Large area mosaic films of graphene–titania: self-assembly at the liquid–air interface and photo-responsive behavior†
Timothy N.
Lambert
*a,
Carlos A.
Chavez
a,
Nelson S.
Bell
b,
Cody M.
Washburn
c,
David R.
Wheeler
d and
Michael T.
Brumbach
e
aDepartment of Materials, Devices and Energy Technologies, Sandia National Laboratories, P.O. Box 5800, MS-0734, Albuquerque, New Mexico 87185, USA. E-mail: tnlambe@sandia.gov; Fax: +1 505 844 7786; Tel: +1 505 284 6967
bDepartment of Electronic & Nanostructured Materials, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA
cDepartment of Organic Materials, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA
dDepartment of Biosensors and Nanomaterials, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA
eDepartment of Materials Characterization, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA
First published on 4th November 2010
Abstract
Photo-responsive graphene–titania composite nanofilms were formed via evaporative induced self-assembly at the air–liquid interface from the UV-photo-reduction of titania–graphene oxide colloidal solutions.
Graphene is attracting much attention due to its unique mechanical, electronic and thermal properties and wide ranging potential applications.1 Combining graphene, with its theoretically high surface area of ∼2630 m2 g−1, and inorganic nanomaterials makes it possible to develop new nanocomposite materials with expanded utilities. For example, coupling the desirable properties of graphene with the well established photo-properties of titania (TiOx)2 has led to enhanced photocatalyst and photovoltaic materials.3 A significant challenge is in developing new synthetic routes to such materials. Routes based on solution processing are often favored due to their expected cost effectiveness and simplicity.4 To date, TiOx–graphene composites have been prepared by solution mixing, e.g. dispersion of nanoparticle titania with colloidal graphene oxide (GO), or by the in situgrowth of TiOx with GO or surfactant dispersed graphene. Chemical, thermal and photo-reduction methods have been employed to convert TiOx–GO to TiOx–reduced graphene oxide (TiOx–RGO) materials. Harvesting the power of self-assembly could provide facile, chemically directed approaches to such composite materials.5
Recently we prepared a series of TiO2–GO composites, with varying TiO2/GO ratios, from the hydrolysis of TiF4 in the presence of GO, and we additionally formed reduced graphene oxide (RGO) composites (i.e.TiO2–RGO) using chemical and thermal reduction methods.4 Under appropriate synthesis conditions, long range ordered stacks of TiO2–GO were formed in the micron sized regime. We rationalized that under carefully controlled conditions we should be able to form similar self-assembled-titanium dioxide–reduced graphene oxide (SA-TiO2–RGO) structures on the nanoscale (∼100 nm thickness). In particular we were attracted to the “on-demand” photo-reduction of GO with TiO2,6 thereby forming soluble TiO2–RGO (sol-TiO2–RGO). We hypothesized that under the appropriate conditions, an evaporation induced self-assembly7 could be used to prepare the target SA-TiO2–RGO films at the fluid–air interface.
We now report that upon UV-photo-reduction of an aqueous solution of TiO2–GO, the sol-TiO2–RGO that is formed can form unsupported self-assembled nanosheets (∼30 to 200 nm thick and up to ∼1 to 2 cm2) at the fluid–air interface under appropriate evaporative conditions. These SA-TiO2–RGO films are formed as a “mosaic assembly” of photo-reduced GO and inter-calated/graphene-bound TiO2. Surface tension and surface wetting measurements support the formation of sheets at the liquid–air interface via an evaporative driven process aided by methanol. These mosaic films were successfully transferred to solid substrates and shown to display photocurrent generation under UV irradiation (λexc = 350 nm, 25–30 mJ cm−2): e.g.SA-TiO2–RGO films deposited onto interdigitated electrodes (IDEs) generated ∼12 to 20 nA cm−2 of current when excited with UV radiation. Exposure of SA-TiO2–RGO films to UV radiation, followed by exposure to deaerated silver (Ag+) salt solutions led to the reduction of Ag+, forming silver nanoparticles on the surface of the graphene film, i.e.Ag0–SA-TiO2–RGO films were formed. It is likely that the UV exposure caused charge transfer, resulting in the storage of electrons in the graphene for subsequent silver reduction.2,3,6
The solution (sol-TiO2–RGO) (ESI†) obtained upon photo-reduction was examined by UV-vis spectroscopy and was found to have a λmax value of 253 nm, compared to aqueous GO with λmax = 230 nm, consistent with an increase in overall conjugation, see Fig. S1a†. Solutions left undisturbed in a loosely covered glass beaker generated numerous films at the air–liquid interface over the course of several days, Fig. 1a and S1†. Leaving the solutions undisturbed for longer times yielded larger (and typically more) films. In general, two different film morphologies were present: square-like shapes and high aspect ratio rectangles, Fig. S1b and c†. If the initial resulting black solution (sol-TiO2–RGO) was stored in a tightly sealed flask, neither obvious precipitate nor film formation occurred, indicative of general colloidal stability and film formation from an evaporation induced self-assembly. A MeOH wash was also found to be critical for effective film formation.
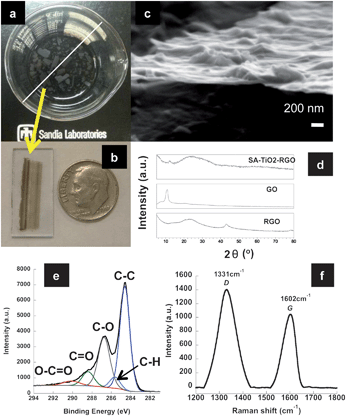 | ||
| Fig. 1 SA-TiO2–RGO thin films (a) after assembly at the air–liquid interface (white bar = 8.5 cm), (b) collected on ITO/glass slide, USA dime is shown for scale; (c) SEM micrograph of exposed edge after deposition on SiOx/Si wafer and partial delamination; (d) XRD patterns comparing SA-TiO2–RGO, GO and RGO; (e) XPS spectra indicating partial de-oxygenation and (f) Raman spectra indicating reduction of the GO sheets. | ||
In order to examine the films, they were carefully transferred to either SiOx/Si wafers, glass or IDEs for characterization, Fig. 1b. Scanning electron microscopy (SEM) determined well formed films ranged from ∼30 to 200 nm in thickness, and were composed of TiO2 nanoparticles/nanoflowers4 and a graphene material, sandwiched together, Fig. 1c and S2†. Electron dispersive X-ray spectroscopy (EDS) confirmed the presence of Ti as expected, Fig. S2d†. Films that were collected shortly after the formation (within 1 day) show the development of the films as a mosaic assembly of smaller pieces of graphene and TiO2, i.e.TiO2–RGO and/or RGO sheets are randomly stacked and overlapped with TiO2 in between sheets or stacks of sheets, Fig. S3†.
The X-ray diffraction (XRD) spectrum was obtained on a collection of films. SA-TiO2–RGO displays one major broad peak at ∼2θ = 24° (002) and a minor sharper peak at ∼2θ = 12°, Fig. 1d. The broadness of the graphene (002) peak is consistent with short ordered domains along the stacking direction and indicative of a material comprised of disordered-like nanosheets, while the minor peak at ∼2θ = 12° is consistent with some GO like character. GO and RGO8 are shown for comparison purposes. Titania is not readily observed, due to its low relative concentration in the film. The XRD is consistent with a GO that is only partially reduced.
X-Ray photoelectron spectroscopy (XPS) was also conducted on a deposited film to determine the extent and type of oxygen functionalities present. Survey XPS spectra gave atomic percentages of 82% C and 17% O for SA-TiO2–RGO, versus 73% C and 25% O for a GO sample. The C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio (as determined by XPS analysis) of ∼5:1 for the SA-RGO–TiO2 film is significantly lower than for the RGO sample obtained by the chemical reduction of GO with hydrazine,8ca. 23:1. Quantification from XPS shows a Ti concentration of less than 1%, which can be partially attributed to the surface sensitivity of XPS as well as a low titanium content in the composite film. The XPS C 1s spectra, for SA-TiO2–RGO film, are shown in Fig. 1e and Fig. S4b†. C 1s peak fitting was performed using a common set of 5 peaks for all C 1s spectra, following previous work.8 For GO, C 1s fitted peak positions, [FWHM], and peak assignments were identified at 284.5 [1.4] eV (C–C), 285.4 [1.2] eV (C–H), 286.5 [1.3] eV (C–O), 288.1 [1.4] eV (C
:O ratio (as determined by XPS analysis) of ∼5:1 for the SA-RGO–TiO2 film is significantly lower than for the RGO sample obtained by the chemical reduction of GO with hydrazine,8ca. 23:1. Quantification from XPS shows a Ti concentration of less than 1%, which can be partially attributed to the surface sensitivity of XPS as well as a low titanium content in the composite film. The XPS C 1s spectra, for SA-TiO2–RGO film, are shown in Fig. 1e and Fig. S4b†. C 1s peak fitting was performed using a common set of 5 peaks for all C 1s spectra, following previous work.8 For GO, C 1s fitted peak positions, [FWHM], and peak assignments were identified at 284.5 [1.4] eV (C–C), 285.4 [1.2] eV (C–H), 286.5 [1.3] eV (C–O), 288.1 [1.4] eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 289.7 [2.0] eV (O–CO),8 see Fig. S4a†. C 1s peak fits for SA-TiO2–RGO gave fitted components at 284.5 [1.2] eV (C–C), 285.5 [1.2] eV (C–H), 286.6 [1.4] eV (C–O), 288.3 [1.4] eV (CO) and 290.0 [2.0] eV (O–CO and carbonate), Fig. 1e and S4b†. There is a ∼45% reduction in the peak height, and a narrowing for the C 1s (C–O) peak for SA-TiO2–RGO (with expected corresponding increase in C–C peak), as compared to GO. In contrast, the CO components are similar in the GO and SA-RGO–TiO2 samples. It has been proposed that ∼50% of the oxygen groups in GO are able to accept electrons, and that the carbonyl groups are not likely to be reduced under such mild photo-reduction conditions.6 The XPS data presented here also support a fairly low level (∼32%) of de-oxygenation from photo-reduction.
O) and 289.7 [2.0] eV (O–CO),8 see Fig. S4a†. C 1s peak fits for SA-TiO2–RGO gave fitted components at 284.5 [1.2] eV (C–C), 285.5 [1.2] eV (C–H), 286.6 [1.4] eV (C–O), 288.3 [1.4] eV (CO) and 290.0 [2.0] eV (O–CO and carbonate), Fig. 1e and S4b†. There is a ∼45% reduction in the peak height, and a narrowing for the C 1s (C–O) peak for SA-TiO2–RGO (with expected corresponding increase in C–C peak), as compared to GO. In contrast, the CO components are similar in the GO and SA-RGO–TiO2 samples. It has been proposed that ∼50% of the oxygen groups in GO are able to accept electrons, and that the carbonyl groups are not likely to be reduced under such mild photo-reduction conditions.6 The XPS data presented here also support a fairly low level (∼32%) of de-oxygenation from photo-reduction.
When examined by Raman spectroscopy the SA-TiO2–GO exhibited slight downfield shifts in the D- and G-bands (1331 and 1602 cm−1, respectively) along with a marked increase in the D/G ratios (ID/IG = 1.34) as compared to that for GO, Fig. 1f. GO prepared by us exhibited D- and G-bands at 1338 and 1603 cm−1 with a D/G ratio of 0.84.4,8 Combined with the XRD and XPS data, this indicates that the graphene in the SA-TiO2–RGO films has undergone a partial reduction, with the carboxylates (and some alcohol or epoxide functionalities) surviving the reduction process.
In order to gain some insight into the self-assembly process, zeta (ζ) potential and dynamic light scattering (DLS) measurements of sol-TiO2–RGO prior to film formation were obtained. The development of the ζ-potential as a function of pH relates to the surface groups in the studied material. We recently demonstrated that GO and TiO2–GO show significant ζ-potentials even under highly acidic conditions (−35 to −40 mV at pH 2.5), which are sufficient for their exfoliation and dispersion in water.4 The surface modification of GO nanosheets with TiO2 presents little change in the development of the ζ-potential beyond a slight reduction in the strength of the measured value. The sol-TiO2–RGO prepared here is similar in nature. ζ-Potentials for sol-TiO2–RGO were in the range of −35 to −45 mV across the entire pH range (Fig. S5†) and these data help to explain the general colloidal stability of sol-TiO2–RGO observed by us and others.6 This wide range pH stability is also consistent with the retention of carboxylate groups during the photo-reduction. DLS data show the average particle size in the solution to be ∼250 nm in size across a wide pH range, Fig. S6†. This is generally consistent with the numerous sheets observed in the mosaic assembly SEM images, Fig. S3†.
Contact angle measurements using known parameter liquids (Table S1†) on the SA-TiO2–RGO films were also performed and analyzed using the surface tension component theory based on the van Oss–Chaudhury–Good (VOCG) model, see ESI†. The VOCG model treats a surface as composed of an apolar component related to Lifshitz–van der Waals interactions (γLW), and the electron acceptor parameter (γ+) and an electron donating parameter (γ−). The treatment results in a relationship between measured contact angle of a probe liquid with the interaction components for the liquid and solid phases (see ESI†).9 Surface energy paramaters for the SA-TiO2–RGO film were determined to be γsLW = 34.2 mJ m−2, γs+ = 0.897 mJ m−2 and γs− = 2.619 mJ m−2. These data indicate that the SA-TiO2–RGO film surface has a low surface polarity component (in addition to the carboxylate groups). While the van der Waals interaction is the major component of the surface energy of the SA-TiO2–RGO, the γs− value indicates that the surface is also a weak electron donor, consistent with the π-bonding character of a reduced graphene surface. Using these parameters, the interaction between the components of the surface energies of the liquid and solid phases (in equilibrium with the atmosphere) can be calculated using eqn (1). Additionally, a free energy of wetting (ΔG131) for the films can also be calculated as ΔG131 = −59.09 mJ m−2 indicating that the material is not hydrophobic (as observed), see ESI†.
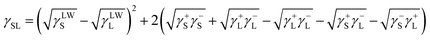 | (1) |
The calculated values of γsLW, γs+ and γs− are used with eqn (2) to determine the total surface energy (γs), which is calculated to be 37.3 mJ m−2.
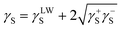 | (2) |
These values (Table S2†) show that the material is not hydrophobic and is wetted by the aqueous fluid phase. The ζ-potentials measured are the relevant dispersion mechanism and explain the colloidal stability prior to evaporation.
The self-assembly process performed here requires the addition of both methanol and subsequent evaporation, which has several effects within the interface and in the interactions between/with the particles. Addition of methanol to water is known to lower the surface tension in a non-linear fashion.10 For example, concentrations as low as 5 mol% methanol lower the surface tension by ∼12 mN m−1. Transfer of a particle or unit surface area from the fluid phase to a second phase is energetically described by the surface energy of transfer, ΔW = γ13 − γ12. The aqueous surface tension of 72 mN m−1 (=γ12) can therefore be lowered to 60 mN m−1 or less by the addition of methanol, and with a solid value γ13 = 37.3 mJ m−2, the nucleation barrier to dewetting is clearly lowered significantly.10
The surface assembly observed here will require nucleation of a stable pore nuclei or pinhole in the growing surface sheet to dewet at the interface. Attempts to measure the contact angle with methanol over the SA-TiO2–RGO films showed rapid spreading and the formation of Moiré fringes before evaporation. This effective zero value for the contact angle is evidence that these films are completely wet by methanol and indicate high surface interaction of methanol with the SA-RGO–TiO2 nanosheets. Thus there is likely a driving force for the association of nanosheets with the excess concentration of methanol at the evaporating interface, leading to thin fluid layers over the nanosheets, and a lower surface tension at the interface which increases the probability of forming a stable pinhole. Local defects leading to the pinhole formation could also be promoted by the methanol due to evaporation induced local cooling or fluid transfer.10
The model for self-assembly therefore is proposed to relate to the nucleation of dewetted nanosheets at the interface based on the surface activity of methanol with both the solid and fluid phases. Growth of these nanosheets into macroscopic structures is a topic which will require additional study, especially with regard to understanding the origin of the two sheet/film morphologies that are observed. It is proposed that additional nanosheets are added by both growth from within the bulk of the solution, as well as lateral capillary forces resulting from the menisci of the dewetted assemblies. Such assembly behavior has been observed in a macroscopic sense, with high aspect rectangular films “annealing” on the surface to form larger films over time, Fig. S1d†.
As shown in recent studies, RGO materials can store and shuttle electrons and have therefore has potential as new 2-dimensional catalyst nanomats.11 Excitation of the TiO2 in a TiO2–GO solution leads to the formation of an excited state (electron–hole pair) which injects the electron into the GO structure, resulting in the reduction of GO and (if in excess) charge storage. This phenomenon is being exploited to develop new devices for photovoltaics, and photoelectrochemical catalysis.3 Very recently, it has been demonstrated that graphene can provide for a ∼90% improvement in photocurrent in TiO2–RGO photoanodes.3a In order to demonstrate that the SA-TiO2–RGO films prepared here could function in a similar manner, films were deposited onto an IDE and dried, Fig. 2a. First, the change in resistance across the IDE device was measured while the sample was exposed to UV radiation (λexc = 365 nm, ∼21.7 mJ cm−2). Fig. 2b shows a representative decrease in resistance of ∼200 kΩ when a ∼0.25 cm2 film is exposed. This drop in resistance is attributed to (1) the generation of an electron–hole pair in the titania, and (2) the subsequent injection/storage of electrons into the graphene structure of the SA-TiO2–RGO sheet. XPS studies indicated that even with extended UV exposure (several hours), no additional reduction or oxidation of the graphene sheet occurred, indicating graphene stability. While oxygen is known to be a scavenger of electrons/holes that are generated in solution, the devices prepared here surprisingly function in air. The amount of photocurrent generated as measured in air was typically ∼12 to 20 nA cm−2. Three repetitive cycles were also performed (e.g. 5 min ON and 30 min OFF), demonstrating the stability of the film to multiple exposures, Fig. 2c. The low current obtained is attributed to the thin nature of the film and also its optical transparency of ∼50%. The ratio of TiO2 to RGO is another variable that will provide an influence over the amount of current generated. The slow return to ground state is also again surprising given that these devices are exposed to air. Some downward drift is also observed and may be due to a build up of charge at the film/substrate interface. In essence, SA-TiO2–RGO films are able to stabilize charge separation generated from photoexcitation. Electron transfer and storage for de-aerated colloidal TiO2–GO solutions have been demonstrated;6,11a however, this stability in air is unexpected. Further studies will be required to better understand this phenomenon. Utilization of this charge generation/storage is demonstrated here by the photochemical synthesis of Ag0 nanoparticles11a on the surface of the SA-TiO2–RGO film. Briefly, SA-TiO2–RGO films were irradiated with a UV-lamp (365 nm, ∼10000 µW cm−2) in air for 30 min, the lamp was then removed and 200 µL of a degassed solution of 10 mM Ag(NO3) was pipetted on top of the film. After 10 min, the solution was removed and the film rinsed with DI water and dried. SEM micrographs of the resulting film show that the surface of the SA-TiO2–RGO film is now decorated with 10–15 nm sized nanoparticles, i.e. a Ag0–SA-TiO2–RGO film is formed, Fig. 2d. Selected area electron diffraction (SAED) studies from Ag0–SA-TiO2–RGO films prepared similarly on transmission electron microscopy (TEM) grids indicate that the nanoparticles index to JCPDS No. 004-0783, Silver-3C, syn, Fig. 2d (inset). The Ag0 nanoparticles are primarily located on the graphene sheet and not co-localized with the TiO2 nanoflowers/nanoparticles, suggestive of a charge transfer process from the TiO2 to the graphene11a prior to the reduction of Ag+, resulting in the nanoparticle growth and deposition on the graphene surface of the SA-TiO2–RGO film. Control studies demonstrate that UV excitation is required for the Ag+ reduction to occur. While it is possible that these films are behaving in a capacitive nature with charge build up at the interface between the film and substrate, the localization of silver nanoparticles on top of the film suggests the storage of charge in the film itself.
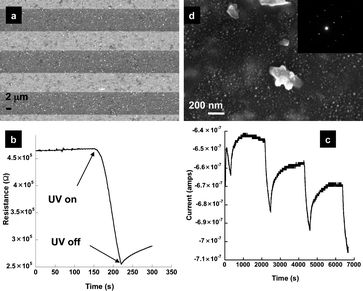 | ||
| Fig. 2 (a) SEM micrograph of SA-TiO2–RGO thin film deposited onto an IDE; representative (b) change in resistance across IDE (λexc = 365 nm), and (c) photocurrent cycling (∼0.25 cm2 film) in air at 5 min ON and 30 min OFF intervals, with exposure to UV-radiation (λexc = 350 nm); (d) SEM micrograph of ternary Ag0–SA-TiO2–RGO film. Smaller Ag0 nanoparticles are visible on the graphene surface. Several larger TiO2 particles/flowers4 are also visible. (Inset) SAED pattern from TEM indexes to Silver-3C, syn, JCPDS (004-0783). | ||
Conclusions
In summary, we have demonstrated that photo-reduction of aqueous TiO2–GO solutions can lead to the formation of unsupported SA-TiO2–RGO films at the air–liquid interface. Assembly occurs upon lowering of the surface tension (by adding methanol) which leads to de-wetting of the sol-TiO2–RGO and ultimately self-assembly at the air–liquid interface. These films were shown to generate ∼12 to 20 nA cm−2 of current when excited by UV radiation (λexc = 350 nm, 25–30 mJ cm−2). The graphene sheets could store charge for brief periods of time (in air) allowing for the synthesis of Ag0 nanoparticles on the graphene surface. Future studies into the self-assembly of the sheets could provide for even larger optimized structures with enhanced photocurrent and electron storage properties. Evaporative driven self-assembly is a useful tool for preparing titanium–graphene composites and is likely to be applicable to other inorganic–graphene composite and chemically modified graphene, in general.Acknowledgements
This work was supported (in part) by the Laboratory Directed Research and Development (LDRD) program and the National Institute for Nano-Engineering at Sandia National Laboratories. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy's National Nuclear Security Administration under Contract DE-AC04-94AL85000. Dr Ping Lu and Bonnie M. McKenzie are thanked for their technical assistance with electron microscopy.Notes and references
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- (a) Y. H. Ng, I. V. Lightcap, K. Goodwin, M. Matsumura and P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 2222–2227 Search PubMed; (b) Y.-B. Tang, C.-S. Lee, J. Xu, Z.-T. Liu, Z.-H. Chen, Z. He, Y.-L. Cao, G. Yuan, H. Song, L. Chen, L. Luo, H.-M. Cheng, W.-J. Zhang, I. Bello and S.-T. Lee, ACS Nano, 2010, 4, 3482–3488 CrossRef CAS; (c) H.-B. Yao, L.-H. Wu, C.-H. Cui, H.-Y. Fang and S.-H. Yu, J. Mater. Chem., 2010, 20, 5190–5195 RSC; (d) H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380–386; (e) O. Akhavan and E. Ghaderi, J. Phys. Chem. C, 2009, 113, 20214–20220 CrossRef CAS; (f) K. K. Manga, Y. Zhou, Y. L. Yan and K. P. Loh, Adv. Funct. Mater., 2009, 19, 3638–3643 CrossRef CAS; (g) W. Q. Peng, Z. M. Wang, N. Yoshizawa, H. Hatori and T. Hirotsu, Chem. Commun., 2008, 4348–4350 RSC.
- T. N. Lambert, C. A. Chavez, B. Hernandez-Sanchez, P. Lu, N. S. Bell, A. Ambrosini, T. Friedman, T. J. Boyle, D. R. Wheeler and D. L. Huber, J. Phys. Chem. C, 2009, 113, 19812–19823 CrossRef CAS.
- (a) Z. H. Tang, J. Zhuang and X. Wang, Langmuir, 2010, 26, 9045–9049 CrossRef CAS; (b) Y. Zhu, W. Cai, R. D. Piner, A. Velamakanni and R. S. Ruoff, Appl. Phys. Lett., 2009, 95, 103104–103103 CrossRef; (c) D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907–914 CrossRef CAS; (d) S. Biswas and L. T. Drzal, Nano Lett., 2009, 9, 167–172 CrossRef CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- C. Chen, Q. H. Yang, Y. Yang, W. Lv, Y. Wen, P. X. Hou, M. Wang and H. M. Cheng, Adv. Mater., 2009, 21, 3007–3011 CrossRef CAS.
- T. N. Lambert, C. C. Luhrs, C. A. Chavez, S. Wakeland, M. T. Brumbach and T. M. Alam, Carbon, 2010, 48, 4081–4089 CrossRef CAS.
- X. Ma, B. Wigington and D. Bouchard, Langmuir, 2010, 26, 11886–11893 CrossRef CAS.
- G. Vazquez, E. Alvarez and J. M. Navaza, J. Chem. Eng. Data, 1995, 40, 611–614 CrossRef CAS.
- (a) I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577–583 CrossRef CAS; (b) P. V. Kamat, J. Phys. Chem. Lett., 2009, 1, 520–527 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and figures as mentioned in the text. See DOI: 10.1039/c0nr00638f |
| This journal is © The Royal Society of Chemistry 2011 |