Silicon speciation by hyphenated techniques for environmental, biological and industrial issues: A review†
Fabien
Chainet
a,
Charles-Philippe
Lienemann
a,
Marion
Courtiade
a,
Jérémie
Ponthus
a and
Olivier François
Xavier Donard
b
aPhysics and Analysis Division, IFP Energies Nouvelles-Lyon, F-69360, Solaize
bLCABIE-IPREM, UMR 5254, CNRS-UPPA, Hélioparc, 2 av. Pr. Angot, 64053, Pau
First published on 8th December 2010
Abstract
Silicon speciation in environmental, biological and industrial matrices is of considerable importance due to its wide use in many consumer and personal care products and industry. In addition, the entry of silicones in various compartments like wastes, soils, air and water highlights the need to perform exposure studies, toxicological surveys and to measure negative effects. Due to possible contamination and trace level presence of silicon compounds, challenges to determination, identification and quantification are presented. The principal species of concern include siloxanes, silanols, silanediols and silanes. State of the art of analytical methods for total silicon determination and silicon speciation are established. Atomic spectroscopic methods are mainly used to measure total Si at trace concentration levels. On the opposite, hyphenated techniques are performed for Si speciation. Particular attention is paid to chromatographic methods coupled to sensitive and selective detectors (MS, AED and ICP) allowing structural information. Liquid and gas chromatography emerge as the most widespread separation techniques. However, other procedures such as MS, NMR, IR and XRF enable a better knowledge of these species. The potential and limitations of hyphenated techniques are highlighted, particularly concerning sensitivity and selectivity. Furthermore, potential sources of contamination and analytical artifacts in silicon determination are reviewed.
 F. Chainet | Fabien Chainet graduated from the University of Orleans with a master degree of Chemical Pollution and Environmental Risk (CPRE) in 2009. He is a Ph.D. student working on ultra and trace analysis in petroleum and derived products and more precisely in silicon speciation in hydrotreatment feeds at IFP Energies Nouvelles. |
 C.-P. Lienemann | Charles-Philippe Lienemann graduated from the University of Geneva, Switzerland, in 1993 and obtained his Ph.D. from the University of Lausanne, Switzerland, in 1997. He then worked at the Wolff Environment Labs in Lyon, France, before joining the IFP Energies Nouvelles in 2000 as the head of the elemental analysis lab. His research is focused on the behaviour, fate and analysis of trace metals in petroleum and related samples. |
 M. Courtiade | Marion Courtiade graduated from the School of Chimie Physique and Electronique (CPE) in 2004. She obtained her Ph.D. from Lyon University, which deals with trace analysis of pesticides in the environment. Since 2007, Dr Courtiade has been at the head of the GC laboratory at the IFP Energies Nouvelles and is an expert in characterization by comprehensive gas chromatography. |
 J. Ponthus | Jérémie Ponthus graduated in 2002 and obtained his Ph.D. dealing with Metastable Atom Bombardment Mass Spectrometry for petroleum related molecules characterisation from the University of Paris (UPMC), France, in 2005. He then joined the IFP Energies Nouvelles where he is now at the head of the Mass Spectrometry laboratory. His research is focused on the FT/MS analysis of complex mixtures from heavy crude oil, biomass and coal. |
 O. F. X. Donard | Olivier François Xavier Donard is a research director at the French CNRS (Centre National de la Recherche Scientifique). He is the vice president of the analytical chemistry division of the French Société Chimique de France. His research interests are related to the speciation and isotopic signature of elements in the environment, and industrial processes and their environmental chemistry. |
Introduction
Organosilicon compounds such as siloxanes and polydimethylsiloxanes (PDMS) are widely used in a variety of industrial applications and consumer goods since their commercial introduction in 1943.1 These molecules find applications in a wide array of common consumer products such as cosmetics, textiles, medicinal implants, pharmaceuticals, chemistry and in industry (lubricants, coatings, gels, adhesives, antifoaming agent…).2–4Silicon species used in these applications are polymeric silicon organo compounds formulation consisting of a backbone of alternating Si–O units with organic side chains attached to each silicon atom.3 These structures provide the species with unique high thermal stability, low surface tension, hydrophobicity, electric insulation and lubrification properties.4 These specificities resulted in the fact that some siloxanes have been identified as high production volume (HPV) chemicals by the US Environmental Protection Agency (USEPA)5 and by the Organisation for Economic Co-operation and Development (OECD).6 HPV products are defined as compounds produced or exported exceeding 1000 tons per year in at least a region or a country.6 As a result of their ever wide usage, these different products are dispersed in the environment and are present in a large variety of biological and industrial matrices (petroleum and derived petroleum products) most frequently at trace levels. Their ubiquitous occurrence in the environment requires the silicon species to be investigated to assess their environmental fate.3,7 Both the ecological8 and toxicological impact of the silicon9,10 have been recognized to be of concern, therefore, it is necessary to carry out exposure and effect studies of the species.11–13 In order to improve the general knowledge of the occurrence, pathways and toxicity assessment of silicon species, speciation of silicon and their species must be undertaken to improve the understanding of their impact and translocation in the environment.
According to the International Union of Pure and Applied Chemistry (IUPAC), speciation is defined as the analytical process that leads to measure the distribution of an element specific chemical species in a sample.14 Silicon speciation is gaining in interest and addresses the identification and quantification of the physical and chemical forms of silicon.15
The developing domain of silicon speciation is being reviewed and Fig. 1 presents the number of publications for Si determination and speciation in its different application domains. This figure shows that environment, medicine, biology, polymer synthesis and petroleum applications are the most investigated fields of interest. Two main books deal with the analytical chemistry of silicones1,16 but more specifically on total Si determination. Also a review was published in 2006 by Varaprath et al. on silicone analysis and its artifacts in environmental and biological samples.17
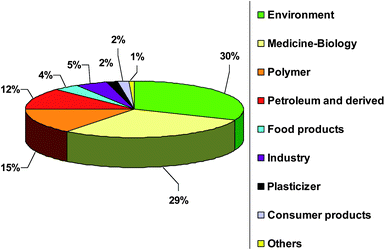 | ||
| Fig. 1 Publication distribution in percentage for Si determination and speciation in different application domains. | ||
However, silicon speciation as such is not well developed and the number of articles is small. Silicon is in general present at very low concentrations in most of the investigated matrices.18,19 Further, contamination problems during the entire analytical procedure20 as well as in the artifacts,21,22 generates a real analytical challenge. The reason being that due to their widespread use of silicon species in many products, their stability and of the high reactivity of certain silicon species which may alter the analytical process. Therefore, a wide variety of high-performance analytical techniques have been developed and applied. Fig. 2 summarises the results of a general survey of more than 120 articles published over the last 30 years dealing with total silicon determination methods as well as the hyphenated techniques applied for silicon speciation.
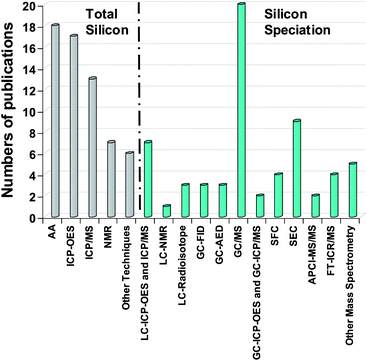 | ||
| Fig. 2 Number of publications for Silicon determination and speciation by different methods. | ||
The objective of this article is to review the main analytical methods used for silicon determination and silicon speciation in environmental, biological and industrial applications. One chapter is focused on contaminations and analytical artifacts which in the case of silicon is of special concern for the validity of the data and has been a major drawback for the development of this analytical domain. In general, direct analytical methods based on atomic spectroscopic methods (AAS, ICP-OES and ICP-MS) and also NMR techniques give access to total silicon measurement. All the direct spectrometric methods do not provide information about silicon speciation (Fig. 2). Silicon speciation relies on hyphenated techniques combining the high separation potential of chromatography (e.g., LC or GC) with identification possibilities offered by mass spectrometry (MS) or the sensitivity and selectivity of an element specific detector (AED, ICP-OES and ICP-MS). This review is mainly focused on silicon speciation by hyphenated techniques particularly at trace concentrations compared to classical total determination methods for silicon. Special attention is given to the contamination problems encountered during the entire analytical process which in this case is of particular relevance for the data quality.
1 Molecules, fields of interest and analytical methods
Organosilicon compounds
The present paper deals with siloxanes (cyclic and linear), silanols (trimethylsilanol), silanediols (dimethylsilanediol) and silanes. These correspond to the four main families of organosilicon species23 (Table 1) found in the fields of interest reported in Fig. 1. As siloxanes have long names, abbreviations created by Hurd24 are employed for several species such as Dn (Table 1). Siloxanes are typically represented by the following letters “M, D, T, Q” which correspond to the number of oxygen atoms linked to an atom of silicon. Indeed, M indicates a silicon linked to one oxygen, while D, T, Q indicate a silicon linked to two, three and four oxygens respectively. Moreover, Table 1 lists formula, molecular mass, boiling point and structure of species according to the fields of interest presented in Fig. 1.| Silicon species (Hurd Abbreviations) | Formula | M M/g mol−1 | B.P/°C | Structure | Fields of Interest a |
|---|---|---|---|---|---|
| Siloxanes | |||||
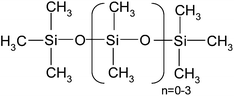
| Polydimethylsiloxane (MDnM) | PDMS viscosity: 12,500 cSt | 67,700 | — |
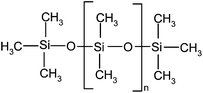
|
EN |
| BI | |||||
| Hexamethylcyclotrisiloxane (D3) | C6H18O3Si3 | 222 | 134 |
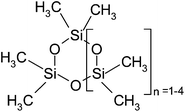
|
BF |
| Octamethylcyclotetrasiloxane (D4) | C8H24O4Si4 | 296 | 175 | ||
| Decamethylcyclopentasiloxane (D5) | C10H30O5Si5 | 370 | 211 | CP | |
| Dodecamethylcyclohexasiloxane (D6) | C12H36O6Si5 | 382 | 245 | ||
| Hexamethyldisiloxane (MM) or L2 | C6H18OSi2 | 150 | 100 |
|
P |
| Octamethyltrisiloxane (MDM) or L3 | C8H24O2Si3 | 236 | 153 | ||
| Decamethyltetrasiloxane (MD2M) or L4 | C10H30O3Si4 | 310 | 194 | PP | |
| Dodecamethylpentasiloxane (MD3M) or L5 | C12H36O4Si5 | 382 | 210 | ||
| Silanols | |||||
|---|---|---|---|---|---|
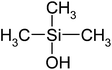
| Trimethylsilanol (TMS) | C3H10SiO | 90 | 99 |
|
EN |
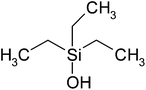
| Triethylsilanol (TES) | C6H16Osi | 132 | 158 |
|
PP |
| Silanediols | |||||
|---|---|---|---|---|---|
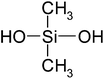
| Dimethylsilanediol (DMSD) | C2H8O2Si | 92 | 102 |
|
EN |
| PP | |||||
| Tetramethyl-1,3-disiloxanediol (TMSD) | C4H14O3Si2 | 166 | — |
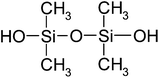
|
EN |
| Silanes | |||||
|---|---|---|---|---|---|
| a EN: Environmental samples (air, soil, water, sediments); BI: Breast implants; BF: Biological fluids; CP: Consumer products; P: Polymers; PP: Petroleum and derived products. | |||||
| Tetramethylsilane | C4H12Si | 88 | 27 |
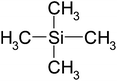
|
EN |
| PP | |||||
| Triethylsilane | C6H16Si | 116 | 107 |
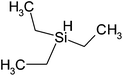
|
PP |
| Diethoxydimethylsilane | C5H14O2Si | 134 | 95 |
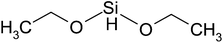
|
PP |
1.2 Fields of interest
PDMS, defined as a high molecular weight (HWM) polymer is the most important silicone used for industrial and consumer applications (80%).3 Other silicones are mainly of low molecular weight (LMW) materials, also referred to as volatile methyl siloxanes (VMS) with significant vapour pressures under ambient environmental conditions.13,25 Consequently, the behaviour of HMW and LMW siloxanes has gained in interest in many matrices. The following paragraphs report problems caused by these species.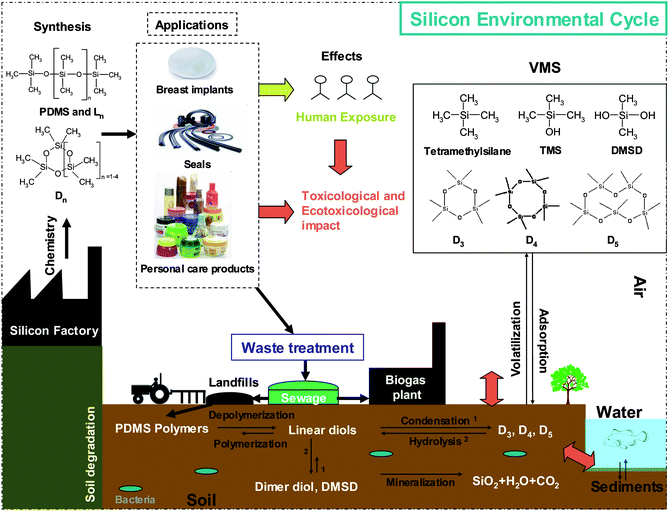 | ||
| Fig. 3 Fate of silicon in the environment. | ||
LMW hydrolysis products are water soluble and volatile. Thus, these induce silicon molecules which can partition from the soil to the water and into the atmosphere (Fig. 3).7 In addition, combustion of silicon containing biogas produces the abrasive silica and causes serious damages to gas engines,2,43 heat exchangers,44 and catalytic exhaust gas treatment.45
Long et al. have developed a novel on-site method for siloxane detection in biogas based on microcantilever array use.46 Performance like high sensitivity, portability, inexpensive and less energy consuming than GC-MS promise to facilitate in-field siloxane analysis and reduce biogas cost.46 Several applications are performed to reduce and eliminate silicon species in biogas.47,48 Silicon speciation in the environment contributes to estimate the environmental fate and the ecological and toxicological risks occurred by siloxane compounds in vulnerable ecosystems (Fig. 3).8
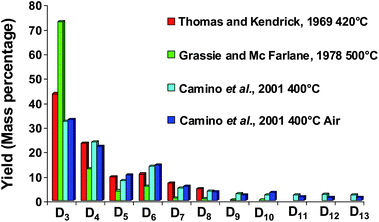 | ||
| Fig. 4 Low molecular cyclic siloxane (Dn) distribution generated during PDMS degradation at different temperatures (Mass percentage). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 damaged cars due to the formation of a silica layer on an O2 probe in automobile sensors.
000 damaged cars due to the formation of a silica layer on an O2 probe in automobile sensors.
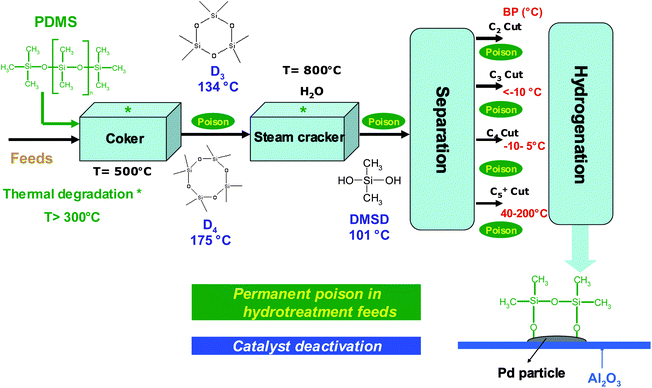 | ||
| Fig. 5 Silicon poisoning in petroleum process. | ||
Silicon presence in feedstocks is derived from antifoaming agents (PDMS), added in the lighter fractions of coker or visbreaker operations.67,68Fig. 5 presents a petroleum process constituted of feed thermal cracking (Coker and Steam cracker), separation and hydrogenation units. Due to the PDMS degradation, silicon compounds (Table 1) are found at trace levels69,70 in feeds and occur as poison which induce severe catalytic deactivation by adsorption on the catalyst surface (Fig. 5).68,71–75 According to theoretical explanations and GC analysis of heated silicon oil, several authors suggest that cyclic oligomers (Dn) are the major breakdown products of PDMS released in petroleum products.67,76 These explanations are in agreement with results presented in Fig. 458–60 due to the high temperatures in petroleum processes.67 According to Breivik and Egebjerg cyclosiloxanes are rapidly adsorbed on the catalyst surface of hydrotreatment and caused catalyst deactivation.76 In addition, Molnar et al. demonstrated a poisoning of olefin hydrogenation by triethylsilane between 200 and 400 °C.75 Furthermore, recent studies have shown poisoning by hexamethyldisiloxane (L2) used as a model poison compound during combustion of volatile organic compounds (VOC) in gas treatment.45,77–79 These phenomena give rise to untimely catalyst replacement and economic loss. Consequently, the petroleum industry must continuously propose sensitive analytical methods in order to reach the specifications in petroleum products.80 As far as demonstrated in the literature, only a total determination of Si is achieved in petroleum matrices.81,82 However, speciation (identification and quantification) is necessary to give access to the chemical nature of poison species and hence, a better understanding of the poisoning processes.
1.3 Analytical methods for silicon determination and speciation
As previously mentioned, silicon compounds result in numerous problems in the environment (fate and exposure), medicine and biology (breast implants), consumer products (exposure, toxicity), polymers (stability) and catalysts (poisons) in petroleum processes and gas treatment. Consequently, two different groups of techniques used for silicon determination, are presented in Fig. 6. These techniques consisted of direct analysis to achieve total silicon determination as well as, hyphenated techniques for silicon speciation. Before selecting an analytical method, it is essential to define the matrix, the species and compound concentration ranges especially at trace levels.17 Generally, detection limits greatly depend on the preparation and one concentration. Thus, comparison between different technique performances is difficult.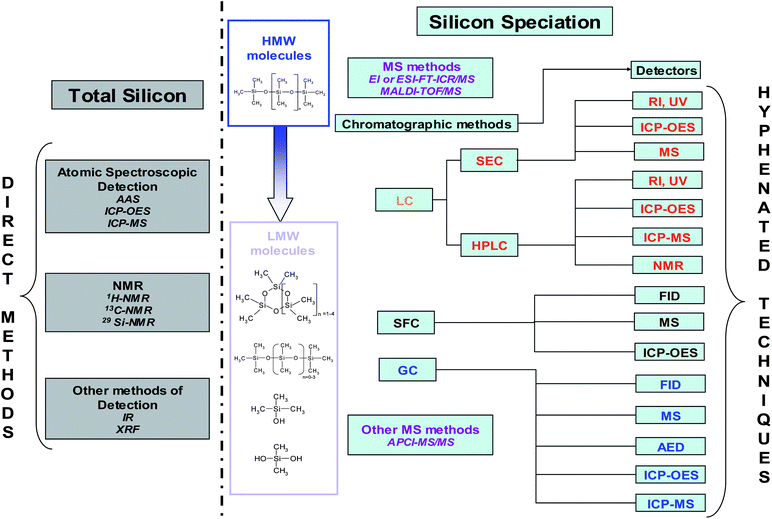 | ||
| Fig. 6 Analytical methods for silicon determination and speciation. | ||
Most total silicon determinations are performed by atomic spectroscopy, NMR and other methods by infra-red spectroscopy (IR) and X-Ray fluorescence (XRF) (Fig. 2). For silicon speciation, MS methods and SEC separation have been usually employed for HMW molecules. On the other hand, the methods on the right side of Fig. 6 are used for LMW compounds. Silicon speciation requires chromatographic separation (LC, SFC and GC) coupled to a suitable detector depending on the expected detection limit. The survey presented in Fig. 2 clearly shows that GC-MS is the most commonly employed hyphenated technique for trace level Si speciation. Flame ionisation detector (FID) can also be applied but it does not provide very low detection limits. The coupling of GC and atomic detection such as AED or ICP (inductively coupled plasma) allows high sensitivity and selectivity therefore, giving access to structural information. The combination of LC and ICP is also used for the determination of a wide variety of silicon compounds as well. MS methods can also be applied to LMW compounds (Fig. 6) providing identification compared to direct methods.
2 Direct methods for total silicon determination
Total silicon can be determined by atomic spectroscopic methods and NMR (Fig. 2 and Fig. 6). The major techniques employed for silicon trace analysis are AAS, ICP-OES and ICP-MS. This chapter is mainly focused on environmental, biological, crude oil and derived samples and more particularly on papers published since 2006 for the first two fields. Other studies in environmental and biological fields have also been reviewed in The Analytical Chemistry of Silicones16 and Organosilicon Materials1 but also in a comprehensive review on the analytical aspects of silicones published in 2006.172.1 Atomic spectroscopic methods
These methods can be applied with a decomposition step before silicon determination in order to use it in aqueous matrices or directly by injection of organic phases without initial sample digestion. This preparation step is mainly used for solid samples83–87 and to limit carbon introduction in the plasma69 allowing minimum mass interferences especially with ICP-MS.The detection of organosilicon materials in river sediments has been reported by AAS after extraction and concentration.36 More recently, Mukhtar and Limbeck developed an accurate procedure for trace silicon detection in solid environmental samples such as soils or airborne particulate matter using GFAAS.88 Based on a preliminary treatment of samples leading to mineralization, and with the use of 20 μL sample injection volume, an instrumental LOD of 52 μg L−1 was obtained, which translated to method detection limits of approximately 0.52 μg m−3 when considering collected air volumes.88
AAS is also applied in medical fields in order to measure concentrations in tissues, plasma and human blood exposed to breast implants.89 Indeed, the concentration of Si in blood of women with silicone gel-filled breast implants was found to be double (33.5/17.1 μg L−1) that a women with no implants.90 Lugowski et al. detected Si in blood and urine of individuals exposed to breast implants91 and in human tissues20 with μg L−1 levels by GFAAS. Detection limit (2SD) of 0.5 mg kg−1 of tissue was obtained by GFAAS after heptane extraction.92 GFAAS has also been employed for silicon measurement in biological tissues as well.93–96 Hornung et Krivan showed that pre-ashing solid samples improved detection limits (0.2 to 0.03 mg kg−1), sample homogeneity and precision compared to direct sampling by this previous technique.95 Moreover, a sensitive (LOD of 1.5 μg L−1), simple and accurate method for the routine determination of trace silicon by Zeeman GFAAS has been reported.97
The first technique used in metal trace analysis of petroleum products is GFAAS with detection limits usually reported in the range of 10 μg L−1.69 Amaro and Ferreira determined silicon determination in naphta (C4–C15) by GFAAS after sample dilution in toluene.81 An experimental design has been achieved to determine optimal conditions for Si determination to obtain detection limit of 15 μg L−1.81 Nevertheless, problems related to the formation of thermally stable silicon carbide (SiC(s)) and volatile SiO(s) during pyrolysis by GFAAS can occur.81,98–100 These constraints can be resolved by using chemical modifiers83,101,102 or treated Zr tubes88 to avoid analyte reactions with graphite.
Finally, several studies in food products102–104 with detection limits ranging between 7 μg L−1 and 1.8 mg L−1 and industrial applications (LOD of 30 μg kg−1)83 have been carried out by AAS with pre-digestion in order to measure silicon levels.
ICP-OES is a useful tool for environmental samples105,106 such as water107 and agriculture matrices.108 For instance, the detection of total silicon has been achieved in rivers and sediments by ICP-OES after an extraction of organosiloxanes in a mixture of petroleum ether/MIBK with detection limits of 100 μg L−1109 and 10 μg kg−1110 of Si respectively. Masson et al. reported quantification of Si in plant samples after digestion using samples (2 mg) issued from an inter-laboratory test with detection limit of 30 μg kg−1.105 More recently, silicon concentration was measured by this technique in marine sediments as a function of pH and salinity.106 The results have shown a reduction of silicon levels while the pH increase.
This spectroscopic method was also applied for silicon determination in biological samples such as urine,111,112 blood,112 tissues and organs,84,85 plasma112 and serum.112 Detection limits of 2 mg kg−1 and 3.7 mg kg−1 obtain by ICP-OES for tissue after digestion using 0.1 g of sample have been reported by Hauptkorn et al. and McConnell et al. respectively.84,85 In 2003, Jia et al. obtained a detection limit of 0.2 mg kg−1 during the analysis of silicone oil extracted in a mixture of toluene/ACN in pharmaceutical matrices by ICP-OES coupled with an ultrasonic nebulization.113 Low silicon concentrations in foods and soils are determined using the association of a microwave dissolution and an ICP-OES.114 Detection limits have been improved by a factor of 2 with the incorporation of tertiary amines in the acid digestion procedure.114 Silicon measurement was also achieved by ICP-OES after microwave acid digestion of foods and beverages due to the importance of Si in bone formation and connective tissue metabolism.115
In 1988, Carduner et al. determined the presence of silicon in petroleum samples and more particularly in unleaded gasolines by ICP-OES with detection limit of 100 μg L−1 for Si.66 Botto reported LOD and RSD precision for Si by ICP-OES using two nebulization systems pneumatic and ultrasonic nebulization (USN).82 The use of ultrasonic nebulization improved the detection limit by a factor of 10. Silicon has been detected at 2.7 μg kg−1 in toluene with a USN system and at 27 μg kg−1 with a pneumatic system. Toluene was an excellent diluent for oil and derived samples. Sanchez et al.116,117 recently worked on silicon compounds determination by ICP-OES in xylene matrices. The effect of silicon chemical form was evaluated for sixteen different silicon molecules with sensitivity varying by a factor of up to 20.116 It was due to the liquid sample introduction system configuration, in particular the spray chamber design. These studies have shown that the application of two introduction systems, heated torch integrated sample introduction system (h-TISIS)116 and demountable direct injection high efficiency nebulizer (d-DIHEN) drastically reduces the influence of the chemical compound form.117 In addition, the response of silicon compounds also depends on the matrix but it less pronounced for high dilution factors.118 Finally, the determination of silicon, iron and vanadium was carried out in petroleum coke by microwave plasma torch atomic optical spectrometry (MPT-OES) after dissolution in nitric acid.119 This method allows a better control of trace elements which is very important when using petroleum coke as an electrode.
A procedure using a chromatographic purification coupled to multi collector (MC) ICP-MS has been described to enable dissolved silicon measurement in natural waters.122 Silicon isotopic measurement was also achieved by MC-ICP-MS in lake waters.123 The determination of Si by laser ablation (LA) ICP-MS and X-ray fluorescence (XRF) are also compared for airborne particulate matter.124 These results emphasized that LA-ICP-MS is the most preferable choice regarding detection limits and matrix limitations.
Several workers have carried out surveys using a microwave digestion before ICP-MS analysis of silicon in biological solid samples.125–127 Direct determination of Si by a double focusing magnetic sector inductively coupled plasma mass spectrometer without preparation step was reported in serum.128 Si measurements were carried out using the major 28Si isotope and with a resolving power of 3000, which is sufficient to avoid spectral interferences from 14N2+ and 12C16O+ during organic product injection. An interlaboratory trial has been achieved by many co-workers for the determination of silicon in biological samples.127 Various techniques (ETAAS, XRF, ICP-OES and ICP-MS) were compared considering LOD, RSD and statistical results for each technique and for each matrix.
Due to numerous matrix effects with the introduction of organic samples in ICP-MS, the use of high dilution is recommended.70 The detection of silicon in naphta and petroleum matrices after dilution by 10 in toluene has been achieved using a near cold plasma ICP-MS.129 Sub-μg kg−1 detection limits120,129 were obtained but solvent purity and volatility species are pointed out as significant issues.120
Takaku et al. have determined trace silicon in ultra high purity water for the manufacture of semiconductors by ICP-MS.121 A preconcentration procedure was applied, allowing quantification down to sub-ng L−1 of Si. The variation of element concentrations such as B, Si, P and S in steels has a significant influence on the mechanical and physical properties of this material.87 Thus, Si determination was achieved by ICP-MS130 or DRC-ICP-QMS.86,87 Detection limits varied from 0.2 μg L−186 and 2 μg L−187 for the introduction of acid solution and precision was better than 6.3%.86 These LOD are low enough for analysis of nitric acid steel samples.130 Finally, electrothermal vaporization (ETV) ICP-MS was applied to quantify silicon in solid polyamide samples.131 According to a comparison between other spectroscopic methods, Resano et al. concluded that ETV-ICP-MS and ETAAS have approximately the same performance concerning detection limits in the solid sample (0.3 μg g−1).131 De Schrijver et al. have shown that the LODs considerably improved with direct solid sampling methods compared to those attainable after dissolution.132 ICP provides a very sensitive determination of total silicon but these do not provide structural information if it is not coupled to a separation technique (see chapter 4).
2.2 NMR methods
NMR has the potential to achieve both total silicon determination and molecular identification133 mainly in biological fluids and tissues, environmental and petroleum products. The application of three NMR methods (1H-NMR, 13C-NMR and 29Si-NMR) has been widely reported for silicon determination in the Analytical Chemistry of Silicones134 and in Organosilicon Materials.19 Varaprath et al. have also described numerous works by NMR for the determination of silicon in environmental and biological samples.17 Nevertheless, 29Si NMR could not detect concentrations below 50 mg kg−1 in these matricesIn 1990, Fux determined the presence of PDMS in extracts of chemicals after a soxlhet extraction with pentane. A quantification limit of 0.1 mg kg−1 for PDMS was obtained by 1H-NMR.135 Other works by 1H-NMR and 29Si-NMR for determination of PDMS degradation products in animal models and human tissues have been related.136–138 Indeed, Garrido et al. have shown that silicon migrates from the implant to local and distant sites, and were located in tissues136 and blood.137 The biodegradation of PDMS and D4 as model compounds in lymph nodes of rats has also been studied by 29Si-NMR.138 In addition to the resonance associated with the PDMS injected, the NMR spectra showed new resonance, compared with lymph nodes control that are attributed to partially hydrolysed polysiloxanes and silica. Moreover, a critical review article of works by NMR has also reported on this problem as well.139
The natural abundance of 29Si isotope is only 4.7% and the NMR signal is weak. However, this technique is attractive for silicon speciation because of the wide distribution of 29Si chemical shifts (120 ppm) from silicon nuclei. In the petroleum domain, Carduner et al. determined the presence of Si and identified the chemical nature of the molecules by 29Si-NMR.66 Indeed, a resonance was observed at −19.55 ppm that corresponded to octamethylcyclotetrasiloxane (D4) in unleaded gasoline. Hamilton also reported the detection of PDMS hydrolysis products by 29Si-NMR in the environment.133 The different chemical shifts were specified for silicon compounds (D3-D6, HMDS, DMSD…). D4 chemical shifts fitted the value reported by Carduner et al.66
In spite of specified identification of chemical structure and non-destructive technique, NMR is not adapted to trace analysis of several contaminants.140 Indeed, the ratio of LOD calculated between AAS and 29Si-NMR is approximately 104. Carduner et al.66 and Bellama et al.141 also noted that concentrations lower than 60 mg kg−1 of Si in unleaded gasolines and 45 mg kg−1 of Si in environmental samples could not be detect by this technique.
2.3 Other methods
Considering the fact that X-ray fluorescence (XRF) and infra red spectroscopy are not adapted to trace determination of silicon, less attention is paid. However, there were applied in petroleum products, industrial and biological samples with detection limits generally above 10 mg kg−1. The determination of silicon by XRF142 and IR143 was widely reported in The Analytical Chemistry of Silicones. The application of XRF for silicon analysis in heavy oil samples allows quantification with detection limit of 12 mg kg−1 and a reproducibility above 97%.144 A comparison between XRF and FTIR applied to silicon determination in paper coating has shown that XRF is the more convenient combination between sensitivity and samples preparation.145 However, FTIR can be possibly used as an alternative of XRF. FTIR has been particularly applied in the determination of silicon in human tissues from breast implant degradation.146–1483 Silicon contamination and analytical artifacts
As seen in the previous chapters, the important use of silicon compounds in a wide variety of applications (cosmetics, personal care products and consumer goods) increases the potential contamination of samples during the analysis.12,13 The purpose of this part is to review the different sources of contamination and the analytical artifacts induced during silicon speciation due to various contamination sources.21 A relative estimation of silicon contamination based on publication numbers and pollution concentration is illustrated in Fig. 7. The importance of each source is discussed throughout this chapter. Generally, contamination through human way has to be minimized by taking care of the use of cosmetic and personal care products during Si determination by laboratory personal.21 Moreover, numerous authors described contamination problems arising from organosiloxanes present in different sources like spare parts made of silicones in various parts of the analyzer or contamination from different sources during sampling and preparation. Thus, extreme care must be taken to minimize the sources of artifacts and siloxane contaminations for speciation analysis at trace levels.149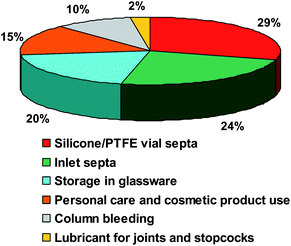 | ||
| Fig. 7 Relative estimation of silicon contamination based on literature publications in percent. | ||
3.1 Contamination in gas chromatography
Contamination in GC mainly concerned cyclic siloxanes generated by the inlet septa and bleeding column during analysis. According to Fig. 7, the degradation influence of the inlet septa is greater than the column bleeding regarding the siloxane levels. Moreover, the contamination augments with the injection temperature. The presence of septa particles located in the liner also contributes to this pollution.150 Nevertheless, this phenomenon can be reduced by using specific gas chromatography tools.Septa are made of pure heavily cross-linked PDMS and of phthalates added as stabilizers.150 At high temperatures (200–300 °C), PDMS starts to degrade and produces cyclic siloxanes (D3-D13) (Fig. 4).58–60 The trimer is reported to be the most abundant product with decreasing amounts of tetramer, pentamer, hexamer and higher oligomers.58 In order to minimize the impact of septum bleeding, De Zeeuw recommends several points:150
• Avoid the use of a septum and use valve injection such as merlin microseal valve22,151
• Reduce the risk of septum scoring by using tapered needles and a prepierced septum (BTO septum)
• Clean the liner (ultrasonic bath) and replace in a time
• Replace the septum frequently
According to this work,150 Horii and Kannan have studied five different septa between 100 and 250 °C for injection temperatures.12Fig. 8 shows Dn concentrations (μg L−1) as a function of septa used during GC analysis of 1 μL of hexane spiked with an internal standard (M4Q) at 200 °C. The concentrations of D4, D5, D6 measured with an Agilent advanced green septum are respectively 6, 1.6, 4.6 μg L−1 at 250 °C and 1.7, 0.6 and 0.3 μg L−1 at 200 °C. It corresponds to a drastic reduction (79%) of Dn concentrations. The use of BTO septum at 200 °C allows a balance between low levels of Dn and a correct vaporization of compounds (Fig. 8). Overall, the background of D6 in blanks is at least 20 times lower than the lowest concentrations found in consumer products.12 Considering this work, Sparham et al. have applied an injection temperature at 150 °C to reduce contamination by septum.152
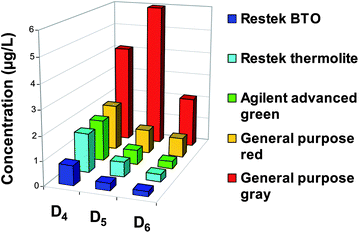 | ||
| Fig. 8 Instrumental background levels of D4, D5, D6 in n-hexane injected into several septa of GC-MS at 200 °C (Adapted from Horii et Kannan, 2008).12 | ||
Septum particles can also be deposited in the liner from 200 °C to 300 °C and caused contamination.17 One inlet septum particle into the sample extract is almost half of the total amount stationary phase coating of the capillary GC column (a few milligrams for a VF-5 MS).12,150 Consequently, a method is proposed to determine if the septum produces cyclic siloxane compounds based on the simulation of different splitless injection time with variation from 0 to 20 min.150
The generation of these molecules also originated from column bleeding because most of the capillary columns are covered with PDMS. Horii et al. examined cyclic oligomer backgrounds of two low bleed columns, a DB-5 MS (5% phenyl and 95% PDMS) and a DB-1 MS (100% PDMS).12 Another column (DB-XLB) has also been studied by Kala et al. in 1997 to minimize hexamethylcyclotrisiloxane (D3) levels51 in blanks. The results suggested that the release of organosiloxanes from a low bleed column is minor in comparison with the release from septa (Fig. 7). The fact that Dn contamination mainly originates from septum degradation has been confirmed by De Zeeuw150 and Wang et al.13 For example, DB-5 MS and DBWAX (phase without silicon) columns produce similar background levels of siloxanes during blank analysis. However, Varaprath et al. have also focused their research on the analytical artifacts related to siloxane analysis. For example, D4 can be generated by interactions between water contained in biological or environmental samples and PDMS stationary phase in GC.153 Higher cyclic siloxanes can also be produced at trace levels.
3.2 Mass spectrometry detection and artifacts
In 1982, Ende and Spitleller reported different contaminations in Mass Spectrometry.149 Several elements present in laboratory (syringe, lubricants, flasks, pump fluids, septa…) contained silicones.17,149 For instance, Carter et al. suggested that plastic pasteur pipettes contain a silicon lubricant with a molecular weight of approximately 16500 g mol−1.154 General precautions to limit contamination are described but even with the highest degree of precaution, these authors suggest that analysts are not able to exclude contamination in trace silicon analysis.149 Indeed, they can only minimize the risk of contamination.
In association with GC, the degradation products formed by column stationary phase decomposition are similar to the products generated by septa degradation.150 These compounds induce the formation of artifact ions (m/z 207, m/z 281 and m/z 355) in MS that correspond to major fragment ion of our interest compounds as D3 (222-CH3), D4 (296-CH3) and D5 (370-CH3).150 Other ions m/z 73 (Me3Si+), m/z 147 (Me3SiOSiOMe2)+m/z 149 (plasticizer), m/z 295 (silicones with M<400) can also appear as contamination sources in the background17 although, low bleeding and MS quality columns are recommended.150
3.3 Other contaminations, storage and conservation
Several authors have focused on the storage and conservation of organosilicon compounds such as silanols and silanediols because of their high reactivity.22 Sample storage in vial septa made of silicones must be avoided and silicon compounds (DMSD) need storage in plastic containers to eliminate condensation enhanced by alkaline surface.Various authors focused their works on the contamination induced by the use of silicone/PTFE vial septa.155–157 Chambers et al.155 and Wang et al.157 demonstrated that silicone/PTFE septa generate Dn compounds when they are in direct contact with the sample. Chambers et al. observed the generation of cyclic siloxanes (D3-D7) during MTBE (methyl tertiary butyl ether) analysis in blood at ng L−1 levels by gas chromatography coupled to mass spectrometry (GC-MS) in single ion monitoring mode (SIM).155 Moreover, the results obtained by Wang et al. showed that sample analysis must be made immediately after they are introduced in vials with silicone/PTFE septa157 in order to avoid the contamination by Dn. On the other hand, Pattinson et al. tested nine vial septa with different solvents by GC-MS.156 They concluded that septa which have a PTFE layer minimize silicon contamination. Thus, vials with butyl/PTFE caps used by Sparham et al. have shown a reduction of Dn generation and a minimization of the sample volatilization more observed with PTFE septa.152
Storage and conservation play an important role during trace analysis. Silicon materials, such as PDMS, have a great affinity for glass and are often adsorbed on the surface.18 Thus, it is more safe to keep them at lower temperatures in teflon or plastic containers (preferentially high density polyethylene).17 This precaution is particularly true for silanols that condense and siloxanes that react in the presence of alkaline surface, acids or strong bases.18 Indeed, glass surfaces, moisture, temperature and acidity will improve condensation.22 Several authors suggest to store samples in polypropylene vials before their analysis.12,31,158
Varaprath and Lehmann have also studied the conservation and stability of the dimethylsilanediol (DMSD) by 29Si NMR and HPLC (high pressure liquid chromatography) at different concentrations.22 This compound is unstable even in pure form159 and must be stored in a freezer between 4 and 8 weeks in plastic container with a drying agent in order to avoid polymerization and the presence of dimerdiol at high concentration.22 At a lower concentration (100 mg L−1), HPLC results confirmed that DMSD is stable for one year.
4 Hyphenated techniques for Silicon speciation
Several hyphenated techniques (Fig. 6) have been developed for Si speciation using chromatographic methods. These separation techniques are based on different physico-chemical properties, such as volatility, molecular size, degree of aromaticity or polarity. This chapter is mainly focused on the combination of chromatographic methods (LC, GC and SFC) with different detectors reported in Fig. 6. However, other techniques such as mass spectrometry have been employed for Si speciation.4.1 Liquid chromatography
For silicon speciation, LC has been employed in two separation techniques: high pressure liquid chromatography (HPLC) for LMW compounds and size exclusion chromatography (SEC) for HMW molecules (Fig. 6). HPLC can be performed in normal phase (NP) or reverse phase (RP). For silicon speciation, HPLC applications by reverse phase are more important than by normal phase.Several researchers have employed RP-HPLC for LMW silicon molecules in environmental and biological samples using classical detectors (UV, RI, radioisotope)160,161 or 29Si NMR162 to provide molecular identification. However, most of the works have been performed by the coupling of RP-HPLC and atomic detectors (ICP-OES and ICP-MS) allowing structural information with the LC separation and selective detection (Fig. 6).31,154,158,163–166 Generally, a considerable improvement of LOD is observed when ICP-OES (20–500 μg L−1) is replaced by ICP-MS detection (0.1–4 μg L−1). However, according to Ebdon et al., ICP-OES is a more attractive detector for silicon speciation when compared to ICP-MS because mass interferences (m/z 28) are deleted.166 RP-HPLC analyses for Si speciation are summarized in Table 2.
| Matrices | Molecules | Solvent | Injection volume/μL | Column | Mobile Phase | Flow rate/mL min−1 | Detection | LOD | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Soil | Dimethylsilanediol | THF | — | C18 | H2O (100) | — | Refractive index | — | 22 |
| 25 cm × 4.6 mm × 5 μm | H2O (100) to ACN (100) | ||||||||
| ACN (100) to H2O (100) | |||||||||
| Biogas | Inorganic silicon | H2O | 100 | C18 | ACN/H2O (20/80) | 0.3 | ICP-OES | 31 | |
| Dimethylsilanediol | 25 cm × 2 mm | ACN/H2O (5/95) | 500 μg L−1 of Si | ||||||
| 1,3-tetramethylsilanediol | ACN/H2O (60/40) for Flushing | 30 μg L−1 of Si | |||||||
| Trimethylsilanol | |||||||||
| Environmental | Methylsilanediols | Xylene | 25 | C18 | ACN/H2O (1:10) to ACN/H2O (4:10) |
0.2 | ICP-OES | 40–150 μg L−1 of Si | 158 |
| 25 cm × 2 mm | |||||||||
| Biological | L2 D5 and its metabolites | mobile phase | — | C18 | H2O (100) | — | Radioisotope | — | 161 |
| 25 cm × 4.6 mm × 5 μm | H2O (100) to ACN (100) | ||||||||
| ACN (100) | |||||||||
| ACN (100) to H2O (100) | |||||||||
| D4 and its metabolites | mobile phase | — | C18 | H2O (100) | — | Radioisotope | — | 160 | |
| 25 cm × 4.6 mm × 5 μm | H2O (100) to ACN (100) | Refractive index | |||||||
| ACN (100) to H2O (100) | |||||||||
| Medical | Inorganic silicon | mobile phase | 100 | C18 | MeOH–H2O (30/70) | 1 | ICP-OES | 100 μg L−1 of Si | 166 |
| 1,3-tetramethylsilanediol | 25 cm × 2 mm | 400 | |||||||
| L2 | 500 | ||||||||
| Inorganic silicon | mobile phase | 100 | C18 | MeOH–H2O (20/80) | 0.15 | ICP-MS | 0,1 μg L−1 of Si | 154 | |
| Dimethylsilanediol | 25 cm × 2.5 mm × 6 μm | 4 | |||||||
| 1,3-tetramethylsilanediol | 4 | ||||||||
| Polymer | L2-L5, D3-D5 | AcOEt | 5 | C18 | MeOH–H2O (95/5) to MeOH (100) | 1 | ICP-OES | 20–50 μg L−1 of Si | 163 |
| 15 cm × 4.6 mm × 5 μm | |||||||||
| L2 | Acetone | 5 | Carbosphere 30 DS | ACN/Acetone (80/20) 30–130 °C at 2 °C min−1 130 °C (8 min) | 0.20 | ICP-OES | 40 μg L−1 of Si | 164 | |
| PDMS | |||||||||
| Hexamethyldisilane | ACN | 60 | C18 | ACN/CDCl3: (9:1) to ACN/CDCl3 (1:9) |
0.5 | 1H NMR | — | 162 | |
| L3 and L5 | CDCl3 | 15 cm × 4.6 mm × 5 μm | 29Si NMR | ||||||
| PDMS | |||||||||
| Organic matrices | D4 | Xylene | — | C18 | MeOH–H2O (7:10) to MeOH(10) |
1 | ICP-OES | 500 μg L−1 of Si | 165 |
| Hexane | 30 cm × 10 μm |
Varaprath et al. detected several metabolites of hexamethyldisiloxane (L2) and decamethylcyclopentasiloxane (D5)161 and also metabolites of D4160 in animal urine by RP-HPLC with a radioisotope detection. Metabolites were eluted on a C18 column using a ACN/H2O mobile phase. The main structures were confirmed by synthesizing 14C-labeled standards and GC-MS was applied in order to further confirm their identities.160,161 DMSD was detected by refractive index detector during PDMS degradation in soils.22,167 These previous detectors provide a non-destructive technique but without the sensitivity to allow trace analysis.17
As previously mentioned, Si speciation in the environment by RP-HPLC-ICP-OES has been reported (Fig. 2). Firstly, the detection and the linearity response of D4 in xylene and hexane matrices have been achieved with detection limit of 500 μg L−1.165 Similarly, Biggs et al. carried out silicon molecules separation (L2-L5 and D3-D5) with improved LODs ranging from 20 and 50 μg L−1 of Si with this combination.163 According to gradient and column optimization, elution on a C18 column (15 cm × 4.6 mm × 5 μm) with a gradient from MeOH–H2O (90/10) to MeOH (100) over 8 min produces the best results.163 Nevertheless, chromatographic separation observed respectively between D4 and L3 and between D5 and L4 is not optimal. The same authors have also indicated the improvement of thermal gradient by RP-HPLC-ICP-OES for PDMS solution in acetone with a ACN/Acetone gradient.164
In 1994, the speciation of silanediols in water, sludge extracts and soils was carried out by RP-HPLC-ICP-OES with detection limits of 40 μg L−1 of Si for trimerdiol and 150 μg L−1 of Si for monomer and dimerdiol.158 It was also shown that low injection volume and use of xylene minimize the instability of the plasma to organic solvents. Grumping and Hirner performed the separation of two silanediols and trimethylsilanol (TMS) in leachate samples by the same hyphenated technique.31 Detection limits of 30 μg L−1 of Si for TMS and 500 μg L−1 of Si for DMSD were calculated for 100 μL injection volume. Additionally, a modification of eluent by a more polar gradient (Table 2) allowed the separation of the DMSD and the silicate.31 Dorn and Kelly158 obtained DMSD lower detection limits than Grumping and Hirner31 because of the different gradient application and the use of a special ICP interface and micro HPLC system.
More recently, Ebdon et al. compared the performance of RP-HPLC with a radial ICP and an axial ICP for polar silicon compounds.166 Generally, it is well known that detection limits are improved using axial ICP compared to radial ICP.168 However, radially viewed ICP-OES gave similar detection limits (100, 400 and 500 μg L−1 of Si for inorganic silicon, TMSD and L2 respectively) to axially viewed ICP-OES with improved chromatographic peak reproducibility for all three compounds.166 In 2004, Carter et al. replaced the ICP-OES with HR-ICP-MS coupled to RP-HPLC for Si speciation.154 Consequently, using similar conditions to previous study,166 an improvement of detection limits for inorganic silicon compound with a 1:1000 ratio and for TMSD with a 1:100 ratio was indicated (Table 2).154 These results show that ICP-MS provides better detection limits than ICP-OES for Si speciation when mass interferences are resolved. Finally, reversed-phase LC-NMR with 1H and 29Si has been developed using a gradient from ACN/CDCl3 (9:1) to ACN/CDCl3 (1:9) in order to determine several organosiloxane structures.162
Hausler and Taylor have achieved PDMS separation by SEC-ICP-OES with detection limits ranging between 0.03 to 3 mg L−1 of Si.171,172 This hyphenated technique was applied in water and sludge extracts with LOD of 160 μg L−1 of Si.158 PDMS separation with molecular mass between 550 and 500000 g mol−1 using THF and xylene as mobile phases has been established. Xylene was chosen because it enables to easily maintain a stable plasma and allows the solubilisation of most of the PDMS polymers. Nevertheless, using THF iso-response as a function of molecular mass and upgrade in sensitivity by a factor 4 are observed. This is due to the better solubility of PDMS polymer in THF, particularly for high weight mass polymer.158
Carter et al. reported a similar separation of three PDMS polymers (162, 1500 and 16500 g mol−1) by SEC-ICP-MS with detection limits of 12, 26 and 30 μg L−1 of Si respectively.154 Extraction efficiencies of these compounds from spiked human plasma were performed with xylene because of its relatively low vapour pressure (plasma stability 158) and compatibility as an elution solvent for the SEC separation.154
Maziar et al. have studied HB-PDMS (α,ω-bis(4-hydroxybutyl) polydimethylsiloxane) behaviour by SEC-MALDI-TOF/MS in order to obtain detailed information during synthesis.173 A comparative study between automated SEC-MALDI-TOF/MS and on-line SEC-ESI-TOF/MS for PDMS characterization demonstrated an improvement of chromatographic resolution and an enhancement in low molecular weight polymer (M<550 g mol−1) by this latter coupling.174 In opposition, the first technique effectively reported the HMW oligomers and underestimated the LMW oligomers.174
4.2 Gas chromatography
Gas chromatography has been widely used for Si speciation (Fig. 2). An enhancement in separation compared to HPLC based on the volatility and the polarity of species can be achieved by GC. Fig. 6 summarizes various detectors for silicon reported in the literature. GC is preferred to LC because of higher resolution and lower detection limits. For instance, chromatographic resolution between silicon molecules like D4 and L3 is better by GC-ICP-OES31 than by LC-ICP-OES.175 GC chromatography applied for Si speciation in environmental (Table 3), biological and industrial samples (Table 4) are presented. Most of the studies is carried out on non polar columns (95% methylPolysiloxanes-5% Phenyl) with 1 μL injection volume in splitless mode.| Matrices | Molecules | Solvent | Injection | Column | Detection | LOD; Si Eq.a | Ref. |
|---|---|---|---|---|---|---|---|
| a Si Eq.: Silicon Equivalent. b Estimated. | |||||||
| Soil | D4-D6 | THF | 1 μL | HP5 | MS SIM | 1 μg L−1; 0.4 μg L−1 of Si | 22 |
| Silanediols (1–5) | Acetone | 250 °C | 30 m × 0.25 mm × 0.25 μm | 0.1–1 mg L−1; 0.04–0.4 mg L−1 of Si | |||
| Silicon degradation products | THF | Merlin system | HP5 | MS Full Scan | — | 151 | |
| Acetone | 2μL 250 °C | 30 m × 0.25 mm × 0.25 μm | |||||
| Hexane | |||||||
| Dimethylsilanediol | THF | 1μL | HP5 | MS SIM | — | 33 | |
| 250 °C | 30 m × 0.25 mm × 0.25 μm | ||||||
| Dimethylsilanediol | THF | 2μL | DB1 | MS SIM | — | 35 | |
| silanols | Acetone | 250 °C | 60 m × 0.32 mm × 0.25 μm | ||||
| Dn | |||||||
| Water | D5 | Hexane | 3 ml in HS | DB-FFAP | MS SIM | 3 ng L−1; 1.1 ng L−1 of Si | 152 |
| 220 °C | 30 m × 0.25 mm × 0.25 μm | ||||||
| Air | D3-D5 | — | — | HP5-MS | MS SIM | — | 179 |
| 30 m × 0.25 mm × 0.25 μm | |||||||
| D3-D4 | — | 180 °C | 200 f t OV-17 | MS Full Scan | — | 178 | |
| L5 | 200 ft OV-101 | ||||||
| Tetramethylsilane | 400 ft OV-101 | ||||||
| Alkoxysilanes | Heptane | 1 μL | DB5 | FID | 1–5 mg L−1; 0.12–0.62 mg L−1 of Si | 56 | |
| 30 m × 0.32 mm × 0.25 μm | |||||||
| Air, water, soil, sediments, biota | D3 | Hexane | 1 μL | CP-Sil8CB | MS SIM | 50 μg L−1; 18.9 μg L−1 of Si | 8 |
| D4-D6 | 200 °C | 30 m × 0.25 mm × 0.5 μm | 5 μg L−1; 1.9 μg L−1 of Si | ||||
| L2-L5 | 0.3–0.5 μg L−1; 0.11–0.18 μg L−1 of Si | ||||||
| Biogas | L2-L4, D3-D6 et Trimethylsilanol | Pentane | 200 °C | RTx-1 | AED | 9.5 μg L−1 of Si | 32 |
| 47 m × 0.32 mm × 1.5 μm | MS Full Scan | — | |||||
| Waste sludge | L2 et D3-D6 | Hexane | 1 μL | VF-1MS | FID | — | 29 |
| 125 °C | |||||||
| Waste | D3-D5; L2-L4 | Pentane | 5 μL | HP1 | ICP-OES | 0.1 μg L−1 of Sib | 175 |
| Trimethylsilanol | — | ||||||
| D3-D6 | Hexane | 1μL | SE-54 | FID | — | 30 | |
| 240 °C | 50 m × 0.32 mm × 0.25 μm | MS Full Scan |
| Matrices | Molecules | Solvent | Injection | Column | Detection | LOD; Si Eq. a | Ref. |
|---|---|---|---|---|---|---|---|
| a Si Eq.: Silicon Equivalent. b Estimated. | |||||||
| Biological | D4 | Tetrahydrofuran | Merlin system 2 μL 250 °C | HP5-MS | MS SIM | 1 μg L−1; 0.4 μg L−1 of Si | 153 |
| 30 m × 0.25 mm × 0.25 μm | |||||||
| Blood, Plasma | D3-D6 | Hexane | — | HP5 | MS SIM | 2 μg L−1; 0.76 μg L−1 of Si | 50 |
| 200 °C | 30 m × 0.25 mm × 5 μm | ||||||
| D4 | Tetrahydrofuran | 2 μL | ND5-MS | MS SIM | 4.9 μg kg−1; 1.9 μg kg−1 of Si | 11 | |
| 250 °C | 30 m × 0.25 mm × 5 μm | ||||||
| Tissues | D3-D6 | Hexane | — | HP5 | MS SIM | — | 52 |
| 200 °C | 30 m × 0.25 mm × 5 μm | ||||||
| Breast Implants | PDMS | Ethyl acetate | 1 μL | DB1-HT | AED | 80 μg L−1 of Si | 53 |
| Dn | 320 °C | 30 m × 0.32 mm × 0.2 μm | MS SIM | 100 μg kg−1; 38 μg kg−1 of Si | |||
| Dn and Ln | Ethyl acetate | — | Extra Low bleed DR-XLE | MS SIM | — | 51 | |
| AED | — | ||||||
| Medical | Silylated alcool | Pyridine | 1 μL | FS-Suprem-5 | ICP-MS | 3 μg L−1 of Si b | 185 |
| 25 m × 0.25 mm × 0.25 μm | |||||||
| Consumption products | D4-D7 | Ethyl acetate | 1 μL 200 °C | Rxi5-MS | MS SIM | 117 μg kg−1; 44 μg kg−1 of Si | 12 |
| L5-L14 | Hexane | 30 m × 0.25 mm × 0.25 μm | 18 μg kg−1; 6.8 μg kg−1 of Si | ||||
| D3 | Methanol | 280 °C | DB5-MS | MS SIM | 120 μg kg−1; 45.3 μg kg−1 of Si | 13 | |
| D4-D6 | Acetone hexane | 30 m × 0.25 mm × 0.25 μm | 80 μg kg−1; 30.2 μg kg−1 of Si | ||||
| Polymer | D3-D8 | Toluène | 1 μL | MDN-5S | MS Full Scan | — | 184 |
| 250 °C | 30 m × 0.25 mm × 0.25 μm | ||||||
| Dn et Ln | Dichloromethane chloroform | 200 °C | DB5 | MS Full Scan | — | 177 | |
| 30 m × 0.25 mm × 0.25 μm | |||||||
| D3-D10 | — | — | CP-Sil 8 | MS Full Scan | — | 183 | |
| 30 m × 0.25 mm × 0.25 μm | |||||||
| Degradation resin products | Tricholormethane | 250 °C | Anabond 225 | MS Full Scan | — | 182 | |
| 50 m × 0.32 mm × 1 μm | |||||||
| D3-D6 | — | 200 °C | 100% methylsilicone | MS Full Scan | — | 181 |
Dn (D4-D6) and silanediols (mono-pentamer) were extracted from soils with polar solvent (THF, acetone) and identified using SIM mode via two ionization methods.22 Detection limits of 1 μg L−1 with a signal to noise ratio of 5 for Dn and between 100 μg L−1 and 1 mg L−1 for diols have been calculated. In order to differentiate these compounds which have the same fragmentation patterns in their mass spectrum, some authors22,177 have preferred to work using chemical ionization (CI) instead of electron impact ionization (EI).
Siloxanes lose a methyl group (M-15) under EI conditions. On the other hand, silanediols (excepted DMSD and dimerdiol) lose a water molecule (18) in addition to a methyl group (M-33), trimer, tetramer and pentamer diols have a mass difference of 18 with D3, D4, D5 respectively. Thus, their mass spectrum is similar (M-15).22 Although these compounds can be distinguished by their retention time in GC, CI helps to identify these molecules. For instance, the identification of D4 (m/z 314) and tetramerdiol (m/z 332) with a mixture of reagent gas CH4:NH3 (90:10) [M + NH4]+22 and Dn compounds using isobutane [M + H]+177 were carried out.
Other VMS (D3-D5, L5 and tetramethylsilane) were detected by GC-MS in ambient atmospheric extracts.178,179 More recently, D5 analysis was performed in river water and treated waste water by hexane extraction and head space (HS) coupled to GC-MS SIM using a DB-FFAP column.152 The use of internal standard in SIM mode (m/z 73, m/z 267, m/z 355 for D5 and m/z 360 for 13C5-D5) provides a quantification limit of 10 ng L−1 for D5 in water. In 2005, concentrations of cyclic and linear oligomers were reported by GC-MS SIM in the nordic environment (water, sediments, air, sludge, biota…) of six countries.8 Detection limits have been calculated for various matrices after an hexane extraction. For instance, detection limits for D3, D4-D6 and L2-L5 were found to be 50, 5 and between 0.3 and 0.5 μg kg−1 in biota samples respectively.
Analysis of polar silicon compounds, such as silanols or silanediols may require derivatization by sylilating agents before GC analysis due to their instability (see chapter 3).17,22 Basically, if stationary phase contents siloxanes, the formation of reactive hydrogen atoms can appear in presence of moisture17 and promotes silanol condensation into the GC column.180 Consequently, this reaction compromises severely the sensitivity and degenerates the peak shapes.22 Several workers have capped OH functions of the molecules with trimethylsilyl groups,180 by usually using BSTFA (bis(trimethylsilyl)-trifluoroacetamide).151 However, Varaprath et al. have shown that species like DMSD can be analyzed without derivatization reaction.22 The procedure described a 1 μL BSA (bis(trimethylsilyl)acetamide) injection volume into GC column before analysis in order to deplete all reactive surfaces.22 In opposition, Lehmann et al. have achieved the direct detection of DMSD, silanols and Dn without derivatization step by GC-MS SIM during PDMS degradation in soil.33,35
Cyclic and linear compounds were also analyzed by GC-MS in FS mode for qualitative analysis and in SIM mode for quantitative study in biological matrices. After extraction by ethyl acetate and THF, detection limits of 100 μg kg−1 for tissue53 and 5 μg kg−111 for plasma samples have been respectively reported by SIM mode. Several workers quantified Dn in blood and plasma using a widely internal standard M4Q (tetrakis(trimethylsiloxy)silane) for Si speciation.11,12,50,153 Extraction yields above 90% with THF and a 2μL injection volume gave access to very low detection limits around 1 μg L−1.153 In addition, linearity range between 1 to 16 μg L−1 of D4 compared to internal standard was obtained. Considering the same analytical method, Flassbeck et al.50 calculated detection limit of 2 μg L−1 after an hexane extraction of Dn from blood and plasma, which is similar to the values reported in the literature.11,22,153
As previously mentioned in chapter 1, silicon molecules are used in consumer products. Horii and Kannan measured high VMS (Dn and Ln) levels in 76 cosmetic and personal care products by SIM mode.12 After an extraction by a mixture of ethyl acetate–hexane, 1 μL of analyte injected in a Rxi-5 MS column at 200 °C has allowed the acquisition of detection limits ranging between 18 (Ln) and 120 μg kg−1 (Dn). Details in contamination problems were also given (see chapter 3 for more details).12 More recently, cVMS have been analyzed in 252 consumer products by GC-MS (fragrances, hair care products, antiperspirants, lotions…)13 using similar conditions to Horii and Kannan.12 Detection limits between 120 μg kg−1 (D3) and 80 μg kg−1 (D4-D6) and calibration curve ranging from 50 μg kg−1 to 10 mg kg−1 were reported.13 According to a comparison between D5 and D6 levels in skin lotions by Horii and Kannan12 (35.3 mg g−1 and 6.3 mg g−1) and Wang et al.13 (47.3 mg g−1 and 6.5 mg g−1), these studies are completely in agreement.
GC-MS has also been applied for Si speciation mainly in FS mode for qualitative analysis. Silicon compounds (siloxanes and chlorosilanes) were separated by GC and identified by MS in EI and in CI for silicon rubbers and silicon resins respectively.181,182 Furthermore, Dn characterization was performed by GC-MS when PDMS was submitted to corona discharges.183 Wacholz et al. have also analyzed a mixture of cyclic and linear siloxanes by GC-MS and GC-FTIR.177 More recently, thermal stability of polysiloxanes has been studied by HS-GC-MS in toluene.184 These results have shown the presence of Dn, phthalates and ethylhexanoic acid. In addition, Hall et al.184 have reported a non-linear response for D5 and L5 contrary to Sparham et al. researches.152
In 1997, a novel and highly sensitive method for detection, quantification and characterization of LMW siloxanes in biological tissues by GC-AED and GC-MS for further identification has been reported.53 After an extraction by ethyl acetate, detection limit of 100 μg of Si per kg of tissue was calculated by GC-AED at 251.6 nm using a DB-1 column. Similarly, Lykissa et al. identified cyclic compounds (D3-D7) and L7 in silicon gel implants51 with extra low bleed column in order to minimize Si contamination (see chapter 3).
The development of canister sampling and GC-MS-AED was performed by Schweigkofler and Niessner for the determination of siloxanes (Dn and Ln) and silanols.32 Detector signals obtained are linear over more than 4 orders of magnitude (R2>0.99) and detection limit of 9.5 μg L−1 of Si was determined in pentane. After thermodesorption and analysis, L2-L4, D3-D6 and TMS are the major compounds found in biogas with detection limit of 342 ng m−3.32 As mentioned in section 1.3, LOD depend on sample extraction and concentration before analysis. Consequently, the comparison of sensitivity between two different techniques is accurate in biogas analysis especially considering collected sampling volume.
Grumping et al. identified TMS, L2 and cyclic oligomers (D3-D5) in biogas sample by LT-GC-ICP-OES.175 For example, VMS (L2, D3-D5) were measured at concentrations ranging between 0.1 and 1.1 μg of Si per litre of waste water samples. In addition, resolution between L2 and TMS was non-optimal using a non-polar column (100% PDMS). Different responses between compounds of interest have also been observed due to molecule volatility or condensation phenomena.175 According to Grumping et al., the choice of ICP-OES avoids mass interferences (m/z 28) occurring in ICP-MS when collision/reaction chamber is not available (previously mentioned in section 2.1.3). The second study concerns the application of GC-HR-ICP-MS to silylated alcohols quantification.185 Sylilation was achieved by reaction between four alcohols (C4–C7) with N-methyl-N-trimethylsilyltrifluoroacetamide (MSTFA) in pyridine. Medium resolution (m/Δm = 4000) gives access to the resolving of mass interferences between 28Si+, 12C16O+ and 14N2+121,154,185 in order to obtain detection limit of 0.1 μmol L−1, which correspond to 3 μg L−1 of Si for C4 silylated alcohol.185 Nevertheless, column bleeding can limit these detection limits and contributes to the background pollution.185
4.3 Supercritical Fluid Chromatography (SFC)
Silicon is dispersed in very low levels for several fields such as environment,22 biology50 and petroleum and derived products.70,80 SFC method is not very adapted to trace analysis and the combination between SFC and detectors like ICP-OES, FID or MS don't lead to sufficient sensitivity. Indeed, SFC coupled to an atomic detector (ICP-OES) was achieved to separate various siloxanes with detection limit of 57.9 mg kg−1 of Si.190 Excellent separation of PDMS with a molecular mass of 2000 g mol−1 has been reported by SFC-FID.191 This method was applied with SFC/MS for the determination and the identification of cyclic siloxanes in technical silicon oils and rubbers.1924.4 Other mass spectrometry techniques
Without chromatographic separation, mass spectrometry avoids contamination due to the GC parts (see chapter 3) and is applied both for LMW and HMW silicon molecules (Fig. 6). These techniques were mainly performed in environment (soil193 and biogas194,195) with variable sensitivity depending on the sample preparation (2 μg m−3195 and 50 mg kg−1193) and in polymer characterization (FT-ICR/MS and TOF/MS) usually as qualitative analysis for industrial applications.The determination of D4 and D5 in biogas by APCI-MS/MS without extraction or prior chromatographic separation was a primer in Si speciation.194 Badjagbo et al. developed and validated a sensitive (4 < LOD (μg m−3 of air) < 6) and selective method for direct analysis of siloxanes in gaseous matrices.194 Indeed, direct MS detection completely avoided background contamination from GC systems (see chapter 3) and allows the direct distinction between VMS and siloxanediols previously discussed (section 4.2.2) due to the soft APCI ionization [M + H]+.194 More recently, Badjagbo et al.195 quantified D4 and D5 with an internal deuterated standard hexamethyldisiloxane (HMDS-d18) using the previous analytical method.194 The use of HMDS-d18 provided effective signal compensation of D4 and D5 and improved the sensitivity and reliability of these compounds in biogas.195 According to Badjagbo et al., a detection limit around 2 μg m−3 has been obtained,195 which is 500 times better than that reported for a GC-FID176 and much more sensitive than that reported in a recent study by a microcantilever array sensor.46 The latter method, recently developed by Long et al., provided linear and cyclic siloxanes analysis in biogas with detection limits of 17 μg L−1 for D5, corresponding to 257 μg m−3.46 After possible adsorption or deposition of silicon in the environment (section 1.2), a method for isotopic determination of 30Si by MS in plants and soils was proposed with detection limit of 50 mg kg−1 for the soil samples.193
MS is applied to have a better control of silicon compounds present in industrial applications such as semiconductors and packaging industries196 and also in some processes.197 For instance, in situ MS was carried out to characterize silicon compounds (tetraethoxysilane and hexamethyldisiloxane) enabling the production of protective layers for semiconductors.196 In addition, Apicella et al. observed siloxane series after an extraction by dichloromethane during the analysis of soot recovered in fuel-rich flames using several burners and different fuels by MALDI/MS.197 In addition, Apicella et al. observed siloxane series after an extraction by dichloromethane during the analysis of soot formed in fuel-rich flames by MALDI/MS.197
For HMW silicon compounds (Fig. 6) that do not elute in GC, mass spectrometry methods without separation such as MALDI-TOF/MS (time of flight mass spectrometry) or ESI-FT-ICR/MS (Fourier transform-ion cyclotron resonance mass spectrometry) were applied to silicon molecules.17 In polymer synthesis, the degree of intramolecular condensation, defined as the number of residual silanol (Si–OH) groups per oligomer, for a variety of silsesquioxane polymer (thermal stability and chemical resistance), was measured by MALDI-TOF/MS.198,199 Indeed, condensation of the Si–OH groups leads to the formation of intramolecular group Si–O–Si bridges accompanied by the loss of water.199 This phenomenon was easily characterized by high resolution MS.198 The use of high resolution MS, like FT-ICR/MS,200 offers a sub-ppm mass measurement accuracy and allows successful identification of polymers which generally provide nearly identical mass spectra at low m/z.201 Murthy et al. studied the kinetics and pathways of the ion-molecule reactions for a mixture of silanes and chlorosilanes (SiHnCl4-n) by FT-ICR/MS using a 1 Tesla magnet and EI ionization.202 By coupling electrospray ionization (ESI) to FT-ICR/MS, the first work on PDMS pointed out several fragmentation patterns by hydrogen bond rupture or by methyl transfer.201 Another study by ESI-FT-ICR/MS in positive ion mode demonstrated the benefit of high resolution in order to characterize resulting molecules, but also to determine fragmentation pathways.203 Note that polymer containing labile hydrogen in terminal groups can easily undergo a fragmentation in gas phase and generate species with silanol groups.203 Tecklenburg et al. compared ESI-FT-ICR/MS and MALDI/MS for the characterization of silsesquioxane polymers.204 Samples are prepared in a mixture chloroform–methanol with addition of ammonium acetate to improve silicone ionization. Mass accuracy down to 5 ppm was reported for each fraction and Si isotopes (28Si, 29Si and 30Si) were identified in one structure.204 In conclusion, MS methods with or without chromatographic separation allow identification and quantitative analysis of Si compounds.
5 Conclusions
Due to their wide use in many applications, silicones are spread in several matrices, mainly in environmental and biological, usually at trace concentrations where they have negative impacts. Many analytical methods improve the identification of silicon compounds in environmental, biological and industrial fields. Two different analytical strategies were investigated for silicon analysis: direct methods for total silicon determination and hyphenated techniques for silicon speciation.However, the unravelling of silicon speciation is relatively scarce, particularly in petroleum and derived samples where complex reactions can occur. Indeed, the matrix complexity associated to contamination, instability and trace level presence makes silicon analysis very hard to achieve. Besides, contamination must be minimized using analyzer specific parts and with care concerning sample conservation and storage.
Atomic spectroscopic methods were usually employed for total Si determination. AAS is one of the first technique used for Si determination with detection limits near equal to 1 mg L−1 for environmental and biological samples. The application of ICP-OES and ICP-MS in environmental, biological and industrial matrices progressed over the decades due to their sensitivity (sub μg L−1 and sub ng L−1 respectively), selectivity and robustness performance. Nevertheless, these analytical methods do not allow molecular separation and identification (speciation) unless they are coupled to a separation technique. On the opposite, NMR methods have the potential to achieve silicon determination but also silicon speciation at high levels (50 mg kg−1) in various matrices.
Hyphenated techniques based on the coupling of a chromatographic separation (GC, LC) giving access to retention time, and a sensitive detection (SIM, AED, ICP) are an established versatile analytical tool for Si speciation. GC-MS in SIM mode has proved to be very effective for silicon speciation with detection levels of nearly 1 μg kg−1 although possible contamination can occur by column and septum bleeding. MS detection (MS/MS, FT-ICR/MS) without previous separation avoids contamination by GC parts and offers bright perspectives both for LMW and HMW molecules respectively in environmental and polymer field respectively. The coupling between SEC and appropriate detection also allows a better understanding of HMW silicon molecules. RP-HPLC-ICP appears as a good alternative for sensitivity with detection limits ranging between 0.1 and 500 μg L−1 for LMW molecules in environmental and biological samples but chromatographic resolution is lower than GC. Thus, GC separation combined to atomic detectors such as AED, ICP-OES and ICP-MS seems to be the more convenient solution with LOD ranging between 0.1 and 10 μg L−1 for Si speciation.
Considering that detection limits greatly depend on sample extraction and concentration, sensitivity comparison is a hard task. However, the more sensitive detection (sub μg L−1) by ICP-MS using as chromatographic detector has been demonstrated. Consequently, the high resolving capacity of GC and the high sensitivity capability of ICP-MS have made this combination the most efficient and attractive for speciation analysis of silicon in the main fields of interest.
This state of art shows different techniques for silicon speciation. Despite the performances of the classical analytical tools, there remains limitations concerning sensitivity and selectivity in complex matrices. For that reason, the coupling of GC-ICP-MS allowing mass interferences resolution offers promising perspectives and deserves further developments to unravel silicon structures.
6 Glossary of abbreviations
7 Acknowledgments
The authors would like to thank J. Castrogeorgi and B. Omais for their useful comments. Special acknowledgments go to the reviewers for their constructive reports, which considerably improved the manuscript.8 References
- G. Chandra., in Organosilicon Materials; The Handbook of Environmental Chemistry, ed. Chandra G, Springer, Berlin, 1997, 1–324p Search PubMed.
- R. Dewil, L. Appels and J. Baeyens, Energy Convers. Manage., 2006, 47(13–14), 1711–1722 CrossRef CAS.
- N. J. Fendinger, R. G. Lehmann, and E. M. Mihaich, in Organosilicon Materials; The Handbook of Environmental Chemistry, ed. Chandra G, Springer, New York, 1997, 1, pp.1–25 Search PubMed.
- A. L. Smith, in The Analytical chemistry of Silicones, ed. Smith A. L., John Wiley & Sons, New York, 1991, 1, pp.1–16 Search PubMed.
- USEPA (United States Environmental Protection Agency), High Production Volume (HPV) Challenge Program, Sponsored Chemicals, 2007, 18-5-2010.
- OECD (Organisation for Economic Co-operation and Development), The 2007 OECD list of high production volume chemicals, oecd secretariat, 2007, 18-1-2010.
- E. F. C. Griessbach and R. G. Lehmann, Chemosphere, 1999, 38(6), 1461–1468 CrossRef CAS.
-
L. Kaj, M. Schlabach, J. Andersson, A. P. Cousins, N.Schmidbauer, and E. Brorström-Lundén, Siloxanes in the Nordic Environment, Report 2005:593, Copenhagen, 2005 Search PubMed.
- A. L. Quinn, J. M. Regan, J. M. Tobin, B. J. Marinik, J. M. McMahon, D. A. Mcnett, C. M. Sushynski, S. D. Crofoot, P. A. Jean and K. P. Plotzke, J. Toxicol. Sci., 2007, 96(1), 145–153 Search PubMed.
- A. L. Quinn, A. Dalu, L. S. Meeker, P. A. Jean, R. G. Meeks, J. W. Crissman, R. H. Gallavan and K. P. Plotzke, Reprod. Toxicol., 2007, 23(4), 532–540 CrossRef CAS.
- M. J. Utell, R. Gelein, C. P. Yu, C. Kenaga, E. Geigel, A. Torres, D. Chalupa, F. R. Gibb, D. M. Speers, R. W. Mast and P. E. Morrow, J. Toxicol. Sci., 1998, 44(2), 206–213 Search PubMed.
- Y. Horii and K. Kannan, Arch. Environ. Contam. Toxicol., 2008, 55(4), 701–710 CrossRef CAS.
- R. Wang, R. P. Moody, D. Koniecki and J. P. Zhu, Environ. Int., 2009, 35(6), 900–904 CrossRef CAS.
- D. M. Templeton, F. Ariese, R. Cornelis, L. G. Danielsson, H. Muntau, H. P. Van Leeuwen and R. Lobinski, Pure Appl. Chem., 2000, 72(8), 1453–1470 CrossRef CAS.
- E. H. Evans, Anal. Bioanal. Chem., 2003, 376(3), 311–312 CAS.
- A. L. Smith, in The Analytical Chemistry of Silicones, ed. Smith A. L., John Wiley & Sons, New York, 1991, 1–551p Search PubMed.
- S. Varaprath, D. Stutts and G. Kozerski, Silicon Chem., 2006, 79, 79–102 CrossRef.
- A. L. Smith and R. D. Parker, in The Analytical chemistry of Silicones, ed. Smith A. L., John Wiley & Sons, New York, 1991, 4, pp.71–91 Search PubMed.
- J. C. Carpenter and R. Gehards, in Organosilicon Materials; The Handbook of Environmental Chemistry, ed. Chandra G, Springer, New York, 1997, 2, pp.29–50 Search PubMed.
- S. J. Lugowski, D. C. Smith, H. Bonek, J. Lugowski, W. Peters and J. Semple, J. Trace Elem. Med. Biol., 2000, 14(1), 31–42 CrossRef CAS.
- C. L. Frye, Environ. Toxicol. Chem., 1987, 6(5), 329–330 CrossRef CAS.
- S. Varaprath and R. G. Lehmann, Journal of Environmental Polymer Degradation, 1997, 5(1), 17–31 Search PubMed.
- R. B. Allen and P. Kochs. G. Chandra, in Organosilicon Materials; The Handbook of Environmental Chemistry, ed. Chandra G, Springer, New York, 1997, 1, pp.1–25 Search PubMed.
- C. Hurd, J. Am. Chem. Soc., 1946, 68, 364 CrossRef.
- J. F. Hobson, R. Atkinson, and W. P. L. Carter, in Organosilicon Materials; The Handbook of Environmental Chemistry, ed. Chandra G, Springer, New York, 1997, 6, pp.138–177 Search PubMed.
- J. T. James, T. F. Limero, J. H. Leano, J. F. Boyd and B. A. Covington, Aviation, Space, and Environmental Medicine, 1994, 851–857 CAS.
- H. C. Shields, D. M. Fleischer and C. J. Weschler, Indoor Air-International Journal of Indoor Air Quality and Climate, 1996, 6(1), 2–17 Search PubMed.
- S. Xu, Environ. Sci. Technol., 1999, 33(4), 603–608 CrossRef CAS.
- R. Dewil, L. Appels, J. Baeyens, A. Buczynska and L. Vaeck, Talanta, 2007, 74(1), 14–19 CrossRef CAS.
- R. Huppmann, H. W. Lohoff and H. F. Schroder, Fresenius J. Anal. Chem., 1996, 354(1), 66–71 CAS.
- R. Grumping and A. V. Hirner, Fresenius J. Anal. Chem., 1999, 363(4), 347–352 CrossRef.
- M. Schweigkofler and R. Niessner, Environ. Sci. Technol., 1999, 33(20), 3680–3685 CrossRef CAS.
- R. G. Lehmann, S. Varaprath and C. L. Frye, Environ. Toxicol. Chem., 1994, 13(11), 1753–1759 CrossRef CAS.
- R. G. Lehmann, S. Varaprath and C. L. Frye, Environ. Toxicol. Chem., 1994, 13(7), 1061–1064 CrossRef CAS.
- R. G. Lehmann, S. Varaprath, R. B. Annelin and J. L. Arndt, Environ. Toxicol. Chem., 1995, 14(8), 1299–1305 CrossRef CAS.
- R. E. Pellenbarg, Environ. Sci. Technol., 1979, 13(5), 565–569 CrossRef CAS.
- R. G. Lehmann, J. R. Miller, S. Xu, U. B. Singh and C. F. Reece, Environ. Sci. Technol., 1998, 32(9), 1260–1264 CrossRef CAS.
- R. Atkinson, Environ. Sci. Technol., 1991, 25(5), 863–866 CAS.
- R. Sommerlade, H. Parlar, D. Wrobel and P. Kochs, Environ. Sci. Technol., 1993, 27(12), 2435–2440 CAS.
- M. J. Whelan, E. Estrada and R. van Egmond, Chemosphere, 2004, 57(10), 1427–1437 CrossRef CAS.
- R. R. Buch and D. N. Ingebrigtson, Environ. Sci. Technol., 1979, 13(6), 676–679 CAS.
- J. C. Carpenter, J. A. Cella and S. B. Dorn, Environ. Sci. Technol., 1995, 29(4), 864–868 CrossRef CAS.
- M. Arnold and T. Kajolinna, Waste Manage., 2010, 30(6), 1011–1017 CrossRef CAS.
- L. Appels, J. Baeyens, J. Degreve and R. Dewil, Prog. Energy Combust. Sci., 2008, 34(6), 755–781 CrossRef CAS.
- M. Rahmani and M. Sohrabi, Kinet. Catal., 2006, 47(6), 891–900 CrossRef CAS.
- Z. Long, J. Storey, S. Lewis and M. J. Sepaniak, Anal. Chem., 2009, 81(7), 2575–2580 CrossRef CAS.
- T. Matsui and S. Imamura, Bioresour. Technol., 2010, 101, S29–S32 CrossRef CAS.
- M. Schweigkofler and R. Niessner, J. Hazard. Mater., 2001, 83(3), 183–196 CrossRef CAS.
- J. J. Barnard, E. L. Todd, W. G. Wilson, R. Mielcarek and R. J. Rohrich, Plast. Reconstr. Surg., 1997, 100(1), 197–203 CrossRef CAS.
- D. Flassbeck, B. Pfleiderer, R. Grumping and A. V. Hirner, Anal. Chem., 2001, 73(3), 606–611 CrossRef CAS.
- E. D. Lykissa, S. V. Kala, J. B. Hurley and R. M. Lebovitz, Anal. Chem., 1997, 69(23), 4912–4916 CrossRef CAS.
- D. Flassbeck, B. Pfleiderer, P. Klemens, K. G. Heumann, E. Eltze and A. V. Hirner, Anal. Bioanal. Chem., 2003, 375(5), 356–362 CAS.
- S. V. Kala, E. D. Lykissa and R. M. Lebovitz, Anal. Chem., 1997, 69(7), 1267–1272 CrossRef CAS.
- L. Lipworth, R. E. Tarone and J. K. McLaughlin, Plast. Reconstr. Surg., 2009, 123(3), 790–793 Search PubMed.
- L. A. Brinton, Plast. Reconstr. Surg., 2007, 120(7), 94S–102S Search PubMed.
- J. Maittala, S. Pennanen and J. Liesivuori, Analyst, 1999, 124(5), 665–668 RSC.
- Dow Corning Corporation, Decamethylcyclopentasiloxane:A 24-month combinedchronic toxicity and oncogenicity whole body vapor inhalation study in Fischer-344 rats, Report 2004-10000-54953, 2005 Search PubMed.
- T. H. Thomas and T. C. Kendrick, J. Polym. Sci., Part A-2, 1969, 7, 537 CrossRef CAS.
- N. Grassie and I. G. Macfarlane, Eur. Polym. J., 1978, 14(11), 875–884 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lageard, Polymer, 2002, 43(7), 2011–2015 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lazzari, Polymer, 2001, 42(6), 2395–2402 CrossRef CAS.
- V. V. Rode, M. A. Verkhotin and S. R. Rafikov, Vysokomol soyed, 1968, 7, 1529–1538 Search PubMed.
- G. Deshpande and M. E. Rezac, Polym. Degrad. Stab., 2001, 74(2), 363–370 CrossRef CAS.
- N. Grassie and K. F. Francey, Polym. Degrad. Stab., 1980, 2(1), 53–66 CrossRef CAS.
- P. Dufresne, Appl. Catal., A, 2007, 322, 67–75 CrossRef CAS.
- K. R. Carduner, R. O. Carter and L. C. Westwood, Appl. Spectrosc., 1988, 42(7), 1265–1267 CrossRef CAS.
- L. N. Kremer and T. G. Hueston, Petrol Tech Q, 2002, 65–69 Search PubMed.
- L. Kellberg, P. Zeuthen and H. J. Jakobsen, J. Catal., 1993, 143(1), 45–51 CrossRef CAS.
- C. P. Lienemann, Oil Gas Sci. Technol., 2005, 60(6), 951–965 CrossRef CAS.
- C. P. Lienemann, S. Dreyfus, C. Pecheyran and O. F. X. Donard, Oil Gas Sci. Technol., 2007, 62(1), 69–77 Search PubMed.
- B. Didillon, J. Cosyns, C. Cameron, D. Uzio, P. Sarrazin and J. P. Boitiaux, Catalyst Desactivation, 1997, 111, 447–454 Search PubMed.
- J. W. Da, J Porus Matter, 2008, 15(199), 204 Search PubMed.
- M. O. G. Souza, P. Reyes and M. C. Rangel, Stud. Surf. Sci. Catal., 1999, 126, 469–472 CAS.
- M. D. Phillips and E. L. Sughrue, Fuel Sci. Technol. Int., 1991, 9(3), 305–319 Search PubMed.
- A. Molnar, I. Bucsi, M. Bartok, F. Notheisz and G. V. Smith, J. Catal., 1986, 98(2), 386–391 CrossRef CAS.
- R. Breivik and R. Egebjerg, Petrol Tech Q, 2008, Q1, 69–75 Search PubMed.
- K. Arnby, M. Rahmani, M. Sanati, N. Cruise, A. A. Carlsson and M. Skoglundh, Appl. Catal., B, 2004, 54(1), 1–7 CrossRef CAS.
- A. C. Larsson, M. Rahmani, K. Arnby, M. Sohrabi, M. Skoglundh, N. Cruise and M. Sanati, Top. Catal., 2007, 45(1–4), 121–124 CrossRef CAS.
- M. Rahmani and M. Sohrabi, React. Kinet. Catal. Lett., 2005, 86(2), 397–405 CrossRef CAS.
- F. McElroy, A. Mennito, E. Debrah and R. Thomas, Spectroscopy, 1998, 13(2), 42 Search PubMed.
- J. A. D. Amaro and S. L. C. Ferreira, J. Anal. At. Spectrom., 2004, 19(2), 246–249 RSC.
- R. I. Botto, J. Anal. At. Spectrom., 1993, 8(1), 51–57 RSC.
- H. M. Dong and V. Krivan, J. Anal. At. Spectrom., 2003, 18(4), 367–371 RSC.
- S. Hauptkorn, J. Pavel and H. Seltner, Fresenius J. Anal. Chem., 2001, 370(2–3), 246–250 CrossRef CAS.
- J. P. McConnell, T. P. Moyer, D. E. Nixon, P. L. Schnur, D. R. Salomao, T. B. Crotty, J. Weinzweig, J. B. Harris and P. M. Petty, American Journal of Clinical Pathology, 1997, 107(2), 236–246 CAS.
- H. T. Liu and S. J. Jiang, Spectrochim. Acta, Part B, 2003, 58(1), 153–157 CrossRef.
- C. H. Yang and S. J. Jiang, Spectrochim. Acta, Part B, 2004, 59(9), 1389–1394 CrossRef.
- A. Mukhtar and A. Limbeck, Anal. Chim. Acta, 2009, 646(1–2), 17–22 CrossRef CAS.
- C. M. Malata, S. Varma, M. Scott, J. C. Liston and D. T. Sharpe, Medical Progress Through Technology, 1994, 20(3–4), 251–260 CAS.
- W. Peters, D. Smith and S. Lugowski, Ann. Plast. Surg., 1995, 35(4), 442–443 CrossRef.
- S. Lugowski, D. Smith and J. Bzdega, Chemia Analityczna, 1998, 43(6), 1011–1019 Search PubMed.
- S. Felby, Forensic Sci. Int., 1986, 32(1), 61–65 CrossRef CAS.
- M. D. Huang and V. Krivan, Spectrochim. Acta, Part B, 2007, 62(3), 297–303 CrossRef.
- F. Y. Leung and P. Edmond, Clin. Biochem., 1997, 30(5), 399–403 CrossRef CAS.
- M. Hornung and V. Krivan, J. Anal. At. Spectrom., 1997, 12(10), 1123–1130 RSC.
- H. J. Gitelman and F. B. Alderman, J. Anal. At. Spectrom., 1990, 5(8), 687–689 RSC.
- Z. E. Huang, Spectrochim. Acta, Part B, 1995, 50(11), 1383–1393 CrossRef.
- W. Frech and A. Cedergren, Anal. Chim. Acta, 1980, 113(2), 227–235 CrossRef CAS.
- C. J. Rademeyer and I. Vermaak, J. Anal. At. Spectrom., 1992, 7(2), 347–351 RSC.
- G. N. Brown, D. L. Styris and M. W. Hinds, J. Anal. At. Spectrom., 1995, 10(8), 527–531 RSC.
- A. F. Shoukry, Y. M. Issa, R. A. Farghaly, M. Grasserbauer, H. Puxbaum and J. Rendl, Fresenius J. Anal. Chem., 1998, 360(6), 650–653 CrossRef CAS.
- P. Bermejo-Barrera, M. C. Barciela-Alonso, R. Dominguez-Gonzalez, A. Bermejo-Barrera, J. A. C. de Juan and J. M. Fraga-Bermudez, Anal. Bioanal. Chem., 2002, 374(7–8), 1290–1293 CrossRef CAS.
- R. D. Parker, Journal of The Association of Official Analytical Chemists, 1990, 73(5), 721–723 Search PubMed.
- D. A. Mccamey, D. P. Iannelli, L. J. Bryson and T. M. Thorpe, Anal. Chim. Acta, 1986, 188, 119–126 CrossRef CAS.
- P. Masson, M. Dauthieu, F. Trolard and L. Denaix, Spectrochim. Acta, Part B, 2007, 62(3), 224–230 CrossRef.
- Y. C. Qin and H. X. Weng, Estuarine, Coastal Shelf Sci., 2006, 67(3), 433–440 CrossRef CAS.
- A. L. Molinero, L. Martinez, A. Villareal and J. R. Castillo, Talanta, 1998, 45(6), 1211–1217 CrossRef.
- J. M. T. Carneiro, A. L. R. M. Rossete, G. S. Oliveira and J. A. Bendassolli, Commun. Soil Sci. Plant Anal., 2007, 38(11–12), 1411–1423 CrossRef CAS.
- N. Watanabe, T. Nakamura, E. Watanabe, E. Sato and Y. Ose, Sci. Total Environ., 1984, 35(1), 91–97 CrossRef CAS.
- N. Watanabe, H. Nagase and Y. Ose, Sci. Total Environ., 1988, 73(1–2), 1–9 CrossRef CAS.
- J. F. Belliveau, W. C. Griffiths, C. G. Wright and J. R. Tucci, Annals of Clinical and Laboratory Science, 1991, 21(5), 328–334 Search PubMed.
- G. M. Bercowy, H. Vo and F. Rieders, Journal of Analytical Toxicology, 1994, 18(1), 46–48 Search PubMed.
- X. J. Jia, T. B. Wang, X. D. Bu and J. Wu, Microchem. J., 2003, 75(2), 103–107 CrossRef CAS.
- A. P. Krushevska and R. M. Barnes, J. Anal. At. Spectrom., 1994, 9(9), 981–984 RSC.
- J. J. Powell, S. A. McNaughton, R. Jugdaohsingh, S. H. C. Anderson, J. Dear, F. Khot, L. Mowatt, K. L. Gleason, M. Sykes, R. P. H. Thompson, C. Bolton-Smith and M. J. Hodson, Br. J. Nutr., 2005, 94(5), 804–812 CrossRef CAS.
- R. Sanchez, J. L. Todoli, C. P. Lienemann and J. M. Mermet, J. Anal. At. Spectrom., 2009, 24(4), 391–401 RSC.
- R. Sanchez, J. L. Todoli, C. P. Lienemann and J. M. Mermet, J. Anal. At. Spectrom., 2009, 24(10), 1382–1388 RSC.
- R. Sanchez, J. L. Todoli, C. P. Lienemann and J. M. Mermet, J. Anal. At. Spectrom., 2010, 25(2), 178–185 RSC.
- J. Zhang, L. Li, J. Zhang, Q. Zhang, Y. Yang and Q. Jin, Pet. Sci. Technol., 2007, 25(3–4), 443–451 CrossRef CAS.
- R. I. Botto and J. Talbott, 2009 Winter Conference on Plasma Spectrochemistry, Graz, Austria, 2009 Search PubMed.
- Y. Takaku, K. Masuda, T. Takahashi and T. Shimamura, J. Anal. At. Spectrom., 1994, 9(12), 1385–1387 RSC.
- E. Engstrom, I. Rodushkin, D. C. Baxter and B. Ohlander, Anal. Chem., 2006, 78(1), 250–257 CrossRef.
- L. Y. Alleman, D. Cardinal, C. Cocquyt, P. D. Plisnier, J. P. Descy, I. Kimirei, D. Sinyinza and L. Andre, J. Great Lakes Res., 2005, 31(4), 509–519 CrossRef CAS.
- C. F. Wang, F. H. Tu, S. L. Jeng and C. J. Chin, J. Radioanal. Nucl. Chem., 1999, 242(1), 97–103 CAS.
- X. B. Feng, S. L. Wu, A. Wharmby and A. Wittmeier, J. Anal. At. Spectrom., 1999, 14(6), 939–946 RSC.
- P. Klemens and K. G. Heumann, Fresenius J. Anal. Chem., 2001, 371(6), 758–763 CrossRef CAS.
- K. Van Dyck, H. Robberecht, R. Van Cauwenbergh, H. Deelstra, J. Arnaud, L. Willemyns, F. Benijts, J. A. Centeno, H. Taylor, M. E. Soares, M. L. Bastos, M. A. Ferreira, P. C. D'Haese, L. V. Lamberts, M. Hoenig, G. Knapp, S. J. Lugowski, L. Moens, J. Riondato, R. Van Grieken, M. Claes, R. Verheyen, L. Clement and M. Uytterhoeven, J. Anal. At. Spectrom., 2000, 15(6), 735–741 RSC.
- J. Riondato, F. Vanhaecke, L. Moens and R. Dams, J. Anal. At. Spectrom., 1997, 12(9), 933–937 RSC.
- J. Talbott and R. I. Botto, Silicon in Naphtas: A near cold plasma approach for sub-ppb detection limits on an ICP/MS, 2008 Winter Conference on Plasma Spectrochemistry, Temecula, California, 2008 Search PubMed.
- H. M. Kuss, D. Bossmann and M. Muller, At. Spectrosc., 1994, 15(4), 148–150 CAS.
- M. Resano, M. Verstraete, F. Vanhaecke and L. Moens, J. Anal. At. Spectrom., 2002, 17(8), 897–903 RSC.
- I. De Schrijver, M. Aramendia, M. Resano, A. Dumoulin and F. Vanhaecke, J. Anal. At. Spectrom., 2008, 23(4), 500–507 RSC.
- R. Hamilton, University of Lakehead, 2001.
- R. B. Taylor, B. Parbhoo, and D. M. Fillmore, in The Analytical chemistry of Silicones, ed. Smith A. L., John Wilery & Sons, New York, 1991, 12, pp.347–414 Search PubMed.
- P. Fux, Analyst, 1990, 115(2), 179–183 RSC.
- L. Garrido, B. Pfleiderer, B. G. Jenkins, C. A. Hulka and D. B. Kopans, Magn. Reson. Med., 1994, 31(3), 328–330 CrossRef CAS.
- L. Garrido, A. Bogdanova, L. L. Cheng, B. Pfleiderer, E. Tokareva, J. L. Ackerman and T. J. Brady, Immunology of Silicones, 1996, 210, 49–58 Search PubMed.
- B. Pfleiderer, A. Moore, E. Tokareva, J. L. Ackerman and L. Garrido, Biomaterials, 1999, 20(6), 561–571 CrossRef CAS.
- W. E. Hull, Magn. Reson. Med., 1999, 42(5), 984–995 CrossRef CAS.
- P. Macdonald, N. Plavac, W. Peters, S. Lugowski and D. Smith, Anal. Chem., 1995, 67(20), 3799–3801 CrossRef CAS.
- J. M. Bellama, S. R. Meyer and R. E. Pellenbarg, Appl. Organomet. Chem., 1991, 5(2), 107–109 CrossRef CAS.
- D. R. Peterson, R. D. Parker, and M. J. Owen, in The Analytical chemistry of Silicones, ed. Smith A. L., John & Wiley Sons, New York, 1991, pp.485–515 Search PubMed.
- A. D. Lipp and A. L. Smith, in The Analytical chemistry of Silicones, ed. Interscience Publication, New York, 1991, pp.305–337 Search PubMed.
- M. Hernandez, P. Diaz and M. Carrillo, UNITAR 5th International Conference, 1991, 57–63 Search PubMed.
- E. D. Lipp, P. S. Rzyrkowski and R. F. Geiger, Tappi J., 1987, 95–98 Search PubMed.
- J. A. Emery, G. Kasnic, N. Hardt and S. S. Spanier, Modern pathology, 1994, 7(7), 728–733 Search PubMed.
- N. S. Hardt, L. T. Yu, G. Latorre and B. Steinbach, Modern pathology, 1994, 7(6), 669–676 Search PubMed.
- L. H. Kidder, V. F. Kalasinsky, J. L. Luke, I. W. Levin and E. N. Lewis, Nat. Med., 1997, 3(2), 235–237 CrossRef CAS.
- M. Ende and G. Spiteller, Mass Spectrom. Rev., 1982, 1, 29–62 CAS.
- J. De Zeeuw, Am. Lab., 2005, 37, 18–19 CAS.
- S. Varaprath and P. S. Larson, J. Polym. Environ., 2002, 10(4), 119–131 CrossRef CAS.
- C. Sparham, R. van Egmond, S. O'Connor, C. Hastie, M. Whelan, R. Kanda and O. Franklin, J. Chromatogr., A, 2008, 1212, 124–129 CrossRef CAS.
- S. Varaprath, M. Seaton, D. McNett, L. Cao and K. P. Plotzke, Int. J. Environ. Anal. Chem., 2000, 77(3), 203–219 CrossRef CAS.
- J. Carter, L. Ebdon and E. H. Evans, Microchem. J., 2004, 76(1–2), 35–41 CrossRef CAS.
- D. M. Chambers, D. O. McElprang, J. P. Mauldin, T. M. Hughes and B. C. Blount, Anal. Chem., 2005, 77(9), 2912–2919 CrossRef CAS.
- S. J. Pattinson and J. P. G. Wilkins, Analyst, 1989, 114(4), 429–434 RSC.
- Y. X. Wang, Am. Lab., 2006, 38(2), 10–12 CAS.
- S. B. Dorn and E. M. S. Frame, Analyst, 1994, 119(8), 1687–1694 RSC.
- C. Eaborn, in Organosilicon Compounds, ed. Butterworth Scientific, London, 1960, 8, pp.227–288 Search PubMed.
- S. Varaprath, K. L. Salyers, K. P. Plotzke and S. Nanavati, Drug Metab. Dispos., 1999, 27(11), 1267–1273 CAS.
- S. Varaprath, J. M. McMahon and K. P. Plotzke, Drug Metab. Dispos., 2003, 31(2), 206–214 CrossRef CAS.
- V. Blechta, M. Kurfurst, J. Sykora and J. Schraml, J. Chromatogr., A, 2007, 1145(1–2), 175–182 CrossRef CAS.
- W. R. Biggs, J. C. Fetzer and R. J. Brown, Anal. Chem., 1987, 59(23), 2798–2802 CrossRef CAS.
- W. R. Biggs and J. C. Fetzer, Anal. Chem., 1989, 61(3), 236–240 CrossRef CAS.
- D. R. Heine, M. B. Denton and T. D. Schlabach, J. Chrom. Sci., 1985, 23(10), 454–458 Search PubMed.
- L. Ebdon, M. Foulkes, K. Fredeen, C. Hanna and K. Sutton, Spectrochim. Acta, Part B, 1998, 53(6–8), 859–865 CrossRef.
- R. G. Lehmann and J. R. Miller, Environ. Toxicol. Chem., 1996, 15(9), 1455–1460 CrossRef CAS.
- J. C. Ivaldi and J. F. Tyson, Spectrochim. Acta, Part B, 1995, 50(10), 1207–1226 CrossRef.
- R. D. Steinmeyer and M. A. Becker, in The Analytical chemistry of Silicones, ed. Smith A. L., John Wiley & Sons, New York, 1991, 10, pp.255–303 Search PubMed.
- D. M. Smith, R. G. Lehmann, R. Narayan, G. E. Kozerski and J. R. Miller, Compost Science & Utilization, 1998, 6(2), 6–12 Search PubMed.
- D. W. Hausler and L. T. Taylor, Anal. Chem., 1981, 53(8), 1223–1227 CrossRef CAS.
- D. W. Hausler and L. T. Taylor, Anal. Chem., 1981, 53(8), 1227–1231 CAS.
- E. P. Maziarz, X. M. Liu, E. T. Quinn, Y. C. Lai, D. M. Ammon and G. L. Grobe, J. Am. Soc. Mass Spectrom., 2002, 13(2), 170–176 CrossRef CAS.
- X. M. Liu, E. P. Maziarz, D. J. Heiler and G. L. Grobe, J. Am. Soc. Mass Spectrom., 2003, 14(3), 195–202 CrossRef CAS.
- R. Grumping, D. Mikolajczak and A. V. Hirner, Fresenius J. Anal. Chem., 1998, 361(2), 133–139 CrossRef.
- S. C. Popat and M. A. Deshusses, Environ. Sci. Technol., 2008, 42(22), 8510–8515 CrossRef CAS.
- S. Wacholz, F. Keidel, U. Just, H. Geissler and K. Kappler, J. Chromatogr., A, 1995, 693, 89–99 CrossRef CAS.
- E. D. Pellizzari, J. E. Bunch, R. E. Berkley and J. Mc Rae, Anal. Chem., 1976, 48(6), 1–4.
- X. M. Wang, S. C. Lee, G. Y. Sheng, L. Y. Chan, J. M. Fu, X. D. Li, Y. S. Min and C. Y. Chan, Appl. Geochem., 2001, 16(11–12), 1447–1454 CrossRef CAS.
- J. J. Kennan, L. L. M. Breen, T. H. Lane and R. B. Taylor, Anal. Chem., 1999, 71(15), 3054–3060 CrossRef CAS.
- H. Homma, T. Kuroyagi, K. Izumi, C. L. Mirley, J. Ronzello and S. A. Boggs, IEEE Trans. Nucl. Sci., 2000, 15(2), 796–803 CAS.
- K. Kappler, U. Scheim, F. Keidel and U. Just, Fresenius J. Anal. Chem., 1996, 354(1), 21–26 CAS.
- H. Hillborg, S. Karlsson and U. W. Gedde, Polymer, 2001, 42(21), 8883–8889 CrossRef CAS.
- A. D. Hall and M. Patel, Polym. Degrad. Stab., 2006, 91(10), 2532–2539 CrossRef CAS.
- M. Edler, D. Metze, N. Jakubowski and M. Linscheid, J. Anal. At. Spectrom., 2002, 17(10), 1209–1212 RSC.
- J. A. Caruso and M. Montes-Bayon, Ecotoxicol. Environ. Saf., 2003, 56(1), 148–163 CrossRef CAS.
- C. A. P. de Leon, M. Montes-Bayon and J. A. Caruso, J. Chromatogr., A, 2002, 974(1–2), 1–21 CrossRef CAS.
- J. C. A. Wuilloud, R. G. Wuilloud, A. P. Vonderheide and J. A. Caruso, Spectrochim. Acta, Part B, 2004, 59(6), 755–792 CrossRef.
- B. Bouyssiere, J. Szpunar and R. Lobinski, Spectrochim. Acta, Part B, 2002, 57(5), 805–828 CrossRef.
- K. A. Forbes, J. F. Vecchiarelli, P. C. Uden and R. M. Barnes, Anal. Chem., 1990, 62(18), 2033–2037 CrossRef CAS.
- D. W. Later, E. R. Campbell and B. E. Richter, J. High Resolut. Chromatogr., 1988, 11(1), 65–69 CAS.
- U. Just, F. Mellor and F. Keidel, J. Chromatogr., A, 1994, 683(1), 105–113 CrossRef CAS.
- J. M. T. Carneiro, A. L. R. M. Rossete and J. A. Bendassolli, Anal. Lett., 2008, 41(9), 1640–1647 CrossRef CAS.
- K. Badjagbo, A. Furtos, M. Alaee, S. Moore and S. Sauve, Anal. Chem., 2009, 81(17), 7288–7293 CrossRef CAS.
- K. Badjagbo, M. Heroux, M. Alaee, S. Moore and S. Sauve, Environ. Sci. Technol., 2010, 44(2), 600–605 CrossRef CAS.
- V. Rouessac, S. Roualdes and J. Durand, Chem. Vap. Deposition, 2002, 8(4), 155–161 CrossRef CAS.
- B. Apicella, A. Ciajolo, M. Milian, C. Galmes, A. A. Herod and R. Kandiyoti, Rapid Commun. Mass Spectrom., 2004, 18(3), 331–338 CrossRef CAS.
- W. E. Wallace, C. M. Guttman and J. M. Antonucci, J. Am. Soc. Mass Spectrom., 1999, 10(3), 224–230 CrossRef CAS.
- W. E. Wallace, C. M. Guttman and J. M. Antonucci, Polymer, 2000, 41(6), 2219–2226 CrossRef CAS.
- A. G. Marshall, C. L. Hendrickson and G. S. Jackson, Mass Spectrom. Rev., 1998, 17(1), 1–35 CrossRef CAS.
- E. P. Maziarz, G. A. Baker and T. D. Wood, Macromolecules, 1999, 32(13), 4411–4418 CrossRef.
- S. Murthy and J. L. Beauchamp, J. Phys. Chem., 1992, 96(3), 1247–1257 CrossRef CAS.
- H. P. Chen, J. Am. Soc. Mass Spectrom., 2003, 14(9), 1039–1048 CrossRef CAS.
- R. E. Tecklenburg, W. E. Wallace and H. P. Chen, Rapid Commun. Mass Spectrom., 2001, 15(22), 2176–2185 CrossRef CAS.
Footnote |
| † This article is part of a themed issue highlighting outstanding and emerging work in the area of speciation. |
| This journal is © The Royal Society of Chemistry 2011 |