Photoluminescence, cytotoxicity and in vitro imaging of hexagonal terbium phosphate nanoparticles doped with europium†
Weihua
Di
*ab,
Jie
Li
c,
Naoto
Shirahata
bd,
Yoshio
Sakka
*ab,
Marc-Georg
Willinger
e and
Nicola
Pinna
ef
aWorld Premier International Research (WPI) Center Initiative on Materials Nanoarchitronics (MANA), National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan. E-mail: DI.Weihua@nims.go.jp; Sakka.Yoshio@nims.go.jp
bFine Particle Processing Group, Nano Ceramics Center, NIMS, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
cNanotechnology Innovation Center, NIMS, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
dPRESTO, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
eDepartment of Chemistry, CICECO, University of Aveiro, 3810-193, Aveiro, Portugal
fWorld Class University (WCU) program of Chemical Convergence for Energy & Environment (C2E2), School of Chemical and Biological Engineering, College of Engineering, Seoul National University (SNU), Seoul, 151-744, Korea
First published on 7th December 2010
Abstract
Luminescent TbPO4 nanoparticles were synthesized via a citric-acid-mediated hydrothermal route. Eu3+ doping of TbPO4 enables an efficient Tb3+-to-Eu3+ energy transfer, leading to a four-fold increase of the absolute emission quantum yield (QY), compared to that of undoped TbPO4. To check the potential of biological use, we conducted in vitro biological experiments on human cervical carcinoma HeLa cells incubated with TbPO4:Eu nanoparticles. TbPO4:Eu nanoparticles can be successfully internalized into the cells, and they show bright intracellular luminescence and very low cytotoxicity. Photoluminescence intensity dependence upon time demonstrates that Eu3+-doped TbPO4 nanoparticles are highly resistant to photobleaching. Our present work represents a demonstration of the use of rare-earth-based nanocrystals as a biological labeling agent because they combine several advantages including high emission quantum yield, long luminescence lifetime, low cytotoxicity and high photostability.
1. Introduction
There is an increasing interest in the development of luminescent labeling materials for bioimaging.1–5 Conventional luminescent labels include organic dyes3,4 and semiconductor quantum dots (QDs) such as CdSe and CdTe.1,2,5 However, these labeling agents also have some intrinsic limitations in their practical biological uses. For example, the main drawback of organic dyes is photobleaching,6 while QDs are still controversial due to their inherent toxicity and chemical instability.7Recently, rare-earth-based nanoparticles have been proposed as a new generation of biological luminescent labels and as viable alternatives to conventional labels,8–14 due to their attractive chemical, optical and biocompatible features, such as tunable emission colors, high photochemical stability, long luminescence lifetime (up to several ms), low photobleaching and low toxicity.11,14 Moreover, these rare-earth-based probes also possess additional distinctive features, such as sharp emission bands and large Stokes shifts (as much as several hundred nanometres),15 leading to an improved sensitivity.
Metal phosphates represent an important family of materials and constitute good host matrixes for luminescent rare earth ions.16–18 They have been used as active components in a wide range of applications such as high performance luminescent devices, magnets, catalysts, fluorescence labels for biological detection, and other functional materials based on the electronic, optical, and chemical characteristics resulting from the 4f shell of their ions.19 Very recently, much effort regarding metal phosphate has been devoted to novel synthetic routes and their new potential in applications. Yan's group20 reported on the synthesis of orderly aligned and highly luminescent monodisperse rare-earth orthophosphate nanocrystals by a limited anion-exchange reaction in solution phase. Luo et al.21 reported the synthesis of ordered cubic mesoporous yttrium phosphates (YPO4) via a hard template method and the corresponding photoluminescence properties upon rare earth doping. Our previous work22 demonstrated luminescent CePO4:Tb nanocrystals as a novel oxygen sensing material based on redox responsive reversible luminescence. Epple's group14,16 claimed that calcium phosphate is a very promising biomaterial for bioimaging, photodynamic therapy and as DNA carriers due to its high biocompatibility and good biodegradability. Wong's group10 used a simple and effective template-mediated protocol to synthesize CePO4:Tb nanowires and demonstrated their potential applications in biological imaging and cellular labeling.
It is well known that nanosized luminescent materials with small particle size show lower luminescence quantum yield than those of materials with larger particle size or the corresponding bulk materials due to a low crystallinity and a number of surface defect states. A high luminescence quantum yield (QY) is required for the use of nanoparticles in biological imaging and labeling. In this work, photoluminescent TbPO4 nanoparticles were synthesized via a citric-acid-mediated hydrothermal route and investigated as luminescent labeling agents. It is found that the undoped TbPO4 nanoparticles show a low photoluminescence QY of only 0.07, while Eu3+ doping in TbPO4 matrix enables an efficient Tb3+-to-Eu3+ energy transfer, leading to a four-fold increase of QY. Such highly luminescent nanoparticles were used to conduct in vitro biological experiments using human cervical carcinoma HeLa cells. The cellular permeability, cytotoxicity, photostability and in vitro imaging of fluorescent TbPO4:Eu nanoparticles were studied. Our work demonstrated that rare-earth-based luminescent nanomaterials have a high potential for biological imaging and labeling.
2. Results and discussion
2.1 Size and morphology
The as-synthesized product corresponds to hexagonal rhabdophane-type TbPO4 crystals, as revealed by X-ray diffraction studies (JCPDS No. 20-1244; Fig. S1 in the ESI†). The size and morphology of the product were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) observations. A SEM image (Fig. 1a) reveals that the sample consists of hexagonal prism nanoparticles. High resolution TEM and the corresponding Fourier transform images provide deep insight into the crystalline size and quality, and preferential growth axis (the insets of Fig. 1c). The products are single-crystalline nanoparticles, ranging from 60 to 80 nm in diameter. They are highly crystalline without visible defects and dislocations. Generally, lanthanide phosphates with hexagonal rhabdophane-type structure prefer to grow along the [001] direction in the absence of any organic additive acting as chelating agent, leading the formation of rod-shaped particles with high aspect ratio.17,18 In our synthesis, citric acid acts as chelating agent of Tb3+. Moreover, citrate ions can also selectively bind to specific crystallographic facets, inhibiting or promoting the growth along specific directions, finally leading to dramatic modifications of the final morphologies of the particles.17,23 In this case, one of the consequences of the presence of citrate ions is the inhibition of the [001] growth direction and the resulting relative enhancement of the growth sideways along the [100] and [010] directions, which gives rise to highly symmetric hexagonal-prism-shaped nanoparticles with well-defined crystallographic facets. Similar results were observed in the synthesis of other types of inorganic materials.24,25 | ||
| Fig. 1 SEM (a) and TEM (b and c) images of TbPO4 synthesized by a citric-acid-mediated hydrothermal route. | ||
2.2 Photoluminescence
Fig. 2 presents the emission spectra of TbPO4 without and with Eu3+ doping. For undoped TbPO4 nanoparticles excited at 377 nm, the spectrum displays a series of emission lines ascribed to the Tb3+ intra-4f85D4 → 7F6–3 electronic transitions. Upon doping of a small amount of Eu3+, we can observe additional emission lines appearing from 585 to 710 nm ascribed to the Eu3+ intra-4f65D0 → 7F0–4 transitions. The simultaneous observation of the emission arising from Tb3+ and Eu3+ excited states suggests the possible existence of Tb3+-to-Eu3+ energy transfer.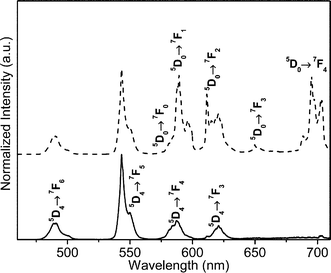 | ||
| Fig. 2 Room temperature emission spectra of pure (full line) and 0.3 mol% Eu3+-doped TbPO4 (dashed line) excited at 377 nm. | ||
To verify whether the energy transfer of Tb3+-to-Eu3+ is present, we measured the excitation spectra of TbPO4 without and with Eu3+ doping (Fig. S2 in the ESI†). The excitation of the Eu-doped samples were monitored within the Tb3+5D4 → 7F5 (544 nm) and Eu3+5D0 → 7F4 (697 nm) transitions, respectively. The former excitation spectra is similar to the one acquired for the undoped TbPO4 sample. The spectra monitored within the Eu3+ emission lines exhibit a series of Tb3+ related transitions and a low-relative intensity peak attributed to the Eu3+7F0 → 5L6 transition. The observation of the Tb3+ excited levels in the excitation spectra monitored within the Eu3+ intra-4f6 transitions is a clear evidence of the occurrence of Tb3+-to-Eu3+ energy transfer.
The existence of the energy transfer from Tb3+ to Eu3+ can be also evidenced by the dependence of the 5D4 lifetime values on the Eu3+ concentrations. The luminescence decay of pure TbPO4 and doped TbPO4 by monitoring the Tb3+ emission at 543 nm were measured. All the curves are well-reproduced by a single exponential function, yielding to lifetime values of 0.495 ± 0.001 ms for the pure TbPO4 sample and 0.354 ± 0.001, 0.230 ± 0.001 and 0.109 ± 0.001 ms for TbPO4 doped with 0.001, 0.003 and 0.005 mol% Eu3+, respectively. The presence of the acceptor Eu3+ increases the transition rate of Tb3+ excited states and thus decreases the Tb3+5D4 lifetime, proving the existence of Tb3+-to-Eu3+ energy transfer.
The dynamics of the Eu3+ luminescence (monitored at 697 nm, 5D4 → 7F6 transition of Eu3+) under pulsed excitation at 355 nm also confirms the occurrence of Tb3+-to-Eu3+ energy transfer in TbPO4:Eu sample (Fig. 3). It is observed that the population loading presents a longer rise time following the excitation laser pulse, as marked by a circle in Fig. 3. The slow rise time is due to an increase of population of the Eu3+:5D0 state via energy transfer from Tb3+. Eu3+-doped TbPO4 shows a luminescence lifetime of Eu3+ as long as 2.5 ms.
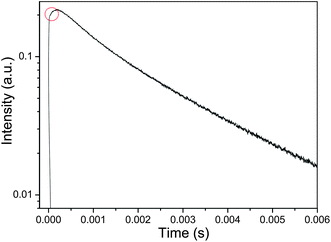 | ||
| Fig. 3 Room temperature emission decay curves of TbPO4:5%Eu3+ monitored with Eu3+ emission at 697 nm (5D0 → 7F4 transition). | ||
Fig. 4 shows the Eu3+ emission intensity of samples as a function of Eu3+ concentration. It can be seen that the luminescence of Eu3+ increases with Eu3+ concentration and reaches a maximum value around 5 mol% Eu3+. At this concentration range, the Tb3+-to-Eu3+ energy transfer is dominant to the luminescence of materials and Tb3+ emissions are almost completely quenched. For Eu3+ concentration higher than 5%, the emission intensity starts to decrease. The increase of Eu3+ concentration above 5% reduces the distance between the luminescent centers and thus increases the probability of nonradiative energy migration between Eu3+ ions leading to nonradiative deexcitation, which is known as the concentration quenching.
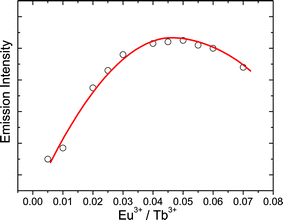 | ||
| Fig. 4 Emission intensity of Eu3+ as a function of Eu3+ concentration. | ||
The photoluminescence features were further quantified through the estimation of the absolute emission QY as a function of the Eu3+ concentration. For all the samples, the maximum quantum yield value was attained under excitation at 377 nm. Table 1 records the QY values of undoped TbPO4 and TbPO4 doped with various concentrations of Eu3+. The undoped TbPO4 shows a low QY value of only 0.07, while a small amount of Eu3+ doping improves the QY significantly, that is, a two-fold increase of QY (0.14) is acquired for TbPO4:Eu0.003. The QY value continues to increase with the increase of Eu3+ concentration up to 5 mol%, where a maximum QY value of 0.30 was acquired. This is in good agreement with the tendency observed in Fig. 4. The remarkable increase of the QY of samples with Eu3+ doping is attributed to an efficient Tb3+-to-Eu3+ energy transfer. Under direct intra-4f6 excitation (7F0 → 5L6, 393 nm) the quantum yield value of the TbPO4:Eu samples lies below the detection limits of our equipment (0.01), confirming that the preferential path for the Eu3+ excitation is via the Tb3+ excited levels. Luminescent nanoparticles with high luminescence QY and long excited-state lifetime are preferred for their biological application, as will be demonstrate below.
| Tb1−xPO4:Eux | x = 0 | x = 0.003 | x = 0.01 | x = 0.03 | x = 0.05 | x = 0.06 |
|---|---|---|---|---|---|---|
| QY | 0.07 | 0.14 | 0.19 | 0.26 | 0.30 | 0.24 |
2.3 Cellular uptake and imaging
Generally, the cellular permeability and cytotoxicity characteristics of fluorescent nanomaterials are critical to their biological applications. To check the possibility of the use of TbPO4:Eu nanoparticles as luminescent biological labels, we conducted in vitro biological experiments using human cervical carcinoma HeLa cells. We choose TbPO4 doped with 5 mol% Eu3+ for all biological experiment since it shows the highest luminescence QY. As a control experiment, HeLa cells alone show almost no background fluorescence (Fig. 5a). Nonetheless, upon incubation, incorporation of TbPO4:Eu nanoparticles into HeLa cells was confirmed by a fluorescence microscopy. Fig. 5b and c show fluorescence and bright-field optical microscopy images of HeLa cells after incubation with 0.1 mg mL−1 of TbPO4:Eu nanoparticles for 8 h. As observed, the nanoparticles can permeate into the cell membrane and display bright intracellular luminescence from Eu3+ (Fig. 5b). These inorganic phosphate nanoparticles clearly retained their intrinsic fluorescent properties upon cellular internalization. Moreover, the corresponding bright-field measurements taken after incubation with the nanoparticles confirmed that the cells were viable throughout the imaging experiments (Fig. 5c) and that there were no evident regions of cell death. To check the spatial localization of foreign materials within the cells, we took a series of Z-stack images of the cell (e.g., top to bottom) at 1 µm “slice” intervals of the HeLa cells stained with TbPO4:Eu nanoparticles. A representative fluorescence image (Fig. 5d) taken from an internal slice clearly shows a red fluorescence within the cells. This indicates that TbPO4:Eu nanoparticles are localized within the interior environment of the HeLa cells. Very recently, Wong's group10 and our previous work26 have obtained similar results for CePO4:Tb nanomaterials with green luminescence, which also shows intense intracellular luminescence after incubated with HeLa cells. This indicates the high potential of rare-earth-based luminescent materials toward applications in bioimaging and biolabeling because of their successful cellular internalization and bright intracellular luminescence.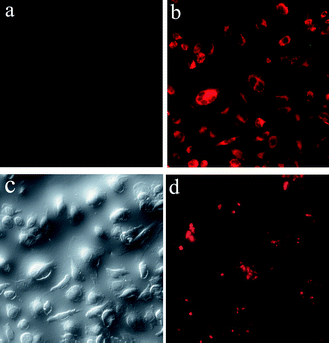 | ||
| Fig. 5 Fluorescence images of HeLa cells alone (a); fluorescence (b) and bright-field optical images (c) of HeLa cells after incubation for 8 h with 0.1 mg mL−1 of TbPO4:Eu nanoparticles; a representative fluorescence image of HeLa cells stained with TbPO4:Eu nanoparticles from an internal slice (d). | ||
To reveal the cellular internalization process of nanoparticles upon time, the intracellular luminescence intensity was recorded as a function of incubation time (Fig. 6). It can be seen that the luminescence intensity increases with time during the first 6 h, and then reached a plateau at 8–10 h. This indicates that more and more nanoparticles were able to permeate into the cell membrane with time since the intracellular luminescence intensity is proportional to the number of nanoparticles taken up by cells. The uptake of nanoparticles significantly increases for the first 6 h. Subsequently, the uptake gradually slowed and reached the saturation state.
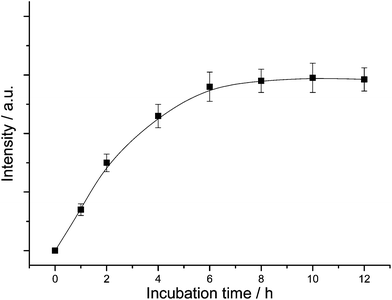 | ||
| Fig. 6 Intracellular luminescence intensity as a function of incubation time. | ||
The surface of the as-synthesized TbPO4:Eu nanoparticles is negatively charged due to the presence of citrate groups. Generally, the anionic molecules and structures are difficult to enter the membrane with the negative charged surface because of electrostatic repulsion. The mechanism of cellular internalization of nanoparticles is not clear so far. Some researchers stated that the cellular uptake of nanoparticles involves a receptor-mediated endocytosis.10,27,28 It has been reported that serum proteins from biological media, such as bovine serum albumin (BSA), can nonspecifically coat the surfaces of nanoparticles, leading to all nanoparticles surface bearing the same effective charge, regardless of their initial surface charge.26,27 On this basis, it was considered that the surface adsorbed BSA facilitated the uptake of particles into human cancer cells, such as HeLa or HT-29, viareceptor-mediated endocytosis arising from cellular recognition of these proteins. To support this speculation, we observed the cellular internalization of nanoparticles at low temperature (4 °C). Endocytosis is known to be energy dependent and could be hindered at low temperature.29 It is found that the fluorescence intensity of cells incubated at 4 °C decreases by about one order of magnitude as compared with that of cells treated at 37 °C (Fig. S3, ESI†), indicating a significant hindrance of cellular internalization at 4 °C. The above result confirms that the receptor-mediated endocytosis dominates over the cellular internalization of the as-synthesized TbPO4:Eu nanoparticles, while the passive diffusion process of migration of nanoparticles seems a minor contribution.
2.4 Photostability
It should be pointed out that one of the advantages of rare-earth-doped luminescent nanoparticles as biological labels is their long fluorescence lifetime and stable photoluminescence features. The photostability of Eu3+-doped TbPO4 nanoparticles was investigated by means of time-sequential scanning of the cells. After 300 s of continuous illumination with a laser, no obvious change was observed in the photoluminescence intensity (Fig. 7). In fact, no photobleaching of our present Eu3+-doped TbPO4 nanoparticles was observed under further long-term illumination. These findings prove that the Eu3+-doped TbPO4 nanoparticles are highly resistant to photobleaching compared to conventional luminescent labels such as organic dyes.6 Long luminescence lifetime and high photostability are advantageous for long-time observations in biological cases.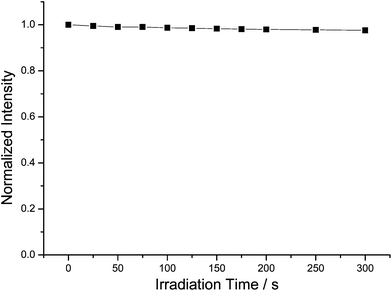 | ||
| Fig. 7 Photoluminescence intensity as a function of illumination time. | ||
2.5 Cytotoxicity
Only nontoxic nanomaterials are suitable for bioimaging and cell labeling. To verify whether these luminescent TbPO4:Eu nanoparticles are biologically nontoxic and biocompatible, the particle-induced cytotoxic effects on HeLa cells were evaluated by conducting MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays. The viability of untreated cells was assumed to be 100%. Fig. 8a shows the concentration effect of TbPO4:Eu nanoparticles on cell viability after incubation for 8 h. It can be seen that, TbPO4:Eu nanoparticles show almost no toxicity upon incubation with HeLa cells. The cell viability remains as high as 92% even after incubation with 0.1 mg mL−1TbPO4:Eu for 8 h. Fig. 8b shows the incubation time dependence on the cell viability at a given TbPO4:Eu concentration (0.1 mg mL−1). The observed cell viability shows only a slight decrease with the incubation time and remains as high as 90% even after incubation with 0.1 mg mL−1TbPO4:Eu nanoparticles for long as 72 h. Low toxicity of rare-earth-based inorganic nanoparticles further demonstrated the potential of their use as biological labels.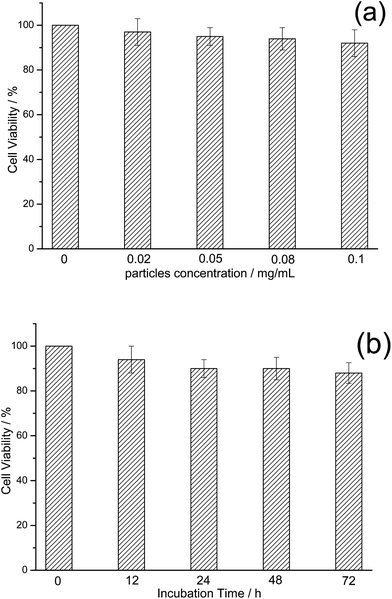 | ||
| Fig. 8 In vitro cell viability of HeLa cells incubated with TbPO4:Eu nanoparticles at different concentrations (a) for periods ranging from 0 to 72 h (b). | ||
3. Experimental
3.1. Synthesis of materials
Rare earth nitrate (Tb(NO3)3 and Eu(NO3)3) stock solutions of 0.2 M were prepared by dissolving the corresponding metal oxide in concentrated HNO3 at elevated temperature. In a typical procedure using citric acid (CA) as ligand, 20 mL of Tb(NO3)3 (0.2 M) solution was added to 20 mL of aqueous solution containing 8 mmol of citric acid. The solution, turning turbid due to the formation of the terbium citrate complex, was kept under vigorous stirring for 30 min. Subsequently, an aqueous solution containing 4.4 mmol of (NH4)2HPO4 was added. The final pH of the mixture was adjusted to about 1–2 by adding aqueous ammonia (NH4OH). After additional agitation for 15 min, the solution was transferred into a Teflon bottle held in a stainless steel autoclave, which was sealed and heated at 150 °C for 20 h. After cooling down the autoclave to room temperature, the precipitation was separated by centrifugation, washed with deionized water and ethanol several times, and finally dried overnight under air at 60 °C. Eu3+-doped terbium phosphate samples were obtained under the same conditions by replacing a fraction of Tb(NO3)3 by Eu(NO3)3.3.2. In vitro biological experiments
3.3. Structure and morphology characterization
The X-ray powder diffraction (XRD) data were collected on an X'Pert MPD Philips diffractometer (CuKα X-radiation at 40 kV and 50 mA). The patterns were measured in the 2θ range from 10° to 60° with a scanning step of 0.02°. Scanning electron microscopy (SEM) images were recorded using a FEG-SEM Hitachi SU-70 microscope operating at 4 kV at a working distance of 2–3 mm. For the SEM observations, samples were prepared without any carbon coating simply by depositing some powder onto a double gluing tape. High resolution transmission electron microscopy (HRTEM) investigations were carried using a JEOL 2200FS microscope. Samples for TEM investigations were prepared by first dispersing the particles in ethanol under assistance of ultrasonication and then dropping 1 drop of the suspension on a copper TEM grid coated with a holey carbon film.3.4. Photoluminescence characterization
The luminescence in the ultraviolet/visible (UV/vis) spectral range was recorded at room temperature with a modular double grating excitation spectrofluorimeter with a TRIAX 320 emission monochromator (Fluorolog-3, Jobin Yvon-Spex) coupled to an R928 Hamamatsu photomultiplier, using the front face acquisition mode. The excitation source was a 450 W Xe arc lamp. The emission spectra were corrected for detection and optical spectral response of the spectrofluorimeter and the excitation spectra were corrected for the spectral distribution of the lamp intensity using a photodiode reference detector. Fluorescent lifetime measurements were made under a pulsed excitation at 355 nm from the third harmonic of a Nd:YAG laser. It was with a line width of 1.0 cm−1, pulse duration of 10 ns, and repetition frequency of 10 Hz. The absolute emission quantum yields were measured at room temperature using a quantum yield measurement system C9920-02 from Hamamatsu with a 150 W xenon lamp coupled to a monochromator for wavelength discrimination, an integrating sphere as a sample chamber, and a multichannel analyzer for signal detection. Three measurements were made for each sample so that the average value is reported. The method is accurate to within 10%.4. Conclusions
Luminescent TbPO4 nanoparticles were synthesized via a citric-acid-mediated hydrothermal route. The as-synthesized nanoparticles are hexagonal prisms with a diameter of about 60–80 nm. The excitation spectra and lifetime measurements confirmed the presence of Tb3+-to-Eu3+ energy transfer in Eu3+-doped TbPO4. The efficient Tb3+-to-Eu3+ energy transfer leads to four-fold increase of the absolute luminescence quantum yield, compared to undoped TbPO4. We conducted in vitro biological experiments using human cervical carcinoma HeLa cells. The cellular uptake of TbPO4:Eu nanoparticles based on receptor-mediated endocytosis was observed, leading to bright intracellular luminescence. The cellular uptake dependence on the incubation time was investigated, revealing that the cellular uptake significantly increases first and then reaches a plateau. The cell viability assay and the photoluminescence intensity dependence upon irradiation time reveal very low cytotoxicity and high photostability of the luminescent particles. This work demonstrates the advantages of the use of rare-earth-based nanocrystals in biological labeling agents because they combine high emission quantum yield, long luminescence lifetime, low cytotoxicity and high photostability.Acknowledgements
This study was partly supported by the Grant-in Aid for Scientific Research of the JSPS and the World Premier International Research (WPI) Center Initiative on Materials Nanoarchitronics (MANA), MEXT, Japan, the WCU (WorldClass University) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R31-10013) and FCT project (PTDC/CTM/73243/2006). NS wishes to thank the JST PRESTO program for financial support.References
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS.
- G. Seisenberger, M. U. Ried, T. Endress, H. Buning, M. Hallek and C. Brauchle, Science, 2001, 294, 1929 CrossRef CAS.
- M. Zhang, M. X. Yu, F. Y. Li, M. W. Zhu, M. Y. Li and Y. H. Gao, J. Am. Chem. Soc., 2007, 129, 10322 CrossRef CAS.
- R. Hu, K. T. Yong, I. Roy, H. Ding, W. C. Law, H. X. Cai, X. H. Zhang, L. A. Vathy, E. J. Bergey and P. N. Prasad, Nanotechnology, 2010, 21, 145105 CrossRef.
- F. Wang, W. B. Tan, Y. Zhang, X. P. Fan and M. Q. Wang, Nanotechnology, 2006, 17, R1 CrossRef; Z. L. Zhang, L. Yao, J. B. Pan and X. M. Yan, J. Mater. Chem., 2010, 20, 1179 RSC.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11 CrossRef CAS.
- K. Wong, G. Law, M. Murphy, P. Tanner, W. Wong, P. Lam and M. Lam, Inorg. Chem., 2008, 47, 5190 CrossRef CAS.
- S. F. Lim, R. Riehn, W. S. Ryu, N. Khanarian, C. K. Tung, D. Tank and R. H. Austin, Nano Lett., 2006, 6, 169 CrossRef CAS.
- F. Zhang and S. S. Wong, ACS Nano, 2010, 4, 99 CrossRef CAS.
- S. Setua, D. Menon, A. Saok, S. Nair and M. Koyakutty, Biomaterials, 2010, 31, 714 CrossRef CAS.
- G. K. Das and T. T. Y. Tan, J. Phys. Chem. C, 2008, 112, 11211 CrossRef CAS.
- S. F. Lim, R. Riehn, C. K. Tung, W. S. Ryu, R. Zhuo, J. Dalland and R. H. Austin, Nanotechnology, 2009, 20, 405701 CrossRef.
- S. P. Mondejar, A. Kovtun and M. Epple, J. Mater. Chem., 2007, 17, 4153 RSC.
- F. Meiser, C. Cortez and F. Caruso, Angew. Chem., Int. Ed., 2004, 43, 954.
- M. Epple, K. Ganesan, R. Heumann, J. Klesing, A. Kovtun and V. Sokolova, J. Mater. Chem., 2010, 20, 18 RSC.
- W. H. Di, M. G. Willinger, R. A. S. Ferreira, X. G. Ren, S. Z. Lu and N. Pinna, J. Phys. Chem. C, 2008, 112, 18815 CAS.
- Y. P. Fang, A. W. Xu, R. Q. Song, H. X. Zhang, L. P. You, J. C. Yu and H. Q. Liu, J. Am. Chem. Soc., 2003, 125, 16025 CrossRef CAS.
- W. H. Di, X. J. Wang, B. J. Chen, H. S. Lai and X. X. Zhao, Opt. Mater. (Amsterdam), 2005, 27, 1386 Search PubMed.
- H. X. Mai, Y. W. Zhang, L. D. Sun and C. H. Yan, Chem. Mater., 2007, 19, 4514 CrossRef CAS.
- L. Luo, S. D. Shen, G. Z. Lu, X. Z. Xiao, D. S. Mao and Y. Q. Wang, J. Mater. Chem., 2009, 19, 8079 RSC.
- W. H. Di, X. J. Wang and X. G. Ren, Nanotechnology, 2010, 21, 075709 CrossRef.
- C. X. Li, Z. Y. Hou, C. M. Zhang, P. P. Yang, G. G. Li, Z. H. Xu, Y. Fan and J. Lin, Chem. Mater., 2009, 21, 4598 CrossRef CAS.
- G. K. Zhang, S. J. Yu, Y. Q. Yang, W. Jiang, S. M. Zhang and B. B. Huang, J. Cryst. Growth, 2010, 312, 1866 CrossRef CAS.
- L. Xu, C. L. Lu, Z. H. Zhang, X. Y. Yang and W. H. Hou, Nanoscale, 2010, 2, 995 RSC.
- W. H. Di, J. Li, N. Shirahata and Y. Sakka, Nanotechnology, 2010, 21, 455703 CrossRef.
- A. M. Alkilany, P. K. Nagaira, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662 CrossRef CAS.
- J. Y. Chen, S. Y. Chen, X. R. Zhao, L. V. Kuznesova, S. S. Wong and I. Ojima, J. Am. Chem. Soc., 2008, 130, 16778 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD pattern of the as-synthesized TbPO4 (Fig. S1), room temperature excitation spectra of pure TbPO4 and Eu3+-doped TbPO4 (Fig. S2), and the luminescence intensity of cells incubated with TbPO4:Eu nanoparticles at 37 °C and 4 °C, respectively (Fig. S3). See DOI: 10.1039/c0nr00673d |
| This journal is © The Royal Society of Chemistry 2011 |