 2784 CrossRef CAS.
- P. Bogdanoff, I. Herrmann, M. Hilgendorff, I. Dorbandt, S. Fiechter and H. Tributsch, J. New Mater. Electrochem. Systems, 2004, 7, 85.
- X. Li, C. Liu, W. Xing and T. Lu, J. Power Sources, 2009, 193, 470 CrossRef CAS.
- S. L. Gupta, D. Tryk, I. Bae, W. Aldred and E. B. Yeager, J. Appl. Electrochem., 1989, 19, 19 CrossRef CAS.
- H. Wang, R. Côté, G. Faubert, D. Guay and J. P. Dodelet, J. Phys. Chem. B, 1999, 103, 2042 CrossRef CAS.
- A. Van der Putten, A. Elzing, W. Visscher and E. Barendrecht, J. Electroanal. Chem., 1986, 205, 233 CrossRef CAS.
- J. A. R. Van Veen, H. A. Colijn and J. F. Van Baar, Electrochim. Acta, 1988, 33, 801 CrossRef CAS.
- K. Wiesener, Electrochim. Acta, 1986, 31, 1073 CrossRef CAS.
- M. R. Tarasevich and K. A. Radyushkina, Mater. Chem. Phys., 1989, 22, 477 CrossRef CAS.
- K. Wiesener, D. Ohms, V. Neumann and R. Franke, Mater. Chem. Phys., 1989, 22, 457 CrossRef CAS.
- B. Wang, J. Power Sources, 2005, 152, 1 CrossRef CAS.
- L. Zhang, J. J. Zhang, D. P. Wilkinson and H. J. Wang, J. Power Sources, 2006, 156, 171 CrossRef CAS.
- Y. Feng and N. Alonso-Vante, Phys. Status Solidi B, 2008, 245, 1792 CrossRef CAS.
- A. L. Bouwkamp-Wijnoltz, W. Visscher, J. A. R. van Veen, E. Boellaard, A. M. van der Kraan and S. C. Tang, J. Phys. Chem. B, 2002, 106, 12993 CrossRef.
- I. T. Bae, D. A. Tryk and D. A. Scherson, J. Phys. Chem. B, 1998, 102, 4114 CrossRef CAS.
- M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2000, 104, 11238 CrossRef CAS.
- M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705 CrossRef CAS.
- P. H. Matter, E. Wang, M. Arias, E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. B, 2006, 110, 18374 CrossRef CAS.
- P. H. Matter and U. S. Ozkan, Catal. Lett., 2006, 109, 115 CrossRef CAS.
- V. Nallathambi, J.-W. Lee, S. P. Kumaraguru, G. Wu and B. N. Popov, J. Power Sources, 2008, 183, 34 CrossRef CAS.
- R. Franke, D. Ohms and K. Wiesener, J. Electroanal. Chem., 1989, 260, 63 CrossRef CAS.
- F. Jaouen and J. P. Dodelet, Electrochim. Acta, 2007, 52, 5975 CrossRef CAS.
- N. P. Subramanian, X. Li, V. Nallathambi, S. P. Kumaraguru, H. Colon-Mercado, G. Wu, J.-W. Lee and B. N. Popov, J. Power Sources, 2009, 188, 38 CrossRef CAS.
- P. He, M. Lefèvre, G. Faubert and J. P. Dodelet, J. New Mater. Electrochem. Systems, 1999, 2, 243 CAS.
- G. Lalande, G. Tamizhmani, R. Côté, L. Dignard-Bailey, M. L. Trudeau, R. Shultz, D. Guay and J. P. Dodelet, J. Electrochem. Soc., 1995, 142, 1162 CrossRef CAS.
- F. Jaouen, S. Marcotte, J. P. Dodelet and G. Lindbergh, J. Phys. Chem. B, 2003, 107, 1376 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63 CrossRef CAS.
- R. Atanasoski, in U.S. Department of Energy–Hydrogen Program, 2007 Annual Progress Report. http://www.hydrogen.energy.gov/pdfs/progress07/v_e_1_atanasoski.pdf Search PubMed.
- T. E. Wood, Z. Tan, A. K. Schmoekel, D. O'Neill and R. Atanasoski, J. Power Sources, 2008, 178, 510 CrossRef CAS.
- G. Wu, Z. Chen, K. Artyushkova, F. H. Garzon and P. Zelenay, ECS Trans., 2008, 16, 159 Search PubMed.
- A. L. M. Reddy, N. Rajalakshmi and S. Ramaprabhu, Carbon, 2008, 46, 2 CAS.
- F. Jaouen, J. Herranz, M. Lefevre, J. P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. Olson, S. Pylypenko, P. Atanassov and E. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71 CrossRef CAS.
- G. Wu, K. Artyushkova, M. Ferrandon, A. J. Kropf, D. Myers and P. Zelenay, ECS Trans., 2009, 25, 1299 Search PubMed.
- K. C. Neyerlin, W. Gu, J. Jorne and H. A. Gasteiger, J. Electrochem. Soc., 2006, 153, A1955 CrossRef CAS.
- S. Ruggeri and J. P. Dodelet, J. Electrochem. Soc., 2007, 154, B761 CrossRef CAS.
- C. Médard, M. Lefèvre, F. Jaouen, J. P. Dodelet and G. Lindbergh, Electrochim. Acta, 2006, 51, 3202 CrossRef CAS.
- F. Charreteur, F. Jaouen and J. P. Dodelet, Electrochim. Acta, 2009, 54, 6622 CrossRef CAS.
- F. Jaouen, M. Lefèvre, J. P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553 CrossRef CAS.
- J. Herranz, M. Lefevre, N. Larouche, B. Stansfield and J. P. Dodelet, J. Phys. Chem. C, 2007, 111, 19033 CrossRef CAS.
- M. Lefevre and J. P. Dodelet, Electrochim. Acta, 2008, 53, 8269 CrossRef CAS.
- G. Faubert, R. Côté, J. P. Dodelet, M. Lefèvre and P. Bertrand, Electrochim. Acta, 1999, 44, 2589 CrossRef CAS.
- G. Faubert, G. Lalande, R. Côté, D. Guay, J. P. Dodelet, L. T. Weng, P. Bertrand and G. Dénès, Electrochim. Acta, 1996, 41, 1689 CrossRef CAS.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. H. Haynes, N. Pernicone, D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739 CrossRef CAS.
- F. Charreteur, F. Jaouen, S. Ruggeri and J. P. Dodelet, Electrochim. Acta, 2008, 53, 2925 CrossRef CAS.
- F. Jaouen and J. P. Dodelet, J. Phys. Chem. C, 2007, 111, 5963 CrossRef CAS.
- F. Jaouen, A. M. Serventi, M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. C, 2007, 111, 5971 CrossRef CAS.
- H. T. Chung, C. M. Johnston and P. Zelenay, ECS Trans., 2009, 25, 485 Search PubMed.
- H. T. Chung, C. M. Johnston, F. H. Garzon and P. Zelenay, ECS Trans., 2008, 16, 385 Search PubMed.
- I. Herrmann, U. I. Kramm, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2009, 156, B1283 CrossRef CAS.
- P. H. Matter, E. Wang, M. Arias, E. J. Biddinger and U. S. Ozkan, J. Mol. Catal. A: Chem., 2007, 264, 73 CrossRef CAS.
- I. Herrmann, P. Bogdanoff, G. Schmithals and S. Fiechter, ECS Trans., 2006, 3, 211 Search PubMed.
- I. Herrmann, U. I. Kramm, S. Fiechter and P. Bogdanoff, Electrochim. Acta, 2009, 54, 4275 CrossRef CAS.
- I. Herrmann, U. I. Koslowski, J. Radnik, S. Fiechter and P. Bogdanoff, ECS Trans., 2008, 13, 143 Search PubMed.
- U. I. Koslowski, I. Herrmann, P. Bogdanoff, C. Barkschat, S. Fiechter, N. Iwata, H. Takahashi and H. Nishikori, ECS Trans., 2008, 13, 125 Search PubMed.
- Y. Nabae, S. Moriya, K. Matsubayashi, S. M. Lyth, M. Malon, L. Wu, N. M. Islam, Y. Koshigoe, S. Kuroki, M. Kakimoto, S. Miyata and J. Ozaki, Carbon, 2010, 48, 2613 CrossRef CAS.
- C. M. Johnston, P. Piela, P. Zelenay, in Handbook of fuel cells: Advances in Electrocatalysis, Materials, Diagnostics and Durability, ed. W. Vielstich, H. Yokokawa, and H. A. Gasteiger, Vol. 5–6, John Wiley & Sons, 2009 Search PubMed.
- M. Lefevre and J. P. Dodelet, Electrochim. Acta, 2003, 48, 2749 CrossRef CAS.
- P. Zelenay, R. Bashyam, E. Brosha, J.-H. Choi, S. Conradson, F. Garzon, C. Johnston, R. Mukundan and J. Ramsey, Non-Platinum Cathode Catalysts, US Department of Energy, Hydrogen, Fuel Cells & Infrastructure Technologies Program; 2006 Annual Progress Report; http://www.hydrogen.energy.gov/pdfs/progress06/v_c_7_zelenay.pdf, 2006 Search PubMed.
- Z. Chen, C. M. Johnston, Y. Yan, P. Zelenay, 213th Meeting of the Electrochem. Soc., vol. 801, The Electrochem. Soc., Phoenix, Arizona, 2008 Search PubMed.
- G. Wu, K. Moore, P. Zelenay, to be submitted in Sept. 2010.
- T. S. Olson, S. Pylypenko, J. E. Fulghum and P. Atanassov, J. Electrochem. Soc., 2010, 157, B54 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. A. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904 CrossRef CAS.
- B. N. Popov, X. Li, G. Liu, J.-W. Lee, Int. J. Hydrogen Energy, in press Search PubMed.
- H. Meng, N. Larouche, M. Lefevre, F. Jaouen, B. Stansfield and J. P. Dodelet, Electrochim. Acta, 2010, 55, 6450 CrossRef CAS.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Electrochim. Acta, 2010, 55, 2853 CrossRef CAS.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Appl. Catal., B, 2009, 93, 156 CrossRef CAS.
- C. M. Johnston, G. Wu, K. Artyushkova, M. Ferrandon, K. L. More, U. I. Kramm, A. J. Kropf, E. Brosha, P. Bogdanoff, D. J. Myers, P. Zelenay, to be submitted in Sept. 2010.
- C. Chen, B. Liang, D. Lu, A. Ogino, X. Wang and M. Nagatsu, Carbon, 2010, 48, 939 CrossRef CAS.
- F. Jaouen and G. Lindbergh, J. Electrochem. Soc., 2002, 149, A437 CrossRef CAS.
- F. Jaouen, Ph.D. thesis, Royal Institute of Technology, Stockholm, Sweden, 2003, http://kth.diva-portal.org/smash/record.jsf%3FsearchId%3D1%26pid%3Ddiva2%3A9303.
- C. Boyer, S. Gamburzev, O. Velev, S. Srinivasan and A. J. Appleby, Electrochim. Acta, 1998, 43, 3703 CrossRef CAS.
- Y. Liu, C. Ji, D. R. Baker, W. Gu, J. Jorne and H. A. Gasteiger, ECS Trans., 2008, 16, 1775 Search PubMed.
- H. Iden, A. Ohma and K. Shinohara, J. Electrochem. Soc., 2009, 156, B1078 CrossRef CAS.
- P. Gode, F. Jaouen, G. Lindbergh, A. Lundblad and G. Sundholm, Electrochim. Acta, 2003, 48, 4175 CrossRef CAS.
- S. C. Davis, S. W. Diegel, R. G. Boundy, in Transportation Energy Data Book-Edition 28, Appendix B, Table B4, http://cta.ornl.gov/data Search PubMed.
- S. Lasher, C. McKenney, J. Sinha, P. Chin, Analysis of Hydrogen Storage Materials and On-board Systems, in U.S. Department of Energy-Hydrogen Program, 2009 Annual Merit Review, http://www.hydrogen.energy.gov/pdfs/review09/st_12_lasher.pdf Search PubMed.
- D. Linden, in Handbook of Batteries, ed. D. Linden and T. B. Reddy, McGraw-Hill Inc., New York, 2002, p. 12 Search PubMed.
- Dortmund Data Bank Software and Separation Technology, DDBST GmbH, http://www.ddbst.com/en/online/Online_Calc_den_Form.php.
- S. C. Davis, S. W. Diegel, R. G. Boundy, in Transportation Energy Data Book-Edition 28, Table 2.12, http://cta.ornl.gov/data Search PubMed.
Footnote |
| † Primary energy is the energy found in nature that has not been subjected to any conversion or transformation process, or the chemical energy contained in raw fuels. The scalar of 140,000 TWh of primary energy corresponds to 45,000 TWh of useful energy given in Schiermeier et al., Nature, 2008, 454, 816 (meaning that the average conversion efficiency from primary to useful energy is, today, 32%). |
| This journal is © The Royal Society of Chemistry 2011 |