CO2 capture by solid adsorbents and their applications: current status and new trends
Qiang
Wang
,
Jizhong
Luo
,
Ziyi
Zhong
* and
Armando
Borgna
Institute of Chemical and Engineering Sciences, Agency for Science, Technology and Research in Singapore (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833. E-mail: Zhong_ziyi@ices.a-star.edu.sg; Fax: +65-83166182; Tel: +65-67963809
First published on 11th October 2010
Abstract
In the last few years there has been a rapid growth in governmental funding and research activities worldwide for CO2 capture, storage and utilization (CSU), due to increasing awareness of the link between CO2 accumulation in the atmosphere and global warming. Among the various technologies and processes that have been developed and are emerging for CSU of CO2, solid CO2-adsorbents are widely applied. In this review, these solid CO2-adsorbents are classified into three types according to their sorption/desorption temperatures: low-, intermediate- and high-temperature adsorbents with temperatures ranging from below 200 °C, between 200–400 °C and above 400 °C, respectively. For each type of solid CO2-adsorbent, the synthesis, interaction mechanism with CO2 and sorption performance, potential applications and problems are reviewed. In the last section, several representative CO2-sorption-enhanced catalytic reactions are discussed. It is expected that this review will not only summarize the main research activities in this area, but also find possible links between fundamental studies and industrial applications.
 Qiang Wang | Qiang Wang is a Research Fellow in the Institute of Chemical and Engineering Sciences (ICES) under A*STAR, Singapore. He received his BSc (2003) and MSc (2005) from Harbin Institute of Technology (HIT) in China, and his PhD (2009) from Pohang University of Science and Technology (POSTECH) in South Korea. His research interests are heterogeneous catalysis and materials chemistry, with a particular focus on CO2 capture and utilization, hydrogen production, vehicle emission abatement, biomass conversion, and synthesis and applications of porous materials and nanocomposites. He was awarded the Chinese Government Award for Outstanding Self-Financed Students Abroad in 2008. |
 Jizhong Luo | Jizhong Luo obtained his PhD from Xiamen University in 1997. After two years of postdoctoral research at Hong Kong Baptist University, he joined the Physics Department of the National University of Singapore as a research fellow in 2000. In 2003, he joined the Institute of Chemical & Engineering Science (ICES), and he is now a senior research follow in the Applied Catalysis group. His research interests include heterogeneous catalysis, alternative energy, microwave chemistry, and the carbon credit market. |
 Ziyi Zhong | Ziyi Zhong received his BSc and MSc degrees from Wuhan University and obtained his PhD from Nanjing University in China in 1995. He spent five years carrying out postdoctoral research at Bar-Ilan University, the University of Washington and finally at the National University of Singapore. In 2002, he was awarded a Visiting Fellowship by NSERC Canada and worked in an institute (BRI) in Montreal. Since 2003, he has been working in the Institute of Chemical and Engineering Sciences in Singapore, researching environmentally related heterogeneous catalysis. So far he has published 90 journal papers, a book chapter and filed two patents. |
 Armando Borgna | Armando Borgna (PhD in Chem. Eng., Universidad Nacional del Litoral, Argentina, 1987) is a Senior Scientist and Programme Manager at ICES (A*STAR, Singapore). He is also an Adjunct Associate Professor at the Department of Chemical and Biomolecular Engineering, National University of Singapore. He was previously a postdoctoral researcher at the Institut de Recherches sur la Catalyse, CNRS (1987–1990), Researcher at the Institute of Research on Catalysis and Petrochemistry, CONICET-Argentina (1991–2001), and Senior Research Associate at TU-Eindhoven (2001–2004). He was a Fulbright Fellow at the University of Oklahoma (1999). His research interest is in heterogeneous catalysis, particularly characterization of working catalysts, kinetic modeling and mechanistic studies. |
Broader contextThere is a growing awareness that anthropogenic CO2 emissions should be reduced as CO2 is a major greenhouse gas in the atmosphere. In order to do that, there are various measures which we can adopt: (1) change part of our current life styles, e.g., by using less fossil fuels and adapting to greener technologies/processes; (2) seek alternative energy and chemical resources; (3) capture, store and utilize (CSU) part of the emitted CO2. For the 3rd option, solid CO2-adsorbents will play an important role. For any practical application of these solid CO2-adsorbents, not only should we take into consideration their sorption capacities, selectivities and recyclibilities, we should also look into other factors, e.g., their working temperatures, which should be compatible to that of the particular process. In addition, in some reactions that produce CO2 as one of its products, CO2-removal is likely to influence the outcome of the reactions. Thus, this will probably widen the application of these materials in catalysis. In light of the increasingly obvious global warming trends worldwide, capture, storage and utilization of CO2 by means of solid CO2-adsorbents via a number of environmentally friendly processes is of increasing importance. Meanwhile, it will create new business opportunities. |
1. Introduction
CO2 is the major anthropogenic greenhouse gas (GHG) in the atmosphere. Its atmospheric concentration has increased to 384 ppm in 2007 from its pre-industrial level of ca. 280 ppm, and is expected to reach 550 ppm by 2050 even if CO2 emission is stable for the next four decades.1 One of the environmental impacts of atmospheric CO2 accumulation is global warming. Indeed, it is evident that a linear warming trend over the last 50 years (from 1956 to 2005) has almost doubled compared to that for the period from 1906 to 2005.2The CO2 emissions associated with human activities are mainly due to the burning of fossil fuels and various chemical processes, e.g., ca. 44% of anthropogenic CO2 emissions come from coal-, oil- or natural gas-fired power plants.2 Unfortunately there will be no big change in the next few decades for the energy consuming spectrum; fossil fuels will still be the dominant source as other energy sources such as biomass-based fuels, solar energy and nuclear energy, which are CO2-neutral or do not emit CO2, still cannot replace fossil fuels on a large scale. Moreover, the energy demand will increase further by 53% by 2030.2 Since an immediate CO2-emission halt is impossible, in recent years worldwide efforts have been devoted to developing new technologies/processes for CO2 capture, storage (sequestration) and utilization (CSU), and to improving the energy utilization efficiency.3 Among them, sorption-based technologies and processes account for the majority of these research activities, and they usually involve solid CO2-adsorbents. For example, in order to remove CO2 from power plant flue gas, which is crucial for large scale CO2-emission reduction, three kinds of technologies have been developed and tested, including post combustion, pre-combustion and oxyfuel.4 Some solid CO2-adsorbents have been tested in the post combustion and pre-combustion capture processes,5,6 though presently almost all commercial processes for capturing CO2 still use liquid alkaline solutions (e.g. amine). Unlike liquid adsorbents, solid adsorbents can be used over a wider temperature range from ambient temperature to 700 °C, yield less waste during cycling, and the spent solid adsorbents can be disposed of without undue environmental precautions.7
In the last few years there has been a rapid growth in publications related to CSU of CO2, and several recent review papers2,4,8 provide very good insights into the progress in this area. However, we feel that a new review focusing on solid CO2-adsorbents, addressing their pros and cons, and potential applications in industry is still needed. Furthermore, in this review, we will not only summarize the latest progress in the synthesis and applications of various solid CO2-adsorbents, but also organize these materials into different types according to their properties and performances, e.g., their working and regeneration temperatures, and associate them with various application processes. In addition, we will also address representative catalytic applications for the solid adsorbents, which is an active research topic with plenty of room for further development. This is based on the fact that since some catalytic reactions generate CO2 as one of the products, integration of a solid CO2-adsorbent with the catalyst is expected to shift the reaction balance. This will probably widen the application of solid CO2-adsorbents. However, some other topics such as the storage of enriched CO2 in proper geologic formations, catalytic conversion of CO2 to some value-added chemicals and utilization of CO2 as a solvent in chemical reactions will not be included.
2. Low-temperature solid adsorbents (< 200 °C)
2.1 Carbon based adsorbents
Owing to their low cost, high surface area, high amenability to pore structure modification and surface functionalization, and relative ease of regeneration, carbon-based materials are considered to be one of the most promising adsorbents for capturing CO2 in integrated gasification combined cycle (IGCC) processes for energy generation and hydrogen production.9 However, the CO2 adsorption on carbon materials is “physical” and weak, which makes these adsorbents sensitive to temperature and relatively poor in selectivity. The CO2 sorption capacity drops dramatically at temperatures associated with power plant flue gas (50–120 °C).10 Thus current research efforts are focused on how to enhance the adsorbate–adsorbent interaction and the selectivity for CO2. One approach is to increase the surface area and tune the pore structure of the carbon adsorbents either by using different precursors,11 or forming different structures such as single-walled carbon nanotubes (CNTs),12 multi-walled CNTs,13–15 ordered mesoporous carbon,16 microporous carbon,17etc. Another efficient approach is to increase the alkalinity by surface modification. This can be achieved either by impregnation of additives such as 3-chloropropylamine-hydrochloride (3-CPAHCl)18 and polyethylene glycol (PEG),10 or by incorporation of basic nitrogen groups into the carbon framework.19–21 Arenillas et al.10 impregnated several organic bases on fly ash-derived carbon materials, and observed a CO2 capture capacity of 1.13 mmol g−1 at 75 °C. It should be noted that, although the impregnation method is often used, the adsorbate–adsorbent interaction is weak thus the recycling ability is not strong. Moreover, the introduction of additives may block the porous carbon structure, thus lowering the adsorption capacity.22,23 Nitrogen can be incorporated into carbon structures by preparing carbon adsorbents from N-containing polymers, or by heat treatment of carbon adsorbents with NH3,23,24 or amines.25–27 Recently Hao et al.28 synthesized a new nitrogen-doped porous carbon monolith, for which a very high CO2 adsorption capacity of 3.13 mmol g−1 was achieved at room temperature and 1 atm. To decrease the cost of the adsorbents, cheaper carbon resources such as fly ash,29 almond shell-derived carbon,30 or anthracite25 can be selected. This is especially the case for the waste recycling of fly ash which cannot be marketed as a cement extender and has to be disposed of.29,31 All of the above mentioned research activities on carbon-based CO2-adsorbents are summarized in Fig. 1.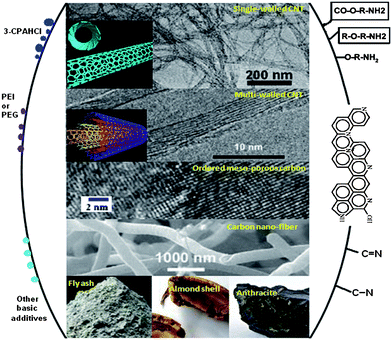 | ||
| Fig. 1 A summary of current research activities on carbon-based CO2-adsorbents. The middle part shows the types of carbons adsorbents that have been studied, the left part shows the impregnation or grafting of basic additives, and the right part shows the incorporation of N-containing groups.13,22,24,26,32 | ||
2.2 Zeolite based adsorbents
Zeolites are porous crystalline aluminosilicates, whose framework consists of interlocking tetrahedrons of SiO4 and AlO4 joined together in various regular arrangements through shared oxygen atoms. They have open crystal lattices containing pores with molecular dimensions, into which molecules can penetrate. The negative charge created by the substitution of an AlO4 tetrahedron for a SiO4 tetrahedron is balanced by exchangeable cations (e.g., Na+, K+, Ca2+, Mg2+), which are located in the channels and cavities throughout the structure. The adsorption and gas separation properties of zeolites are heavily dependent on the size, charge density, and distribution of these cations in the porous structure.33 The CO2 adsorption mechanism on zeolites has been investigated by various groups and it has been revealed that the physisorption of CO2 occurs with CO2 in a linear orientation by an ion–dipole interaction (reaction 1).34,35(metal ion)x+ ⋯⋯ δ−O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) COδ+ COδ+ | (1) |
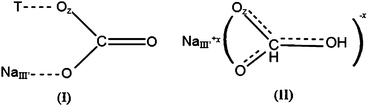 | ||
| Scheme 1 Two types of carbonate species associated with bi-coordination. | ||
Although zeolites have shown promising results for separating CO2 from gas mixtures and can potentially be used in the pressure swing adsorption (PSA) process, their selectivity to CO2 over other gases (N2, CH4, H2O, etc.) is still low, and their adsorption capacities rapidly decline with increasing temperature above 30 °C and become negligible above 200 °C.36–38 Generally speaking, separation of gases by zeolites depends on three factors: structure and composition of the framework, cationic form, and zeolite purity.39 A number of zeolites, especially those which are highly crystalline, with high surface area, and three-dimensional pore structures, have been investigated, including zeolite X,37,40,41 Y,42,43 A,44 β,45 ZSM,46,47 CHA,48 and natural zeolites (e.g. ZAPS, ZNT, ZN-19).39 Harlick et al.49 studied 13 types of zeolite-based adsorbents, and concluded that the most important adsorbent characteristics are a near linear CO2 isotherm and a low SiO2/Al2O3 ratio with cations that have a strong electrostatic interaction with CO2. Siriwardane et al.50 reported that the CO2 adsorption capacity for zeolites 13X and 4A were about 3.64 and 3.07 mmol g−1 respectively at 25 °C and 1 atm of CO2 partial pressure. Inui et al.36 studied the adsorption behavior of different zeolites in the PSA process and claimed that CHA and 13X were the most appropriate for CO2 separation among the zeolites studied. Merel et al.44 investigated the CO2 capture of zeolites 13X and 5A by indirect thermal swing adsorption, and found that zeolite 5A performed better than 13X. Calleja et al.51 reported that the preference of the adsorbent for polar molecules (e.g. CO2) increases as the Si/Al ratio decreases due to the higher surface heterogeneity and the stronger electrostatic field inside the pores of the zeolite.
Another research area is the zeolites exchanged with alkali and alkaline-earth cations. The cations influence the electric field inside the pores as well as the available pore volume, and provide a convenient means for tuning adsorptive properties of these porous materials. Walton et al.52 studied the CO2 adsorption behavior of the Y and X zeolites exchanged with Li, K, Na, Rb, and Cs, and found that the largest CO2 capacity was obtained for the zeolite containing Li. It was suggested that the CO2 molecule possesses a quadrupole moment that can interact more strongly with smaller cations like Li. Díaz et al.42 evaluated the Na- and Cs-treated Y zeolites, and suggested that Cs–Y is a promising material for CO2 adsorption processes at relatively high temperatures. Yamazaki et al.53 and Wirawan et al.46,54 investigated the CO2 adsorption on cation-exchanged MZSM-5 (M = Li, Na, K, Rb, Cs, H, Ba) and observed that, compared to other cation-exchanged zeolites, BaZSM-5 has a higher CO2 adsorption capacity and the adsorbed species also has a higher thermal stability. Zhang et al.48 found that NaCHA and CaCHA have comparative advantages for high temperature CO2 separation whilst NaX shows superior performance at relatively low temperatures.
In terms of practical applications, moisture is another challenge to zeolite based adsorbents. H2O is an important component in boiler flue gas and some other industrial gases, and it may compete with CO2 for the active adsorption sites. Gallei et al.55 have reported that the physical adsorption of carbon dioxide on CaY- and NiY-zeolite is scanty in the presence of water, because water is preferentially adsorbed onto these surfaces. Later Rege et al.56 and Brandani et al.57 observed that H2O has a strong effect on CO2 adsorption on type X zeolites. From a thermodynamic viewpoint, the adsorption of CO2 in the presence of H2O is not favored.58 However, Díaz et al.42 found that the CO2 adsorption on Cs- and Na-treated Y zeolites increased after water treatment, and believed that it was due to the enhancement in the Brønsted acidity in zeolites after treatment with alkali. The number of acid sites presumably increased after alkaline treatment (both with CsOH and NaOH), and the water molecules could coordinate to the Lewis sites and create new Brønsted acid sites. In addition, H2O may have a detrimental effect on the stability of zeolite frameworks. In the presence of CO2, the acidic conditions may cause dealumination of the zeolite structures, leading to a partial or total destruction of the framework. Ertan et al.59 studied CO2 adsorption on acid (HCl, HNO3, H2SO4, H3PO4) treated zeolites, and found that their CO2 adsorption capacities decreased after the acid treatment.
2.3 Metal organic framework based adsorbents
Metal organic frameworks (MOFs), which are constructed from transition metal ions and bridging organic ligands, are a new family of porous materials.60,61 One type of MOF structure (indium soc-MOF) is shown in Fig. 2, in which (a) is a cluster of metal ions (indium-carboxylate trimer, TMBB, In3O(CO2)6(H3O)3), (b) is an organic linker (3,3′5,5′-azobenzenetetracarboxylic acid), (c) and (d) are representations of a cuboidal cage of indium soc-MOF and (e) is a space filling representation of the framework viewed along the y-direction. The CO2 adsorption and desorption in MOFs, which are ascribed to changes in the framework structures, are explained by a “breathing-type mechanism” or a “gate effect mechanism”.62–64 Normally the interaction between adsorbed CO2 and adsorbents is weak, and the species starts to desorb when the temperature is higher than 30 °C. It should be noted that the MOFs used for CO2 capture are not limited to this type of structure.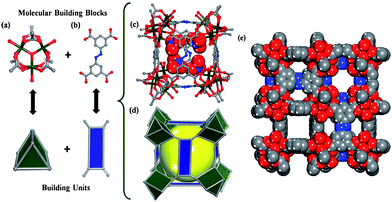 | ||
| Fig. 2 X-ray crystal structure of indium soc-MOF. Color scheme: carbon = gray, oxygen = red, nitrogen = blue, indium = dark green.65 | ||
Owing to their very high porosity and surface area, ordered and well characterized porous structure, and adjustable chemical functionality, MOFs are attracting increasing attention in CO2 capture.66 Millward et al.67 reported that, at 35 bar, a container filled with MOF-177 could capture nine times the amount of CO2 in the container (33.5 mmol g−1) compared to that without the adsorbent. Llewellyn et al.68 obtained a record capacity of 40 mmol g−1 for CO2 capture with MIL-101 at 50 bar and 30 °C.
In order to be applicable to realistic flue streams, MOF-based CO2-adsorbents should fulfil the following requirements: high CO2 capture capacity and selectivity for CO2 over other components in the flue gas, corrosion resistance, and high thermal/hydrothermal stability, etc. To date, although MOFs have shown exceptionally high CO2-storage capacity under equilibrium conditions with pure CO2, most of them show little uptake in the low pressure regime of 0.1–0.2 bar, which is the pressure range used for CO2 capture from flue gas streams.66 Even worse, their capacities reduce dramatically when exposed to mixtures of gases under dynamic conditions, which would be the case in power plant flue gas and methane mining applications.69 Therefore, tremendous efforts have been made to further increase their CO2 capture selectivity while maintaining or improving their capture capacity under equilibrium conditions. The modifications are usually conducted in three aspects: (a) metal ions, (b) organic linkers, or (c) novel combinations of both.
For the metal ions, creating open metal sites has been proven to be an efficient approach, as the vacant coordination sites at metal ions are the primary coordination sites for guest molecules.69–72 Britt et al.69 reported that a MOF replete with open magnesium sites, Mg-MOF-74 (Mg2(DOT); DOT: 2,5-dioxidoterephthalate), has excellent selectivity, facile regeneration, and is ranked among the highest dynamic capacities reported for CO2 in porous materials (Fig. 3(a)). In particular, when Mg-MOF-74 was subjected to a gas stream containing 20% CO2 in CH4, a percentage in the range of industrial separation conditions, it captured only CO2 and not CH4. The porous material retained 89 g of CO2 per kilogram of material before breakthrough (2.23 mmol g−1). Furthermore, these unsaturated metal sites can be functionalized with groups such as amine to further improve their performance.66,69,73–75 Demessence et al.66 functionalized the triazolate-bridged MOF 1 (Cu-BTTri) with ethylenediamine, and obtained a very high uptake of CO2 at low pressures (see Fig. 3(b)). This material also displayed a record isosteric heat of 90 kJ mol−1. Bae et al.75 reported that the CO2/N2 selectivity could also be enhanced by a cavity modification. By replacing the coordinated solvent molecules with highly polar ligands, a high CO2/N2 selectivity of ∼42 was obtained at low pressure. Couck et al.73 demonstrated that the MOF MIL-53(Al) functionalized with amino groups increased its selectivity of CO2/CH4 by orders of magnitude while maintaining a very high capacity for CO2 capture. In addition to amines, the occupation of open metal sites by coordinated water molecules has also been studied (Fig. 3(c)).76,77 Yazaydin et al.76 first predicted by molecular simulations and then validated by experiments that water molecules coordinated to open metal sites could significantly increase CO2 adsorption in Cu-BTC.
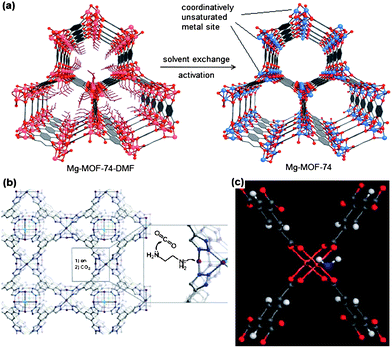 | ||
| Fig. 3 (a) Single crystal structure of Mg-MOF-74, formed by reaction of the DOT linker with Mg(NO3)2·6H2O. C atoms are shown in gray, O atoms in red, 6-coordinate Mg atoms and terminal ligands in pink, and 5-coordinated Mg atoms in blue. H atoms and terminal ligands on the fragment in the top right are omitted for clarity.69 (b) A portion of the structure of the sodalite-type framework of Cu-BTTri (1) showing surface functionalization of a coordinatively unsaturated Cu2+ site with ethylenediamine, followed by attack of an amino group on CO2. Purple, green, gray, and blue spheres represent Cu, Cl, C, and N atoms, respectively; framework H atoms are omitted for clarity.66 (c) Hydrated Cu-BTC (4 wt %) with a coordinated water molecule from DFT. Cu atoms are orange, O red, C gray, and H white. The oxygen atom of the coordinated water molecule is shown in blue.76 | ||
For the modification of organic linkers, Bae et al.60,78 examined the possibility of using MOFs that are carborane-based or coordinated with a mixture of ligands, and obtained a high selectivity for CO2 over CH4 (∼17).78 Carboranes possess several favorable material properties including rigidity, thermal stability, and chemical stability. In addition, carboranes can produce MOFs with smaller pores than phenyl-based MOFs. The use of two different linkers opens up more possibilities to tune pore size and chemical functionality independently. The mixed ligand MOF Zn2(NDC)2(DPNI), 2, (NDC = 2,6-naphthalenedicarboxylate, DPNI = N,N′-di-(4-pyridyl)-1,4,5,8-naphthalene tetracarboxydiimide) exhibited a selectivity of ∼30 for CO2 over CH4.78
Much attention has also been paid to the synthesis of novel MOFs with both high CO2 capture capacity and stability. The ability to synthesize MOFs with various organic linkers and metal joints provides tremendous flexibility in equipping the porous material with specific physical characteristics and chemical functionalities. Recently, a big breakthrough has been made by Yaghi's research group in UCLA.79,80 A new class of MOFs known as zeolitic imidazole frameworks (ZIFs), in which metal atoms such as Zn are linked through N atoms by ditopic imidazolate (C3N2H3− = Im) or functionalized Im links to form neutral frameworks. These compounds have high chemical and thermal stabilities (up to 500 °C).81 Phan et al.82 reported that 1 L of ZIF-69, the best-performing ZIF, could store 82.6 L of CO2 at 0 °C. In addition, ZIFs show greater selectivity than other types of MOFs for CO2 from relevant flue gases. It is believed that only CO2 can slip into the cages within the ZIF, while other gas molecules just pass through them without hindrance.83 Therefore, ZIFs are a very promising type of material for selective adsorption of CO2, and they are twice as efficient as BPL carbon (a commercially available carbon product of Calgon).84 Yaghi's group have also developed another class of new porous crystals, covalent organic frameworks (COFs), by condensation reactions of phenyl diboronic acid (C6H4[B(OH)2]2) and hexahydroxytriphenylene (C18H6(OH)6).85,86 Recent studies indicated that COFs also have a comparable capacity for CO2 adsorption to the most common carbon materials, zeolites, mesoporous solids, MOFs, etc.87
2.4 Alkali metal carbonate based adsorbents
Thermodynamic analysis of dry and re-generable adsorbents has shown that alkali metal carbonates are suitable for the treatment of flue gases at temperatures below 200 °C.88,89 Typically CO2 capture is effective within the temperature range of 50–100 °C, while regeneration occurs in the range of 120–200 °C. This concept is potentially applicable to the capture of CO2 from existing fossil fuel-fired power plants.Liang et al.90 studied the mechanism and revealed that the important reactions involved in the capture of CO2 using Na2CO3 are shown in reactions (2) and (3).
| Na2CO3(s) + CO2(g) + H2O(g) ⇔ 2NaHCO3(s), ΔHr° = −135 kJ mol−1 Na2CO3 | (2) |
| Na2CO3(s) + 0.6CO2(g) + 0.6H2O(g) ⇔ 0.4[Na2CO3·3NaHCO3](s), ΔHr° = −82 kJ mol−1 Na2CO3 | (3) |
The theoretical CO2 capture capacity of Na2CO3, calculated from reactions (2) and (3), is 9.43 mmol g−1 and 5.66 mmol g−1 respectively. Both reactions are reversible and highly exothermic so energy management is an issue that should be considered in any particular application. K2CO3-based adsorbents have also been investigated and showed similar results.91,92 The conversion decreases with an increase in the reaction temperature and pressure, and varying the CO2 and H2O concentrations has little effect, while the maximum reaction rate increases with an increase in temperature and H2O concentration, and with a decrease in pressure. The carbonation can start from 60 °C, and the maximum carbonation temperature is limited by the reaction thermodynamics.90 As a result, a common problem is that the overall carbonation reaction rate for Na2CO3/K2CO3 is rather slow. Many attempts have been made to resolve this, e.g., by dispersing active Na2CO3/K2CO3 on a support such as Al2O3, active carbon (AC), TiO2, SiO2, MgO, ZrO2, etc., so as to enhance the adsorption rate and provide the required attrition resistance in the fluid-bed or transport reactors.7,90,93–98 Nevertheless, these adsorbents have a disadvantage in that the reactivity always decreases with increases in sorption/regeneration operations.99,100 Okunev et al.101 studied various K2CO3/support composite materials and concluded that the host matrix should not be limited to an inert support and can undergo chemical transformations caused by an interaction with the impregnated salt.
So far the issues that should be considered for practical applications of such CO2-adsorbents include durability, carbonation reaction rate and temperature control, and energy management. In addition, contaminants in flue gas such as SO2 and HCl will react irreversibly with Na2CO3 and must therefore be reduced to low levels prior to CO2 capture. Their practical application is probably still a long way off.
2.5 Amine-based solid adsorbents
Some NHx-containing organics or polymers can chemically bind to acidic CO2 molecules at low reaction temperatures, and thus they are potentially good candidates for CO2 capture. CO2 removal from flue gas by sorption and stripping with aqueous amines (commonly monoethanolamine (MEA), diethanolamine (DEA) and methyldiethanolamine (MDEA)) has been established since 1930 and is still believed to be a feasible technology.102,103 However, this process suffers from a series of inherent problems, including the corrosive nature of the amines, high regeneration energy, fouling of the process equipment, etc.104,105 In order to avoid the above problems, intensive efforts have been made to prepare amine-based solid adsorbents by immobilizing organic amines on certain support materials.26,50,106–108 Compared to the aqueous amine solutions, the solid amine adsorbents usually require lower capital cost, lower pressure for gas recovery, and lower energy consumption for regeneration.109 Research activities have been focused on three aspects to improve the CO2-capture properties of amine-based solid adsorbents: (1) supports that are able to bear high amine-loading, (2) amines that can generate high amine density, and (3) effective methods for amine immobilization.Generally speaking, the supports should have a good affinity for the amine molecules, high surface area, proper porosity, good mechanical strength and hydrothermal stability, etc. Silica, carbon, polymers such as resin, glass and metal oxides can be considered, but usually porous materials are preferred due to their high surface areas and large pore size and volume. In other words, nonporous and even microporous materials are rarely selected for the purpose. Among various porous candidates, mesoporous silicas (SBA-15, MCM-41, etc.) are most widely investigated, mainly because of the “silane chemistry”; a number of aminosilanes can react with the silica surface via alkyl–silyl linkages, thus providing vast opportunities for loading amine molecules onto the supports. Harlick et al.110 did systemic work on pore-expanded MCM-41 (PE-MCM-41) grafted with 3-[2-(2-aminoethylamino)ethylamino]propyl trimethoxysilane (TRI), and found that the TRI-PE-MCM-41 adsorbent showed a higher CO2-sorption capacity and kinetics than TRI-MCM-41. One of the drawbacks of these mesoporous silicas is their relatively weak hydrothermal stability, probably limiting their application in aqueous media. Amination of carbon materials is also reported by reacting carbon surface carboxylic groups with halogenated amine molecules.26 This kind of material has been addressed in section 2.1 and is probably better suited to aqueous media.
Various amines tested for CO2 capture include alkanolamines, mono-, di-, and triaminosilanes, aminopolymers such as polyethyleneimine (PEI),110,111 dendrimers,112 hyperbranched aminosilica,113etc. In general, multi-amine-containing molecules are superior due to their higher amino group densities. Chang et al.114 observed a CO2 capture capacity of 2.4–2.7 mmol g−1 at 60 °C on triaminosilane/SBA-15. Sayari et al.115 obtained a value of 2.65 mmol g−1 at 25 °C on TRI-PE-MCM-41. On a tetraethylenepentamine (TEPA)-grafted mesocellullar silica foam (MSF), a capacity as high as 4.5 mmol g−1 adsorbent was achieved at 75 °C by Liu et al.116 For polyethylenimine coated glass fibers, Li et al.117 observed a capacity of 4.1 mmol g−1 at 30 °C. Liang et al.118 synthesized various generations (G0, G1, G2, G3 and G4) of melamine-based dendrimers on SBA-15. However, at 20 °C, a CO2 capacity of only 1.0 mmol g−1 (ca. 4.4 wt% g−1) was observed on G3/SBA-15. On G4/SBA-15, a capacity of only 0.4 mmol g−1 (1.7 wt% g−1) was obtained instead of a theoretical value of 1.36 mmol g−1 (ca. 6 wt% g−1), indicating many amino groups were not accessible by the CO2 molecules. The highest CO2 capture capacity observed was 5.55 mmol g−1 on hyper-branched aminosilica (HAS) on SBA-15 at 25 °C by Hicks et al.,113 which was formed by polymerization of aziridine on SBA-15.119
Methods for loading amines onto supports can be roughly classified as (a) impregnation, (b) post-synthesis grafting and (c) direct condensation and hydrolysis methods.111 The samples prepared by the impregnation method can have high amine-loading but are not usually good in recycling. For the post-synthesis grafting method, the above mentioned grafting of aminosilane molecules onto silica-based supports belongs to this category, and usually takes place in anhydrous solution so as to reduce the self polymerization behavior of the aminosilane molecules.120 In the direct condensation and hydrolysis method, an aminosilane, a surfactant (template) and the silica precursor are mixed and reacted directly, and the amino groups can thus be incorporated into the matrix of the synthesized material. In a recent work by Tang et al.,121 bis((triethoxysilyl)propyl) ethylenediamine (BTEPED) was incorporated into a porous silica and a CO2 capture capacity of 2.3 mmol g−1 was obtained. The materials synthesized by this method should maintain their amines well; however, not all of the amino groups are accessible to CO2 molecules as they are trapped in the silica matrix. In addition, in many cases it is still a challenge to incorporate amine molecules while forming the porous structure, and the uncalcined silica wall should have a poorer hydrothermal stability than the calcined porous silica used in the post-synthesis grafting.
The chemistry of grafted amines with CO2 is similar to that between amines in aqueous solution and CO2.122 Three kinds of carbonates can be formed from primary amines as shown in Scheme 2. From the reaction pathway in Scheme 2, it is understandable that the existence of H2O can lead to the formation of bicarbonate and carbonate and consume more CO2 molecules, thus increasing the CO2 capture capacity. This is the reason why these CO2-adsorbents can tolerate moisture; indeed, the presence of moisture can promote the CO2 capture.110 Since these reactions can run at a fast rate, the CO2-capture process is also fast, unless the CO2 concentration is very low.
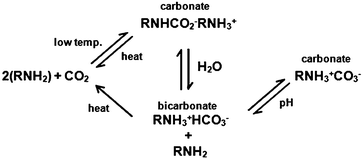 | ||
| Scheme 2 CO2 reaction pathway with a primary amine.110 | ||
The thermal stability of amines is well studied. Degradation of aqueous alkanolamines occurs below 140 °C under atmospheric pressure,123 though a slightly higher temperature has also been reported in some grafted amines.110 The working temperature for CO2-capture should usually be below 60 °C, with better results achieved as the temperature approaches ambient temperature. For regeneration, it is usually between 120–170 °C according to our unpublished data.
3. Intermediate-temperature solid adsorbents (200–400 °C)
Layered double hydroxides (LDHs), also known as hydrotalcite-like compounds (HTs) or anionic clays are layered basic solids. They have been widely used as adsorbents, ion exchangers, base catalysts, and precursors of well-mixed oxides for various catalytic applications.124 Their structure consists of positively charged brucite-like layers with charge compensating anions and water molecules within the interlayer space.125–127 The metal cations occupy the centers of the octahedral structures, whose vertices contain hydroxide ions, and the octahedrons are connected by edge sharing to form an infinite sheet (Fig. 4A). The general formula of the compounds is [M2+1-xM3+x(OH)2][An−]x/n·zH2O, where M2+ and M3+ are divalent (Mg2+, Zn2+, Ni2+, etc.) and trivalent cations (Al3+, Ga3+, Fe3+, Mn3+, etc.) respectively, An− is a non-framework charge compensating anion (CO32−, Cl−, SO42−, etc.) and x is normally between 0.2–0.4. In general, LDH materials possess both high surface area and abundant basic sites at the surface, which are favorable for adsorbing acidic CO2.128–130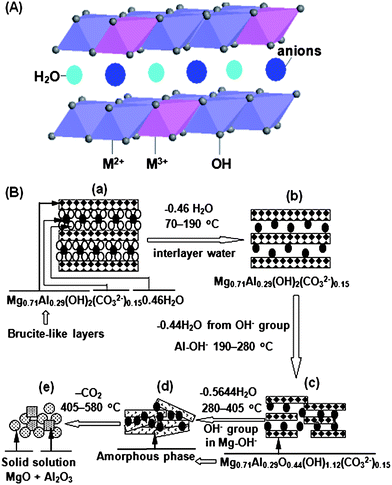 | ||
| Fig. 4 (A) Schematic structural representation of LDHs; M2+ and M3+ represent divalent and trivalent cations. (B) The structural evolution of Mg-Al-CO3 LDH as a function of temperature.131 | ||
Fresh LDH itself does not possess any basic sites. Upon thermal treatment, LDH gradually loses interlayer water, then dehydroxylates and decarbonates to a large extent, leading to the formation of a mixed oxide with a 3D network. The detailed structural evolution of Mg-Al-CO3 LDH as a function of temperature was investigated by Yang et al.,131 as shown in Fig. 4B. Ram Reddy et al.132 reported the effect of calcination temperature on the CO2 adsorption capacity of Mg-Al-CO3 LDH. The sample calcined at 400 °C showed the highest adsorption capacity, which is presumably due to the trade-off between the surface area and availability of active basic sites. This amorphous phase has a relatively high surface area and exposes sufficient basic sites on the surface. These basic sites favor reversible CO2 adsorption in a form as shown in reaction (4).133 The interaction between the adsorbed CO2 and the basic sites is stronger than that in the case of zeolite (see section 2.2) but weaker than that in the case of alkali metal oxide (see section 4), which explains why the working temperature of LDH-based CO2-adsorbents used in flue-gas systems falls in the middle temperate range of 200 °C and above. Normally the regeneration temperature is around 400 °C.132–135
| Mg–O + CO2 → Mg–O⋯CO2(ad) | (4) |
The relatively low CO2 adsorption capacity is a problem for LDH-based adsorbents. Intense efforts have been put into improving the capacity, especially on the mineral Mg-Al-CO3 LDH and its derivatives. For instance, the synthetic conditions,136 presence of SOx and H2O,133,137 operation pressures,127,138,139 alkali (K, Cs) doping,140–143 particle size,144 and the use of supported LDHs145,146 were widely investigated. Sharma et al.136 obtained the optimized synthetic parameters: 37% Al content, room temperature addition of magnesium and aluminium precursors, aging at 65 °C for 18 h, and drying at room temperature. A maximum CO2 adsorption of 0.98 mmol g−1 was obtained. However, it should be noted that, in their work, the CO2 adsorption isotherms were measured at 30 and 60 °C, and not in the high temperature range (200–400 °C). Ram Reddy et al.133,137 reported that the presence of water in the feed gas had a positive effect on the CO2 adsorption capacity, which increased from 0.61 mmol g−1 to 0.71 mmol g−1, whereas the presence of SO2 had a negative effect, as it could competitively interact with the CO2 adsorption sites. The operation pressure has been proven to have a significant effect: CO2 adsorption capacity increases with increasing operation pressure.127,138,139 Doping alkali metal carbonates in LDHs is an effective approach to improving the CO2 adsorption. Oliveira et al.140 reported that the adsorbed CO2 on MG 30 (commercial hydrotalcite from Sasol, Germany) increased from < 0.1 mmol g−1 to 0.76 mmol g−1 after doping with K2CO3. Walspurger et al.143 investigated the K+ promotion mechanism, and suggested that potassium ions could strongly interact with aluminium oxide centers in hydrotalcite, resulting in the generation of basic sites that were able to reversibly adsorb CO2 at high temperatures. Meis et al.146 studied the support and size effects of activated hydrotalcites for pre-combustion CO2 capture, and found that the CO2 adsorption capacity significantly increased with a decrease in the particle size. The adsorption capacity of carbon nanofiber supported hydrotalcites increased by an order of magnitude (1.3–2.5 mmol g−1) compared to that of unsupported hydrotalcites. It was proposed that the CO2 adsorption capacities of LDHs were determined by the amount of low coordination number oxygen sites in the Mg(Al)Ox nanoparticles, which was highest on supported LDHs due to the high distribution.
More work on modifying the composition of Mg-Al-CO3 has also been carried out by either substituting the CO32− anion or Mg2+ divalent cations. Hutson et al.147 investigated CO2 adsorption on various LDHs with different anions including CO32−, Fe(CN6)4−, Cl− and ClO4−, and concluded that Mg-Al-CO3 has the highest basic site density (692 μmol g−1). Lwin et al.148 and Wang et al.149 studied various LDHs with different divalent cations including Mg2+, Co2+, Ca2+, Cu2+, or their combinations, and came to the conclusion that the Ca–Co–Al system had the highest CO2 adsorption capability. Yavuz et al.150 demonstrated that the CO2 capture efficiency could be markedly improved by partially substituting Al with Ga. With 10 mol% substitution for Al, the CO2 capture capacity increased from 0.47 to 1.40 mmol g−1 for K2CO3-promoted Mg-Al-CO3. Recently, our group has carried out a systematic investigation on the Mg-M-CO3 (M = Al, Fe, Ga, Mn) LDHs for the effect of trivalent cations on their CO2 capture performance and found that the M3+ cations actually determine the structure evolution of the LDH derivatives under the thermal treatment, and ultimately influence the CO2 capture capacity. A very different calcination temperature is required for each LDH to obtain the maximum CO2 capture capacity. The optimal calcination temperatures are as follows: Mg3Al1 (400 °C) > Mg3Ga1 (350 °C) > Mg3Fe1 (300 °C) > Mg3Mn1 (250 °C).151Scheme 3 lists the possible methods for modifying the composition of Mg-Al-CO3 LDHs.
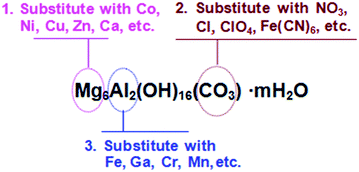 | ||
| Scheme 3 The possible approaches to modifying the composition of Mg-Al-CO3 LDH. | ||
Besides the efforts made to increase the CO2 capture capacity, a more important issue is to enhance the long-term stability of LDHs during CO2 adsorption/desorption cycling operation, which is crucial for the development of practical applications. Though there is still no clear answer from the literature, the solution probably lies in the progress in materials science, either by modifying the current LDHs, or by identifying new systems.
Magnesium oxide is another CO2-adsorbent working in the intermediate temperature range from room temperature up to 200 °C.152 However, due to its moderate CO2 adsorption capacity and poor thermal stability during regeneration, the practical application of MgO as a CO2-adsorbent may be limited. Gregg et al.153 tested the CO2 capture on MgO at 200 °C, and obtained a capacity of only 0.43 mmol g−1. Bhagiyalakshmi et al.154 noted that the complete desorption of adsorbed CO2 could only be accomplished by prolonged heating at 450 °C.
4. High-temperature solid adsorbents (> 400 °C)
4.1 Calcium based adsorbents
It is well accepted that calcium based materials are good adsorbent candidates for capturing CO2 due to their high reactivity with CO2, high capacity and low material cost.155 The reversible reaction between CaO and CO2, as shown in reaction (5), offers a great potential for reducing CO2 from various clean energy systems, e.g. pre-combustion carbon capture from gasification processes and post-combustion carbon capture from zero emission coal (ZEC) processes. The carbonation temperature for CaO-based adsorbents is between 600–700 °C and the regeneration temperature is normally above 950 °C.6,156–160| CaO(s) + CO2(g) ⇔ CaCO3(s), ΔH°873.15 K = −171.2 kJ mol−1 (for carbonation) | (5) |
This carbonation reaction is highly exothermic and it is possible to efficiently recover the large amount of energy released during the CO2 capture. Preliminary economic analyses suggest that this process is economically attractive because limestone (CaCO3) is abundant and low-cost when used at the industrial scale.161–163 The CO2 capture reaction is characterized by an initial rapid and kinetically-controlled phase, followed by an abrupt transition into a slower diffusion-controlled phase, whereas the CO2 release reaction is much faster and always goes to completion in minutes. In practical applications, this reaction is always carried out at temperatures ≥ 850 °C in order to produce a nearly pure stream of CO2.164–166
Despite the simple chemistry involved, the main problem for the calcium-based materials is the loss of reversibility for the carbonation reaction due to the sintering of the adsorbent particles.167,168 This is caused by three major factors: the carbonation process is highly exothermic, the volume increases substantially from CaO to CaCO3 (from 16.9 to 34.1 cm3 mol−1), and the Tammann temperature of CaCO3 (533 °C) is much lower than normal carbonation temperatures.167,169,170 Lysikov et al.171 proposed a simple schematic diagram for the textural transformation of the CaO adsorbent in recarbonation–decomposition cycles. The first decomposition of the calcium carbonate produces highly dispersed, nanosized CaO particles that are highly reactive. The following carbonation is incomplete due to the slow diffusion-controlled reaction with CO2 and pore blocking upon carbonation.6 The amount of unreacted CaO and its particle size increase from cycle to cycle until a rigid interconnected CaO skeleton is formed. This rigid backbone is considered a much less reactive adsorbent compared to the highly dispersed CaO. There are some other restrictions to such processes related to the kinetics and thermodynamics of the reactions, along with undesirable side reactions such as sulfation and processes such as attrition.172–174
Numerous efforts have been undertaken to enhance the durability of CaO-based CO2-adsorbents. Hydration is currently considered to be a promising approach to reactivating the spent adsorbents for CO2 capture.6 Manovic et al.175 and Fennell et al.176,177 observed that the reactivity of the spent adsorbents could be doubled following hydration. The mechanism of reactivation by hydration appeared to be through breakup of particles to increase porosity.178,179 Thermal pre-activation in an inert atmosphere at high temperature (e.g. 1000 or 1100 °C for 6 or 24 h) is another option and was first studied by Lysikov et al.171 in 2007. Later Chen et al.180 found that the activity and durability of thermally pretreated limestone and dolomite were greatly enhanced. Manovic et al.172 proposed a pore-skeleton mechanism to explain the thermal pre-activation. The cycling stability could also be improved by incorporating inert materials such as MgO,170,181,182 CaTiO3,183 Ca12Al14O33,184–187 ZrO2,188etc. One practical problem of this method is the presence of a large amount of undesirable inert material in the adsorbent, since this will increase the capital and operating costs. Some researchers have attempted to synthesize CaO from nano-sized CaCO3 particles155 or organometallic precursors.189 It would make economic sense to use cheap natural adsorbents (e.g. limestone) and to sell the spent adsorbent to the cement industry.177,190–193
4.2 Alkali ceramic based adsorbents
Alkali metal (Li, Na, K, etc.) containing ceramics are another type of high temperature CO2-adsorbent. In 1998, Nakagawa and Ohashi first reported the capture of CO2 using Li2ZrO3 at high temperatures (400–600 °C).194 This material has great potential because it has an excellent CO2 sorption capacity (28 wt%) as well as a small volume change during the CO2 sorption/desorption cycles.195 The reaction and process are described by reaction (6), which can occur mainly due to the lithium mobility in the ceramics (Fig. 5).196,197 During the CO2 sorption, the lithium atoms diffuse from the core of the particles to the surface and react with CO2 to form Li2CO3, and the diffusion of CO2 in the solid Li2CO3 that is produced is the rate-limiting step for Li2ZrO3. The reverse reaction normally happens at temperatures around 750–800 °C.| Li2ZrO3 + CO2 ⇔ Li2CO3 + ZrO2, ΔH298 K = −160 kJ mol−1 | (6) |
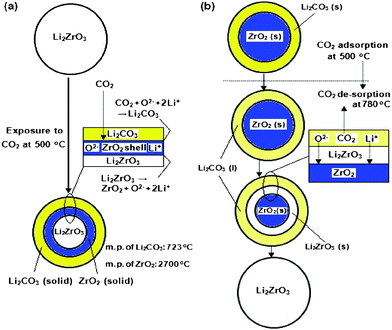 | ||
| Fig. 5 Proposed mechanism for CO2 sorption (a) and desorption (b) on Li2ZrO3.197 | ||
The main obstacle for the practical application of Li2ZrO3 is its kinetic limitation. Various efforts have been made to improve its CO2 capture performance. Nair et al.198 carried out a systematic study of the properties of lithium zirconates with different crystal structures. They compared the properties of three Li2ZrO3 powder samples: one commercial and two others prepared by the solid-state reaction method and the sol–gel method, and found that small particle size and tetragonal phase are crucial for enhancing the performance in CO2 capture. Later, Ochoa-Fernández et al.199,200 successfully synthesized nanocrystalline tetragonal Li2ZrO3 by a novel soft-chemistry route and achieved improved kinetics.
Another method is to substitute or partially substitute Li+ by Na+ or K+. López-Ortiz et al.201 found that Na2ZrO3 achieved a superior CO2 capture ability than Li2ZrO3. Pfeiffer et al.196 explained that Na2ZrO3 has a lamellar structure and the sodium atoms are located among the ZrO3− layers with higher mobility. On the other hand, Li2ZrO3 has a much more packed structure, which limits lithium diffusion. Later lithium–sodium and lithium–potassium metazirconate solid solutions (Li2-xNaxZrO3 and Li2-xKxZrO3) were also investigated.196,202,203 Pfeiffer et al.196 found that LiNaZrO3 presented the best ability for CO2 chemical adsorption, exhibiting a CO2-adsorption capacity of 4.45 mmol g−1 at 600 °C, which means an efficiency of 75.3% (the efficiency is defined as the ratio between the real adsorption capacity to the theoretical value). Furthermore, LiNaZrO3 adsorbed CO2 faster than any other similar ceramics in short time spans. Veliz-Enriqueza et al.203 found that Li2-xKxZrO3 solid solutions adsorbed CO2 between 450 and 730 °C, but the ceramics containing potassium adsorbed CO2 at higher temperatures. The kinetic analyses indicated that Li2-xKxZrO3 solid solutions can adsorb CO2 up to five times faster than Li2ZrO3 in short time spans.
Besides zirconates, some other alkali-metal-containing materials including lithium orthosilicate (Li4SiO4),204–207 Na2SiO3,208 CaSiO3,209 and lithium cuprates (Li2+xCuO2+x/2)210 are also potential high temperature CO2-adsorbents. However, it seems there are still many undiscovered materials in this area, thus a systemic investigation is necessary.
Table 1 summarizes the general CO2 adsorption capacities of the main types of adsorbents mentioned in this paper. However, it is worth mentioning that, once a higher adsorption pressure is applied, the CO2 adsorption capacity might greatly exceed the capacity range listed in this table. This phenomenon is especially significant for low temperature CO2-adsorbents. For instance, Millward et al.67 reported that the amount of adsorbed CO2 could be as high as 33.5 mmol g−1 at 35 atm.
| Adsorbent type | Adsorption temperature/°C | Adsorption pressure/atm | CO2 adsorption capacity/mmol g−1 | References | |
|---|---|---|---|---|---|
| Low temperature adsorbents | Carbon based | ≤ 80 | 1 | ≤ 3.5 | 10–29 |
| Zeolite based | ≤ 100 | 1 | ≤ 4.9 | 34–59 | |
| MOF based | ≤ 100 | 1 | ≤ 4.5 | 62–87 | |
| Alkali metal carbonate based | ≤ 120 | 1 | ≤ 9.4 | 88–101 | |
| Amine based | ≤ 60 | 1 | ≤ 5.5 | 101–122 | |
| Intermediate temperature adsorbents | LDH based | 200–400 | 1 | ≤ 1.4 | 131–150 |
| High temperature adsorbents | Calcium based | 600–700 | 1 | ≤ 11.6 | 155–193 |
| Alkali ceramic based | 500–600 | 1 | ≤ 6.5 | 194–210 | |
5. Catalytic applications of solid CO2-adsorbents
The captured and concentrated CO2 can be catalytically converted to various chemical products, e.g., to methanol via hydrogenation of CO2/CO,211 to syngas (H2 + CO) via drying reforming of CH4 with CO2,212 and to many fine/pharmaceutical chemicals, etc.213 However, here we only focus on some sorption-enhanced reactions that utilize the CO2-capture function of the solid CO2-adsorbents. Besides the CO2-capture capacity, the kinetics and recycling ability of the materials and the temperature gap between the reaction temperature and the sorption/desorption temperature should also be considered. It is better if the temperature gap is small; if not, in some cases it could be overcome by a proper combined configuration of the catalysts and the CO2-adsorbents. Otherwise, the extra energy consumption and cost have to be considered.Several important CO2-sorption enhanced reactions are steam reforming (SR) of hydrocarbons or biomass-derived chemicals, such as glycerol,214 the water gas shift (WGS) reaction, and preferential oxidation (PROX) of CO in H2. In general, these reactions are related to the production of H2-rich gas. The enhanced-WGS reaction has been tested in the pre-combustion process to clear up flue gas of the coal-fueled gasification power plants,156,215,216 and the PROX reaction is used to produce very pure H2 (the CO concentration is required to be less than 100 ppm or even 10 ppm) that can be fed to polymer electrolyte membrane fuel cells (PEMFCs). It is very common for several reactions to occur simultaneously in a single reaction process, thus increasing the complexity for very good control of it. For thermodynamic analyses and process considerations of some reactions and processes, readers can refer to Harrison's review paper and references therein.156 However, we should keep in mind that the effect of CO2 removal is not just limited to these reactions. Theoretically, a reaction having CO2 as a product could be impacted by removal of CO2.
The SR reactions usually take place above 500 °C, so high-temperature CO2-adsorbents are often used in these reactions. For the SR reaction of methane, CaO and Na2ZrO3 exhibit the highest reaction kinetics among various CO2-adsorbents (CaO, Li2ZrO3, K-doped Li2ZrO3, Na2ZrO3, and Li4SiO4). However, the former is easily sintered thus losing its capacity gradually.217 Because of the relatively low reaction kinetics on Li2ZrO3, a long reactor and low space velocity have to be used so as to reach the equilibrium composition.218 The sintering of the adsorbents is not only due to the high reaction temperature, but also to the even higher regeneration temperature, which is over 850 °C for CaO.219 In order to improve the sorption-enhanced SR reaction further, adsorbents with lower sorption/desorption temperature are preferred, or SR catalysts that can catalyze the reaction at lower temperatures should be developed and applied. In the latter case, CO2-adsorbents that have lower sorption/desorption temperature, e.g. LDHs, become applicable.129
The WGS reaction usually takes place between 200–500 °C, so solid CO2-adsorbents with lower sorption/desorption temperatures such as LDHs can be used. In the presence of K2CO3-promoted LDHs, the WGS reaction on a commercial catalyst could produce H2 with a CO concentration as low as 10 ppm, because the CO conversion was increased from 99.87 to 99.999% after addition of the adsorbent.220 Van Selow et al.221 recently reported a good stability for more than 300 cycles of adsorption/reaction and desorption on an WGS reaction system enhanced by a LDH-based CO2-adsorbent, and the CO concentration was kept at ca. 300 ppm, a value much lower than that of the conventional WGS system (usually above 1000 ppm). It should be pointed out that in the SR reactions, the WGS reaction also often occurs, because the reformate contains CO, CO2, H2 and H2O, so the existence of CO2-adsorbents enhances the two reactions.222
It would be ideal to combine the CO2-adsorbents with the PROX reaction catalysts for fuel cell applications. After the WGS reaction, the feed gas contains quite a low amount of CO, which allows a much longer working span for the CO2-adsorbents than that in the sorption-enhanced SR and WGS reactions. The PROX reaction following should be able to further lower the CO concentration significantly. Though some PROX catalysts are efficient, there is limited room for further improvement of the selectivity for CO oxidation over H2 oxidation in a H2-enriched environment. One of the reasons is that, in the PROX process, due to the generation of CO2 and H2O, the WGS and reverse WGS reactions and even the methanation reaction may also occur. The reverse WGS (r-WGS) reaction regenerates CO, and the methanation reaction consumes H2, so both of them should be avoided. Addition of a CO2-adsorbent is likely to shift this WGS reaction towards H2 production and eliminate the methanation reaction. From a thermodynamic viewpoint, the above mentioned reactions favor different reaction temperature ranges, e.g., the r-WGS and methanation reactions occur above 200 °C and 400 °C respectively, while the WGS reaction favors lower reaction temperatures. Theoretically, the enhancement will become more obvious for catalysts with working temperatures above 200 °C as the r-WGS and methanation reactions can be suppressed more significantly. Therefore, some catalysts with high working temperatures may work better and become feasible by combination with a CO2-adsorbent. Of course, the selection of a specific CO2-adsorbent working in low, moderate or high temperature ranges is dependent on the working temperature of the selected catalyst. It is hoped that an efficient PROX process can be developed if their working temperatures are compatible. Unfortunately, so far there are still no promising results reported for this application.
6. Concluding remarks and outlook
There is a growing awareness that anthropogenic CO2 emissions should be reduced as CO2 is a major greenhouse gas in the atmosphere. For the capture, storage and utilization of CO2, solid CO2-adsorbents are widely used. These adsorbents can be roughly classified into three types according to their sorption/desorption temperatures: low-, intermediate- and high-temperature adsorbents with temperature ranges below 200 °C, between 200–400 °C and above 400 °C respectively.(1) The low-temperature CO2-adsorbents include carbon, zeolites, MOFs/ZIFs, alkali metal carbonates and amine-based materials. The first three materials adsorb CO2 mainly by physical interaction while the final two can chemically bind to CO2, and most of them have good CO2 adsorption capacities. However, for practical applications, each of them possesses pros and cons. The carbon-based materials are abundant, cheap, easy to make, and chemically and hydrothermally stable, but their selectivity for CO2 is low in the presence of other gases (e.g. N2, H2, CH4, etc.). Grafting functional amine groups to the carbon surface is a good option to solve this problem. Both zeolites and MOFs/ZIFs have a high capacity for CO2 capture, and some of them are quite stable, but usually not cheap. The main challenge for alkali metal carbonates and amine-based adsorbents lies in their long-term stability. The thermal and hydrothermal stability, especially in the presence of H2O, is a problem to support materials such as mesoporous silica. It is also worthwhile to mention that the CO2 adsorption temperature (normally < 60 °C) of these materials is slightly lower than that of real flue gases (normally ∼90 °C), and the current choice is to first cool down the flue gas, which is also energy consuming. Certain modifications should be made to these materials to increase their adsorption temperatures slightly. The amine-based solid adsorbents have high selectivity towards CO2 and other acidic gases, but their capacities are still lower than that of the corresponding liquid amines. Developing amine molecules with low volatility and high density of amino groups should be a direction to pursue.
(2) Among various intermediate-temperature CO2-adsorbents, LDHs have been well investigated and are believed to be the most promising adsorbent candidate. The main problems of this kind of material are still their insufficient adsorption capacity, kinetics and stability. Doping LDHs with K2CO3 can improve their capacity and kinetics. For LDHs, one critical issue that cannot be bypassed is their stability under real operating conditions, especially during the pressure swing adsorption/temperature swing adsorption (PSA/TSA) cycles and in the presence of H2O and SO2. For the sorption-enhanced hydrogen production reactions, the compatibility between LDHs and WGS/SR catalysts should also be considered, e.g., the temperature gap. In addition, from the viewpoint of fundamental research, the structure of the active sites for CO2 adsorption and the structural evolution of these materials during the CO2 sorption and desorption are still not very clear and should be further investigated.
(3) For the high-temperature CO2-adsorbents, CaO and alkali ceramics are representative materials. These CO2-adsorbents suffer severely from textural degradation during the sorption/desorption operations. Currently, these materials can only run several tens of cycles before any obvious degradation, and are still far from practical applications. Therefore, it is highly desirable to increase their thermal stabilities. Encapsulated or supported CaO-like or LDH adsorbents probably have higher stabilities, but the support or matrix materials must be inert to these active components. Carbon is probably a good choice.
(4) The great success in CSU of CO2 is probably determined not only by exploration of the well-known materials, but also by the discovery and synthesis of new materials that have high capacity for CO2 capture and good thermal and recycling stability. These materials may be new polymers, organic, inorganic materials or their hybrids, and they should bear a high density of basic functional groups or active sites.
(5) For catalytic applications including the WGS, SR and PROX reactions, the first two still suffer from insufficient thermal stability of the CO2-adsorbents (mainly intermediate and high temperature CO2-adsorbents) in the reaction temperature ranges. The combination of the PROX catalysts with low-temperature CO2-adsorbents is promising for practical applications, as both the process temperature and the CO2 concentration are quite low. It is expected that more CO2-adsorbent enhanced catalytic reactions will be identified and studied in the future. However, one should keep in mind that a successful process is not only a scientific issue but also an engineering issue. The proper configuration of the catalyst and the CO2-adsorbent and the whole process will play an important role, and in some cases, become a key performance indicator.
Acknowledgements
This research was supported by the Agency for Science, Technology and Research in Singapore (A*STAR, project code 0921380024). The authors would like to thank Mr Desmond Jia Wei Ng, Ms Hui Huang Tay and Jaclyn Teo for their assistance and Dr P. K. Wong and Dr Keith Carpenter for their support of this project.References
- M. R. Raupach, G. Marland, P. Ciais, C. Le Quéré, J. G. Canadell, G. Klepper and C. B. Field, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10288 CrossRef CAS.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893 CrossRef.
- M. G. Plaza, C. Pevida, B. Arias, M. D. Casal, C. F. Martín, J. Fermoso, F. Rubiera and J. J. Pis, J. Environ. Eng., 2009, 135, 426 CrossRef CAS.
- F. M. Orr, Jr., Energy Environ. Sci., 2009, 2, 449 RSC.
- R. Steeneveldt, B. Berger and T. A. Torp, Chem. Eng. Res. Des., 2006, 84, 739 CrossRef CAS.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennel, Prog. Energy Combust. Sci., 2010, 36, 260 CrossRef CAS.
- D. P. Harrison, Greenhouse Gas Control Technologies 7, 2005, 2, 1101 Search PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS.
- T. C. Dragea, O. Kozynchenkob, C. Pevidac, M. G. Plazac, F. Rubierac, J. J. Pisc, C. E. Snapea and S. Tennisonb, Energy Proc., 2009, 1, 599 Search PubMed.
- A. Arenillas, K. M. Smith, T. C. Drage and C. E. Snape, Fuel, 2005, 84, 2204 CrossRef CAS.
- S. Sircar, T. C. Golden and M. B. Rao, Carbon, 1996, 34, 1 CrossRef CAS.
- M. Cinke, J. Li, C. W. Bauschlicher Jr., A. Ricca and M. Meyyappan, Chem. Phys. Lett., 2003, 376, 761 CrossRef CAS.
- S. C. Hsu, C. Lu, F. Su, W. Zeng and W. Chen, Chem. Eng. Sci., 2010, 65, 1354 CrossRef CAS.
- C. Lu, H. Bai, B. Wu, F. Su and J. F. Hwang, Energy Fuels, 2008, 22, 3050 CrossRef CAS.
- F. Su, C. Lu, W. Cnen, H. Bai and J. F. Hwang, Sci. Total Environ., 2009, 407, 3017 CrossRef CAS.
- D. Saha and S. Deng, J. Colloid Interface Sci., 2010, 345, 402 CrossRef CAS.
- V. Yu. Yakovlev, A. A. Fomkin and A. V. Tvardovski, J. Colloid Interface Sci., 2003, 268, 33 CrossRef CAS.
- M. L. Gray, Y. Soong, K. J. Champagne Jr., P. Toochinda and S. S. C. Chuang, Fuel Chem. Div. Prepr., 2002, 47, 64 Search PubMed.
- A. Arenillas, F. Rubiera, J. B. Parra, C. O. Ania and J. J. Pis, Appl. Surf. Sci., 2005, 252, 619 CrossRef CAS.
- T. C. Drage, A. Arenillas, K. M. Smith, C. Pevida, S. Piippo and C. E. Snape, Fuel, 2007, 86, 22 CrossRef CAS.
- P. J. M. Carrott, J. M. V. Nabais, M. M. L. Ribeiro Carrott and J. A. Pajares, Carbon, 2001, 39, 1543 CrossRef CAS.
- A. Arenillas, T. C. Drage, K. Smith and C. E. Snape, J. Anal. Appl. Pyrolysis, 2005, 74, 298 CrossRef CAS.
- C. Pevida, M. G. Plaza, B. Arias, J. Fermoso, F. Rubiera and J. J. Pis, Appl. Surf. Sci., 2008, 254, 7165 CrossRef CAS.
- J. Przepiorski, M. Skrodzewicz and A. W. Morawski, Appl. Surf. Sci., 2004, 225, 235 CrossRef.
- M. Mercedes Maroto-Valer, Z. Tang and Y. Zhang, Fuel Process. Technol., 2005, 86, 1487 CrossRef CAS.
- M. L. Gray, Y. Soong, K. J. Champagne, J. Baltrus, R. W. Stevens, Jr., P. Toochinda and S. S. C. Chuang, Sep. Purif. Technol., 2004, 35, 31 CrossRef CAS.
- M. G. Plaza, C. Pevida, A. Arenillas, F. Rubiera and J. J. Pis, Fuel, 2007, 86, 2204 CrossRef CAS.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853 CrossRef CAS.
- M. Mercedes Maroto-Valer, Z. Lu, Y. Zhang and Z. Tang, Waste Manage., 2008, 28, 2320 CrossRef CAS.
- M. G. Plaza, C. Pevida, C. F. Martín, J. Fermoso, J. J. Pis and F. Rubiera, Sep. Purif. Technol., 2010, 71, 102 CrossRef CAS.
- Y. Zhang, Z. Lu, M. Mercedes Maroto-Valer, J. M. Andresen and H. H. Schobert, Energy Fuels, 2003, 17, 369 CrossRef CAS.
- V. V. Strelko, V. S. Kuts and P. A. Thrower, Carbon, 2000, 38, 1499 CrossRef CAS.
- D. Zhao, K. Cleare, C. Oliver, C. Ingram, D. Cook, R. Szostak and L. Kevan, Microporous Mesoporous Mater., 1998, 21, 371 CrossRef CAS.
- P. A. Jacobs, F. H. Van Cauwelaert, E. F. Vansant and J. B. Uytterhoeven, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 1056 RSC.
- T. Montanari and G. Busca, Vib. Spectrosc., 2008, 46, 45 CrossRef CAS.
- T. Inui, Y. Okugawa and M. Yasuda, Ind. Eng. Chem. Res., 1988, 27, 1103 CrossRef CAS.
- D. Ko, R. Siriwardane and L. T. Biegler, Ind. Eng. Chem. Res., 2003, 42, 339 CrossRef CAS.
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Microporous Mesoporous Mater., 2003, 62, 29 CrossRef CAS.
- R. Hernández-Huesca, L. Díaz and G. Aguilar-Armenta, Sep. Purif. Technol., 1999, 15, 163 CrossRef CAS.
- P. Xiao, J. Zhang, P. Webley, G. Li, R. Singh and R. Todd, Adsorption, 2008, 14, 575 CrossRef CAS.
- Z. Liang, M. Marshall and A. L. Chaffee, Energy Proc., 2009, 1, 1265 Search PubMed.
- E. Díaz, E. Muñoz, A. Vega and S. Ordoćñez, Ind. Eng. Chem. Res., 2008, 47, 412 CrossRef CAS.
- J. Pires and M. Brotas de Carvalho, J. Mol. Catal., 1993, 85, 295 CrossRef CAS.
- J. Merel, M. Clausse and F. Meunier, Ind. Eng. Chem. Res., 2008, 47, 209 CrossRef CAS.
- P. Li and F. H. Tezel, Microporous Mesoporous Mater., 2007, 98, 94 CrossRef CAS.
- S. K. Wirawan and D. Creaser, Microporous Mesoporous Mater., 2006, 91, 196 CrossRef CAS.
- B. Bonelli, B. Onida, B. Fubini, C. Otero Arean and E. Garrone, Langmuir, 2000, 16, 4976 CrossRef CAS.
- J. Zhang, R. Singh and P. A. Webley, Microporous Mesoporous Mater., 2008, 111, 478 CrossRef CAS.
- P. J. E. Harlick and F. H. Tezel, Microporous Mesoporous Mater., 2004, 76, 71 CrossRef CAS.
- R. V. Siriwardane, M. S. Shen, E. P. Fisher and J. A. Poston, Energy Fuels, 2001, 15, 279 CrossRef CAS.
- G. Calleja, J. Pau and J. A. Calles, J. Chem. Eng. Data, 1998, 43, 994 CrossRef CAS.
- K. S. Walton, M. B. Abney and M. D. LeVan, Microporous Mesoporous Mater., 2006, 91, 78 CrossRef CAS.
- T. Yamazaki, M. Katoh, S. Ozawa and Y. Ogino, Mol. Phys., 1993, 80, 313 CAS.
- S. K. Wirawan and D. Creaser, Sep. Purif. Technol., 2006, 52, 224 CrossRef CAS.
- E. Gallei and G. Stumpf, J. Colloid Interface Sci., 1976, 55, 415 CrossRef CAS.
- S. U. Rege and R. T. Yang, Chem. Eng. Sci., 2001, 56, 3781 CrossRef CAS.
- F. Brandani and D. M. Ruthven, Ind. Eng. Chem. Res., 2004, 43, 8339 CrossRef CAS.
- J. Janchena, D. T. F. Möhlmannb and H. Stach, Stud. Surf. Sci. Catal., 2007, 170, 2116.
- A. Ertan and F. Çakicioǧlu-Özkan, Adsorption, 2005, 11, 151 CrossRef.
- Y. S. Bae, K. L. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592 CrossRef CAS.
- Z. Zhao, Z. Li and Y. S. Lin, Ind. Eng. Chem. Res., 2009, 48, 10015 CrossRef CAS.
- S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2005, 127, 13519 CrossRef CAS.
- D. Li and K. Kaneko, Chem. Phys. Lett., 2001, 335, 50 CrossRef CAS.
- K. S. Walton, A. R. Millward, D. Dubbeldam, H. Frost, J. J. Low, O. M. Yaghi and R. Q. Snurr, J. Am. Chem. Soc., 2008, 130, 406 CrossRef CAS.
- J. Moellmer, E. B. Celer, R. Luebke, A. J. Cairns, R. Staudt, M. Eddaoudi and M. Thommes, Microporous Mesoporous Mater., 2010, 129, 345 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. D. Weireld, J. S. Chang, D. Y. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245 CrossRef.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637 CrossRef CAS.
- P. D. C. Dietzel, R. E. Johnsen, H. Fjellvåg, S. Bordiga, E. Groppo, S. Chavanc and R. Blom, Chem. Commun., 2008, 5125 RSC.
- P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362 RSC.
- B. Chen, M. Eddaoudi, T. M. Reineke, J. W. Kampf, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 11559 CrossRef CAS.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Remy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326 CrossRef CAS.
- B. Arstad, H. Fjellvåg, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755 CrossRef CAS.
- Y. S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131 RSC.
- A. O. Yazaydin, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425 CrossRef.
- P. L. Llewellyn, S. Bourrelly, C. Serre, Y. Filinchuk and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 7751 CrossRef CAS.
- Y. S. Bae, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2008, 4135 RSC.
- H. Hayashi, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501 CrossRef CAS.
- B. Wang, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS.
- A. Phan, C. J. Doonan, F. J. Uribe-romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS.
- O. M. Yaghi and Q. Li, MRS Bull., 2009, 34, 682 CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS.
- J. B. Lee, C. K. Ryu, J. I. Baek, J. H. Lee, T. H. Eom and S. H. Kim, Ind. Eng. Chem. Res., 2008, 47, 4465 CrossRef CAS.
- C. K. Yi, S. H. Jo, Y. Seo, S. D. Park, K. H. Moon, J. S. Yoo, J. B. Lee and C. K. Ryu, Stud. Surf. Sci. Catal., 2006, 159, 501 CAS.
- Y. Liang, D. P. Harrison, R. P. Gupta, D. A. Green and W. J. McMichael, Energy Fuels, 2004, 18, 569 CrossRef CAS.
- S. C. Lee and J. C. Kim, Catal. Surv. Asia, 2007, 11, 171 CrossRef CAS.
- C. Zhao, X. Chen and C. Zhao, Int. J. Greenhouse Gas Control, 2010, 4, 655–658 CrossRef CAS.
- V. E. Sharonov, A. G. Okunev and Y. I. Aristov, React. Kinet. Catal. Lett., 2004, 82, 363 CrossRef CAS.
- C. Zhao, X. Chen and C. Zhao, Chemosphere, 2009, 75, 1401 CrossRef CAS.
- C. Zhao, X. Chen, C. Zhao and Y. Liu, Energy Fuels, 2009, 23, 1766 CrossRef CAS.
- C. K. Yi, S. H. Jo, Y. Seo, J. B. Lee and C. K. Ryu, Int. J. Greenhouse Gas Control, 2007, 1, 31 CrossRef CAS.
- S. C. Lee, H. J. Chae, S. J. Lee, Y. H. Park, C. K. Ryu, C. K. Yi and J. C. Kim, J. Mol. Catal. B: Enzym., 2009, 56, 179 CrossRef CAS.
- C. K. Yi, S. H. Jo, H. J. Ryu, Y. W. Yoo, J. B. Lee and C. K. Ryu, Greenhouse Gas Control Technologies, 2005, 2, 1765 Search PubMed.
- H. Hayashi, J. Taniuchi, N. Furuyashiki and S. Sugiyama, Ind. Eng. Chem. Res., 1998, 37, 185 CrossRef CAS.
- S. C. Lee, B. Y. Choi, T. J. Lee, C. K. Ryu, Y. S. Ahn and J. C. Kim, Catal. Today, 2006, 111, 385 CrossRef CAS.
- A. G. Okunev, V. E. Sharonov, Y. I. Aistov and V. N. Parmon, React. Kinet. Catal. Lett., 2000, 71(2), 355 CrossRef CAS.
- R. R. Bottoms, US patent, 1783901, 1930.
- G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS.
- S. Ma'mum, R. Nilsen, H. F. Svendsen and O. Juliussen, J. Chem. Eng. Data, 2005, 50, 630 CrossRef.
- B. P. Mandal and S. S. Bandyopadhyay, Chem. Eng. Sci., 2006, 61, 5440 CrossRef CAS.
- T. Tsuda, T. Fujiwara, Y. Takteani and T. Saeguas, Chem. Lett., 1992, 21, 2161 CrossRef.
- S. Lee, T. P. Filburn, M. Gray, J. W. Park and H. J. Song, Ind. Eng. Chem. Res., 2008, 47, 7419 CrossRef CAS.
- M. Schladt, T. P. Fiburn and J. J. Helble, Ind. Eng. Chem. Res., 2007, 46, 1590 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2006, 45, 3248 CrossRef CAS.
- T. L. Chew, A. L. Ahmad and S. Bhatia, Adv. Colloid Interface Sci., 2010, 153, 43 CrossRef CAS.
- E. J. Acosta, C. S. Carr, E. E. Simanek and D. F. Shantz, Adv. Mater., 2004, 16(12), 985 CrossRef CAS.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902 CrossRef CAS.
- F. Y. Chang, K. J. Chao, H. H. Cheng and C. S. Tan, Sep. Purif. Technol., 2009, 70, 87 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 46, 446 CrossRef CAS.
- S. H. Liu, C. H. Wu, H. K. Lee and S. B. Liu, Top. Catal., 2010, 53, 210 CrossRef CAS.
- P. Li, B. Ge, S. Zhang, S. Chen, Q. Zhang and Y. Zhao, Langmuir, 2008, 24, 6567 CrossRef CAS.
- Z. Liang, B. Fadhel, C. J. Schneidar and A. L. Chaffee, Microporous Mesoporous Mater., 2008, 111, 536 CrossRef CAS.
- J. H. Drese, S. Choi, R. P. Lively, W. J. Koros, D. J. Fauth, M. L. Gray and C. W. Jones, Adv. Funct. Mater., 2009, 19, 3821 CrossRef CAS.
- A. Simon, T. Cohen-Bouhacina, M. C. Porté, J. P. Aimé and C. Baquey, J. Colloid Interface Sci., 2002, 251, 278 CrossRef CAS.
- Y. Tang and K. Landskron, J. Phys. Chem. C, 2010, 114, 2494 CrossRef CAS.
- P. D. Vaidya and E. Y. Kenig, Ind. Eng. Chem. Res., 2008, 47, 34 CrossRef CAS.
- H. Lepaumier, D. Picq and P. L. Carrette, Ind. Eng. Chem. Res., 2009, 48, 9068 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- G. R. Williams and D. O'Hare, J. Mater. Chem., 2006, 16, 3065 RSC.
- V. Rives and S. Kannan, J. Mater. Chem., 2000, 10, 489 RSC.
- Y. Ding and E. Alpay, Trans IChemE, 2001, 79, 45 Search PubMed.
- Z. Yong and A. E. Rodrigues, Energy Convers. Manage., 2002, 43, 1865 CrossRef CAS.
- H. T. J. Reijers, S. E. A. Valster-Schiermeier, P. D. Cobden and R. W. van den Brink, Ind. Eng. Chem. Res., 2006, 45, 2522 CrossRef CAS.
- M. S. San Román, M. J. Holgado, C. Jaubertie and V. Rives, Solid State Sci., 2008, 10, 1333 CrossRef CAS.
- W. Yang, Y. Kim, P. K. T. Liu, M. Sahimi and T. T. Tsotsis, Chem. Eng. Sci., 2002, 57, 2945 CrossRef CAS.
- M. K. Ram Reddy, Z. P. Xu, G. Q. (Max) Lu and J. C. Diniz da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504 CrossRef CAS.
- M. K. Ram Reddy, Z. P. Xu, G. Q (Max) Lu and J. C. Diniz da Costa, Ind. Eng. Chem. Res., 2008, 47, 2630 CrossRef.
- A. L. Mackenzie, C. T. Fishel and R. J. Davis, J. Catal., 1992, 138, 547 CrossRef.
- T. Horiuchi, H. Hidaka, T. Fukui, Y. Kubo, M. Horio, K. Suzuki and T. Mori, Appl. Catal., A, 1998, 167, 195 CrossRef CAS.
- U. Sharma, B. Tyagi and R. V. Jasra, Ind. Eng. Chem. Res., 2008, 47, 9588 CrossRef CAS.
- M. K. Ram Reddy, Z. P. Xu, G. Q. (Max) Lu and J. C. Diniz da Costa, Ind. Eng. Chem. Res., 2008, 47, 7357 CrossRef.
- S. P. Reynolds, A. D. Ebner and J. A. Ritter, Ind. Eng. Chem. Res., 2006, 45, 4278 CrossRef CAS.
- Y. Ding and E. Alpay, Chem. Eng. Sci., 2000, 55, 3461 CrossRef CAS.
- E. L. G. Oliveira, C. A. Grande and A. E. Rodrigues, Sep. Purif. Technol., 2008, 62, 137 CrossRef CAS.
- A. D. Ebner, S. P. Reynolds and James A. Ritter, Ind. Eng. Chem. Res., 2006, 45, 6387 CrossRef CAS.
- K. B. Lee, A. Verdooren, H. S. Caram and S. Sircar, J. Colloid Interface Sci., 2007, 308, 30 CrossRef CAS.
- S. Walspurger, L. Boels, P. D. Cobden, G. D. Elzinga, W. G. Haije and R. W. van den Brink, ChemSusChem, 2008, 1, 643 CrossRef CAS.
- M. Dadwhal, T. Kim, M. Sahimi and T. T. Tsotsis, Ind. Eng. Chem. Res., 2008, 47, 6150 CrossRef CAS.
- M. R. Othman, N. M. Rasid and W. J. N. Fernando, Chem. Eng. Sci., 2006, 61, 1555 CrossRef CAS.
- N. N. A. H. Meis, J. H. Bitter and K. P. de Jong, Ind. Eng. Chem. Res., 2010, 49, 1229 CrossRef CAS.
- N. D. Hutson and B. C. Attwood, Adsorption, 2008, 14, 781 CrossRef CAS.
- Y. Lwin and F. Abdullah, J. Therm. Anal. Calorim., 2009, 97, 885 CrossRef CAS.
- X. P. Wang, J. J. Yu, J. Cheng, Z. P. Hao and Z. P. Xu, Environ. Sci. Technol., 2008, 42, 614 CrossRef CAS.
- C. T. Yavuz, B. D. Shinall, A. V. Iretskii, M. G. White, T. Golden, M. Atilhan, P. C. Ford and G. D. Stucky, Chem. Mater., 2009, 21, 3473 CrossRef CAS.
- Q. Wang, H. H. Tay, D. J. W. Ng, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, ChemSusChem, 2010, 3, 965 CrossRef CAS.
- B. Feng, H. An and E. Tan, Energy Fuels, 2007, 21, 426 CrossRef CAS.
- S. J. Gregg and J. D. Ramsay, J. Chem. Soc. A, 1970, 2784 RSC.
- M. Bhagiyalakshmi, J. Y. Lee and H. T. Jang, Int. J. Greenhouse Gas Control, 2010, 4, 51 CrossRef CAS.
- N. H. Florin and A. T. Harris, Chem. Eng. Sci., 2009, 64, 187 CrossRef CAS.
- D. P. Harrison, Ind. Eng. Chem. Res., 2008, 47, 6486 CrossRef CAS.
- N. Florin, Chem. Eng. Sci., 2008, 63, 287 CrossRef CAS.
- E. Gorin, US Patent No. 3188179 ( 1965).
- T. Shimizu, T. Hirama, H. Hosoda, K. Kitano, M. Inagaki and K. Tejima, Chem. Eng. Res. Des., 1999, 77, 62 CrossRef CAS.
- J. C. Abanades, E. J. Anthony, J. Wang and J. E. Oakey, Environ. Sci. Technol., 2005, 39, 2861 CrossRef CAS.
- J. C. Abanades, G. Grasa, M. Alonso, N. Rodriguez, E. J. Anthony and L. M. Romeo, Environ. Sci. Technol., 2007, 41, 5523 CrossRef CAS.
- A. MacKenzie, D. L. Granatstein, E. J. Anthony and J. C. Abanades, Energy Fuels, 2007, 21, 920 CrossRef CAS.
- J. C. Abanades, E. S. Rubin and E. J. Anthony, Ind. Eng. Chem. Res., 2004, 43, 3462 CrossRef CAS.
- D. Alvarez and J. C. Abanades, Ind. Eng. Chem. Res., 2005, 44, 5608 CrossRef CAS.
- S. K. Bhatia and D. D. Perlmutter, AIChE J., 1983, 29, 79 CrossRef CAS.
- A. Silaban and D. P. Harrison, Chem. Eng. Commun., 1995, 137, 177 CrossRef CAS.
- P. Sun, J. R. Grace, C. J. Lim and E. J. Anthony, AIChE J., 2007, 53, 2432 CrossRef CAS.
- J. C. Abanades and D. Alvarez, Energy Fuels, 2003, 17, 308 CrossRef CAS.
- H. Lu, A. Khan, S. E. Pratsinis and P. G. Smirniotis, Energy Fuels, 2009, 23, 1093 CrossRef CAS.
- L. Li, D. L. King, Z. Nie and C. Howard, Ind. Eng. Chem. Res., 2009, 48, 10604 CrossRef CAS.
- A. I. Lysikov, A. N. Salanov and A. G. Okunev, Ind. Eng. Chem. Res., 2007, 46, 4633 CrossRef CAS.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2008, 42, 4170 CrossRef CAS.
- E. H. Baker, J. Chem. Soc., 1962, 464 RSC.
- L. Jia, R. Hughes, D. Lu, E. J. Anthony and I. Lau, Ind. Eng. Chem. Res., 2007, 46, 5199 CrossRef CAS.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2007, 41, 1420 CrossRef CAS.
- P. S. Fennell, J. F. Davidson, J. S. Dennis and A. N. Hayhurst, J. Energy Inst., 2007, 80, 116 Search PubMed.
- P. S. Fennell, R. Pacciani, J. S. Dennis, J. F. Davidson and A. N. Hayhurst, Energy Fuels, 2007, 21, 2072 CrossRef CAS.
- P. Sun, J. R. Grace, C. J. Lim and E. J. Anthony, Ind. Eng. Chem. Res., 2008, 47, 2024 CrossRef CAS.
- K. Laursen, W. L. Duo, J. R. Grace and C. J. Lim, Fuel, 2001, 80, 1293 CrossRef CAS.
- Z. Chen, H. S. Song, M. Portillo, C. J. Lim, J. R. Grace and E. J. Anthony, Energy Fuels, 2009, 23, 1437 CrossRef CAS.
- J. E. Readman and R. Blom, Phys. Chem. Chem. Phys., 2005, 7, 1214 RSC.
- K. O. Albrecht, K. S. Wagenbach, J. A. Satrio, B. H. Shanks and T. D. Wheelock, Ind. Eng. Chem. Res., 2008, 47, 7841 CrossRef CAS.
- M. Aihara, T. Nagai, J. Matsushita, Y. Negishi and H. Ohya, Appl. Energy, 2001, 69, 225 CrossRef CAS.
- Z. S. Li, N. S. Cai, Y. Y. Huang and H. Han, Energy Fuels, 2005, 19, 1447 CrossRef CAS.
- Z. S. Li, N. S. Cai and Y. Y. Huang, Ind. Eng. Chem. Res., 2006, 45, 1911 CrossRef CAS.
- C. S. Martavaltzi and A. A. Lemonidou, Ind. Eng. Chem. Res., 2008, 47, 9537 CrossRef CAS.
- B. Feng, W. Liu, X. Li and H. An, Energy Fuels, 2006, 20, 2417 CrossRef CAS.
- H. Lu, E. P. Reddy and P. G. Smirniotis, Ind. Eng. Chem. Res., 2006, 45, 3944 CrossRef CAS.
- H. Lu, A. Khan and P. G. Smirniotis, Ind. Eng. Chem. Res., 2008, 47, 6216 CrossRef CAS.
- D. Alvarez, M. Pena and A. G. Borrego, Energy Fuels, 2007, 21, 1534 CrossRef CAS.
- H. J. Ryu, J. R. Grace and C. J. Lim, Energy Fuels, 2006, 20, 1621 CrossRef CAS.
- A. Bosoaga, O. Masek and J. E. Oakey, Energy Proc., 2009, 1, 133 Search PubMed.
- J. Ewing, D. Beruto and A. W. Searcy, J. Am. Ceram. Soc., 1979, 62, 580 CrossRef CAS.
- K. Nakagawat and T. Ohashi, J. Electrochem. Soc., 1998, 145, 1344 CrossRef CAS.
- E. Ochoa-Fernández, H. K. Rusten, H. A. Jakobsen, M. Rønning, A. Holmen and D. Chen, Catal. Today, 2005, 106, 41 CrossRef CAS.
- H. Pfeiffer, C. Vázquez, V. H. Lara and P. Bosch, Chem. Mater., 2007, 19, 922 CrossRef CAS.
- J. I. Ida and Y. S. Lin, Environ. Sci. Technol., 2003, 37, 1999 CrossRef CAS.
- B. N. Nair, T. Yamaguchi, H. Kawamura and S. I. Nakao, J. Am. Ceram. Soc., 2004, 87, 68 CrossRef CAS.
- E. Ochoa-Fernández, M. Rønning, T. Grande and D. Chen, Chem. Mater., 2006, 18, 1383 CrossRef CAS.
- E. Ochoa-Fernández, M. Rønning, T. Grande and D. Chen, Chem. Mater., 2006, 18, 6037 CrossRef CAS.
- A. López-Ortiz, N. G. P. Rivera, A. R. Rojas and D. L. Gutierrez, Sep. Sci. Technol., 2005, 39, 3559 CrossRef.
- H. Pfeiffer, E. Lima and P. Bosch, Chem. Mater., 2006, 18, 2642 CrossRef CAS.
- M. Y. Veliz-Enriqueza, G. Gonzalezb and H. Pfeiffer, J. Solid State Chem., 2007, 180, 2485 CrossRef.
- M. E. Bretado, V. G. Velderrain, D. L. Gutiérrez, V. GutiérrezCollins-Martínez and A. L. Ortiz, Catal. Today, 2005, 107–108 863.
- M. Kato, S. Yoshikawa and K. Nakagawa, J. Mater. Sci. Lett., 2002, 21, 485 CrossRef CAS.
- C. Gauer and W. Heschel, J. Mater. Sci., 2006, 41, 2405 CrossRef CAS.
- K. Essaki, M. Kato and H. Uemoto, J. Mater. Sci., 2005, 40, 1573.
- M. T. Rodriguez and H. Pfeiffer, Thermochim. Acta, 2008, 473, 92 CrossRef CAS.
- M. Wang and C. G. Lee, Energy Convers. Manage., 2009, 50, 636 CrossRef CAS.
- L. M. Palacios-Romero and H. Pfeiffer, Chem. Lett., 2008, 37, 862 CrossRef CAS.
- Q. Zhang, Y. Z. Zuo, M. H. Han, J. F. Wang, Y. Jin and F. Wei, Catal. Today, 2010, 150, 55 CrossRef CAS.
- C. Song and W. Pan, Catal. Today, 2004, 98, 463 CrossRef CAS.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tang, ChemSusChem, 2008, 1, 893 CrossRef.
- H. Chen, T. Zhang, B. Dou, V. Dupont, P. Williams, M. Ghadiri and Y. Ding, Int. J. Hydrogen Energy, 2009, 34, 7208 CrossRef CAS.
- J. D. Figuero, T. Fout, S. Plasynski, H. Mcilvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9 CrossRef CAS.
- M. van den Broek, R. Hoefnagels, E. Rubin, W. Turkenburk and A. Faaij, Prog. Energy Combust. Sci., 2009, 35, 457 CrossRef CAS.
- E. Ochoa-Fernández, G. Haugen, T. Zhao, M. Rønning, I. Aartun, B. Børesen, E. Rytter, M. Rønnekleiv and D. Chen, Green Chem., 2007, 9, 654 RSC.
- H. K. Rusten, E. E. Ochoa-Fernández, D. Chen and H. A. Jakobsen, Ind. Eng. Chem. Res., 2007, 46, 4435 CrossRef CAS.
- A. A. A. Solieman, J. W. Dijkstra, W. G. Haije, P. D. Cobden, R. W. van den and Brink, Int. J. Greenhouse Gas Control, 2009, 3, 393 CrossRef CAS.
- M. G. Beaver, H. S. Caram and S. Sircar, Int. J. Hydrogen Energy, 2009, 34, 2972 CrossRef CAS.
- E. R. van Selow, P. D. Cobden, P. A. Verbraeken, J. R. Hufton and R. W. van den Brink, Ind. Eng. Chem. Res., 2009, 48, 4148.
- B. Acharya, A. Dutta and P. Basu, Int. J. Hydrogen Energy, 2010, 35, 1582 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |