Phase-controllable synthesis of nanosized nickel phosphides and comparison of photocatalytic degradation ability
Yonghong
Ni
*a,
Lina
Jin
a and
Jianming
Hong
b
aCollege of Chemistry and Materials Science, Anhui Key Laboratory of Functional Molecular Solids, Anhui Normal University, Wuhu, 241000, P. R. China. E-mail: niyh@mail.ahnu.edu.cn; Fax: (+86)553-3869303
bCenters of Modern Analysis, Nanjing University, Nanjing, 210093, P. R. China
First published on 3rd November 2010
Abstract
In this paper, we employed a facile hydrothermal route to successfully synthesize nanosized nickel phosphide particles with controlled phases via selecting different surfactants at different temperatures and times. The phases of the as-obtained products were determined by X-ray powder diffraction (XRD) patterns and Rietveld refinement of XRD data. The morphologies of the products were characterized by (high resolution) transmission electron microscopy (HR/TEM) and field emission scanning electron microscopy (FESEM). Experiments indicated that pure Ni2P phase could be prepared when nontoxic red phosphorus and nickel dichloride were used as starting materials in the presence of polyvinylpyrrolidone (PVP, 30 K), sodium dodecylbenzene sulfonate (SDBS), cetyltrimethylammonium bromide (CTAB) or polyethylene glycol 10000 (PEG-10000) at 160 °C for 10 h. When acrylamide (AM) was selected as the surfactant, however, pure Ni12P5 phase could be prepared by prolonging the reaction time to 20 h at 160 °C, or enhancing the reaction temperature to 180 °C for 10 h. Furthermore, the experiments indicated that the pure Ni2P phase possessed a stronger photocatalytic degradation ability than the pure Ni12P5 phase.
Transition metal phosphides have been of much interest in recent decades owing to their important properties and potential applications in many fields, such as magnetic, photonic, electronic, and data storage devices.1–5 Among them, nickel phosphides with the ratio of Ni/P > 1 exist in a variety of phases (e.g.Ni2P, Ni3P, Ni5P2, Ni8P3 and Ni12P5), and have broad industrial applications, especially in the field of catalysis.6–8
In reported literatures, abundant investigations of nickel phosphides are focused on Ni2P, and few reports are found on the other nickel phosphides. At present, Ni2P used as catalysts is generally prepared by the temperature-programmed H2reduction of ammonium nickel phosphate precursors at relatively high temperature (600–650 °C).9,10 However, the solution synthesis of Ni2P is also developed. In 2005, for instance, Hyeon et al.11 employed trioctylphosphine (TOP) as the phosphorus source to prepare anisotropic nanostructures of magnetic phosphides (including Ni2P) in trioctylphosphine oxide (TOPO) solution via multiple or continuous injection of a metal-TOP precursor solution. Henkes et al.12 prepared metal phosphide (including Ni2P) nanocrystalsvia the direct reaction between freshly formed metal nanoparticles and TOP at below 370 °C. Chiang and co-workers13 obtained hollow Ni2P nanospheres via the reaction between Ni nanocrystals and TOP. Chen et al.14 reported on the synthesis of single-crystalline Ni2P nanowires through using nickel(II) acetylacetonate as a metal precursor and TOP as a phosphorus source. Furthermore, Wang et al.15 and Liu et al.16 developed a solvothermal route for syntheses of Ni2P nanocrystals, using white phosphorus as P3−ion sources. In the above methods, however, precious solvents, highly toxic reagents or/and higher temperatures are often needed. In order to conquer the above shortcomings, some groups attempted to prepare nickel phosphide (Ni2P) nanocrystals in the systems containing water with nontoxic reagents as starting materials,17 or through high temperature solid phase reaction.18 For example, Liu et al.17 employed ethylenediamine as the solvent to prepare Ni2P nanocrystallites by the reaction between nickel dichloride and nontoxic red phosphorus. Shi and Shen18 obtained Ni2P nanoparticlesvia the high temperature solid reaction between Ni2+ and H2PO2− in N2. In recent years, nickel phosphide nanocrystals also attract our research interest. Different from the above mentioned works, however, Ni12P5 hollow micro-/nano-spheres were obtained in our groupvia employing the simple hydrothermal and hydrothermal-microemulsion technology.19,20 Meanwhile, a mixture of Ni2P and Ni8P3 was hydrothermally obtained, too.20 However, it is still a huge challenge to realize phase-controlled synthesis of nickel phosphides in a simple aqueous solution via the hydrothermal technology.
In the current paper, we successfully realized phase-controllable synthesis of nanosized nickel phosphide particles under a simple hydrothermal condition, via selecting various surfactants at different temperatures or times, employing nontoxic red phosphorus and nickel dichloride as P3− and Ni2+ ion sources. It was found that the pure Ni2P phase could be prepared in the presence of polyvinylpyrrolidone (PVP, 30 K) at 160 °C for 10 h. When PVP was replaced by sodium dodecylbenzene sulfonate (SDBS), cetyltrimethylammonium bromide (CTAB) or polyethylene glycol 10000 (PEG-10000), respectively, the phase of the product did not change. However, after acrylamide (AM) was used instead of PVP, a mixture consisted of Ni12P5 and Ni2P was obtained under the same reaction temperature and time. Interestedly, pure Ni12P5 phase could be prepared by prolonging the reaction time to 20 h at 160 °C, or enhancing the reaction temperature to 180 °C for 10 h in the presence of the same amount of AM. Furthermore, we compared the photocatalytic properties of different nickel phosphide phases for the degradation of organic dyes, and found that pure Ni2P possessed better photocatalytic ability.
All reagents were analytically pure, purchased from Shanghai Chemical Company and used without further purification. In a typical experiment, the appropriate amount of NiCl2·6H2O (2 mmol, 0.475 g), PVP (0.100 g) and CH3COONa·2H2O (4 mmol, 0.544 g) were dissolved in distilled water to form 20 mL solution. After the as-prepared solutions were poured into a Teflon-lined stainless steel autoclave of 25 mL capacity, 0.32 g red phosphorus was added. The autoclave was sealed and maintained at 160 °C for 10 h, then allowed to cool down to room temperature naturally. The black precipitates were collected, washed with distilled water and absolute ethanol several times to remove the impurities. Finally, the as-prepared sample was dried in vacuum at 60 °C for 6 h.
The above process was repeated by using acrylamide (AM) of 0.200 g instead of PVP as the phase-controlled reagent at 160 °C for 20 h or at 180 °C for 10 h. The influences of some other surfactants, such as SDBS, CTAB and PEG-10000, on the phase of the product were also investigated.
X-Ray powder diffraction pattern of the product was carried out on a Shimadzu XRD-6000 X-ray diffractometer equipped with Cu-Kα radiation (λ = 0.154060 nm), employing a scanning rate of 0.02° s−1 and 2θ ranges from 20° to 80°. The XRD patterns were analyzed by the Rietveld method as implemented in the Fullprof program. SAED pattern and TEM image of the product were carried out on a JEOL-2010 high resolution transmission electron microscope, employing an accelerating voltage of 200 kV. SEM images of the products were obtained on Hitachi S-4800 field emission scanning electron microscope, employing the accelerating voltage of 5 kV. The holey carbon film was used as the support.
To compare the photocatalytic degradation abilities of two nickel phosphides to organic dyes, 10 mg of the products with various phases were dispersed into 50 mL of solutions with a concentration of 10 mg L−1 of Pyronin B or Safranin T, respectively. Then, the mixed systems were stirred for 30 min in the dark to achieve an adsorption-desorption equilibrium. After that, the systems were irradiated by UV light of 365 nm for given durations. The optical property changes of dyes are recorded on a Hitachi U-3010 UV-Vis absorption spectrophotometer (Tokyo, Japan).
The phases of the products are determined by Powder X-Ray Diffraction analyses. Fig. 1a (upper) gives the XRD pattern of the product prepared from the solution containing PVP of 0.1 g at 160 °C for 10 h. All diffraction peaks can be indexed as the hexagonal Ni2P phase by comparison with the data of the JCPDS Card Files No. 89-4864 (Fig. 1a, down). The average particle size of the crystalline Ni2P is about 14.3 nm as calculated from the Debye-Scherrer formula. No other nickel phosphide peaks are detected. Fig. 1b show the XRD patterns of the products prepared from the solution with AM of 0.2 g at 180 °C for 10 h (upper) and 160 °C for 20 h (middle), respectively. The locations of two diffraction patterns are fully same except the different peak intensity. All diffraction peaks can be indexed as the tetragonal Ni12P5 phase (Fig. 1a, lower; JCPDS Card Files No.74-1381). The average particle sizes of two Ni12P5 products are calculated to be ∼14.9 and 17.5 nm, respectively. The above XRD analyses clearly show that the phase of nickel phosphide has changed when the surfactant and the reaction temperature/time have been changed in the presences of the same initial Ni2+ and P3−ion sources. These facts also indicate that nickel phosphides with different phases can be controllably synthesized in simple aqueous solutions by the hydrothermal route.
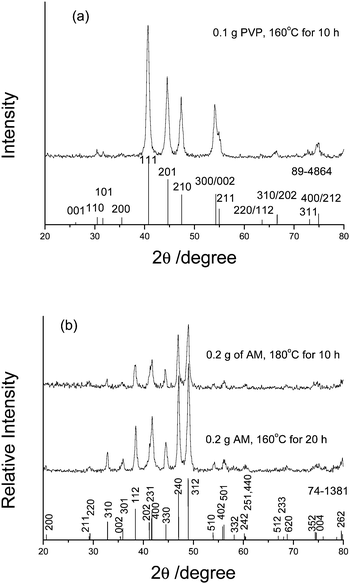 | ||
| Fig. 1 The XRD patterns of the products prepared respectively: (a) in the presence of 0.1 g PVP at 160 °C for 10 h, (b) in the presence of AM at 180 °C for 10 h (upper) or at 160 °C for 20 h (middle). | ||
Rietveld refinement of the XRD data of Ni2P prepared from the system with PVP of 0.1 g at 160 °C for 10 h is shown in Fig. 2a. The calculated XRD pattern is in good agreement with the experimental one. The hexagonal crystal structure can be confirmed. Similarly, Fig. 2b depicts the comparison of experimental and calculated XRD pattern of Ni12P5 prepared from the system with AM of 0.2 g at 160 °C for 20 h. The calculated parameters are a = 8.676 and c = 5.079, which are very close to the reported values (a = 8.646 and c = 5.070, JCPDS cards no.74-1381).
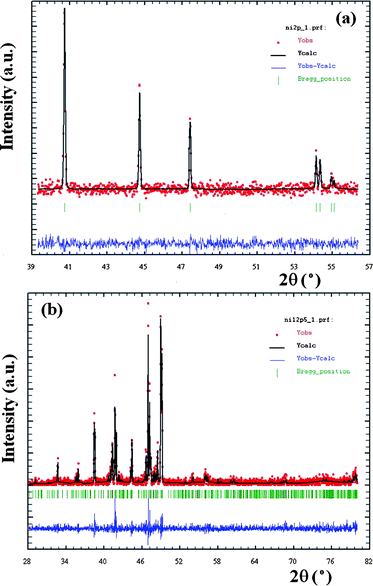 | ||
| Fig. 2 Comparisons of experimental and calculated XRD patterns of the products prepared by different conditions: (a) Ni2P prepared from the system containing 0.1 g of PVA at 160 °C for 10 h and (b) Ni12P5 prepared from the system containing 0.2 g of AM at 160 °C for 20 h. | ||
The morphologies of the products were observed by SEM and HR/TEM. Fig. 3a shows a low-magnification SEM image of the as-obtained Ni2P nanoparticles. A great deal of particles can be clearly seen. These particles are comprised of smaller nanoparticles, which aggregate each other due to big surface energy (see Fig. 3b). The inset in Fig. 3b is the SAED pattern of the product. Concentric rings implies the polycrystalline nature of the product and can be indexed as the hexagonal Ni2P phase. Fig. 3c depicts a HRTEM image of a single nanoparticle with the size of ∼13 nm. The clear stripes imply its good crystallinity and the distance of neighbouring planes is measured to be 0.336 nm, which is very close to 0.338 nm of (001) plane of Ni2P form. Fig. 3d presents a typical SEM image of Ni12P5 prepared from the system containing 0.2 g AM at 160 °C for 20 h. The morphology and size of the product is similar to those of Ni2P shown in Fig. 3a. Some plate-like particles should be attributed to the aggregation of abundant nanoparticles. A representative TEM image is given in Fig. 3e, from which aggregated nanoparticles can be distinctly found. Also, the SAED pattern proves the polycrystalline nature of the product (see the inset in Fig. 3e). A HRTEM image given in Fig. 3f presents regular stripes and the distance between neighbouring planes is 0.217 nm, which is very close to 0.21677 nm of (231) plane of Ni12P5 form. Fig. 3g is a representative SEM image of Ni12P5 prepared from the system containing 0.2 g AM at 180 °C for 10 h. The film-like product is obtained, which should be attributed to the aggregation of nanoparticles. The inset shown in Fig. 3g is a high magnification SEM image, from which one can find that the surface of the film is loose and porous.
 | ||
| Fig. 3 (a–c) FESEM, TEM and HRTEM images of Ni2P prepared at 160 °C for 10 h in the presence of 0.1 g PVP; (d–f) FESEM, TEM and HRTEM images of Ni12P5 prepared at 160 °C for 20 h in the presence of 0.2 g AM. Insets are SAED patterns; (g) SEM image of Ni12P5 prepared at 180 °C for 10 h in the presence of 0.2 g AM. The inset is a high magnification SEM image. | ||
Furthermore, we also prepared nickel phosphide nanoparticles at 160 °C for 10 h in the presences of some other surfactants of 0.1 g such as SDBS, CTAB and PEG-10000. XRD analyses showed that all products were hexagonal Ni2P phase (see Fig. 4a–c). These facts indicate that the above surfactants play the same role in the formation of hexagonal Ni2P phase. However, a mixture composed of Ni2P and Ni12P5 was obtained when AM of 0.2 g was selected as the surfactant under the above temperature and time (see Fig. 4d); and the peaks of Ni12P5 were far stronger than those of Ni2P. This implies that Ni12P5 is the main product in the mixture. Distinctly, AM acts as a different role from the previous four surfactants in the nucleation and growth of nickel phosphides. In order to further ascertain the roles of surfactants on the nucleation and growth of nickel phosphides, we synthesized the product in the absence of any surfactant at 160 °C for 10 h. XRD analyses showed that the as-obtained product was a mixture of Ni2P and Ni12P5, too. Herein, however, the peaks of Ni2P are stronger than those of Ni12P5 (see Fig. 4e), indicating that Ni2P is the main product in the mixture. Comparing Fig. 4d with Fig. 4e, one can easily find that two diffraction patterns are obviously different. Namely, after introducing AM molecules the nucleation and growth of tetragonal Ni12P5 phase are promoted and the growth of hexagonal Ni2P form is restrained. When the reaction temperature is enough high (e.g. 180 °C) or the time is enough long (e.g. 20 h), pure Ni12P5 phase is obtained. Contrarily, PVP, SDBS, CTAB and PEG-10000 can promote the nucleation and growth of hexagonal Ni2P phase.
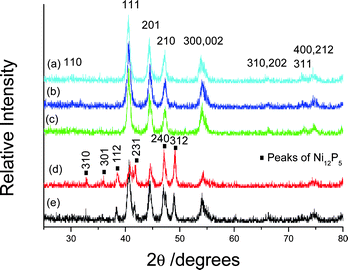 | ||
| Fig. 4 XRD patterns of the products synthesized at 160 °C for 10 h in the presence of various surfactants: (a) 0.1 g SDBS, (b) 0.1 g CTAB, (c) 0.1 g PEG-10000, (d) 0.2 g AM and (e) no surfactant. | ||
In the current work, the reason that AM encourages the nucleation and growth of tetragonal Ni12P5 phase should be attributed to its molecular structure. Different from the other four surfactants, –NH2groups existed in AM molecules have stronger coordination abilities to Ni2+ ions. The presence of abundant complex ions varies the combining fashion of P3− and Ni2+ ions. As a result, the pure Ni12P5 phase is obtained under the proper conditions.
Generally, the properties of materials are related to their shape, size and phase. In our previous works we found that transition metal phosphides could promote the degradation of organic compounds under the irradiation of UV light.19–21 Here, as a case of the relation between properties and phases, we investigated the optical property changes of Pyronin B and Safranin T solutions in the absence/presence of nickel phosphide photocatalysts under the irradiation of the 365 nm UV light for given times, respectively. Fig. 5a shows the degradation rates of Pyronin B irradiated by 365 nm UV light for different times in the presence of different catalysts. When no catalyst was employed, the degradation rate of Pyronin B dye was no more than 15% within 60 min. When pure Ni12P5 was employed as the catalyst, the degradation rate of Pyronin B dye increased to 24% during the same period. This fact shows that Ni12P5 nanoparticles can promote the degradation of Pyronin B dye. While a mixture of Ni12P5 and Ni2P was used as the catalysts, Pyronin B dye of 36% was successfully degraded in 60 min. However, when pure Ni2P nanoparticles were selected as the catalysts, the degradation rate of Pyronin B dye was close to 50% in the same time. The above facts imply that the as-prepared hexagonal Ni2P nanoparticles possess better photocatalytic ability than pure tetragonal Ni12P5 nanoparticles for the degradation of Pyronin B dye. Similar phenomena are also found in the degradation of Safranin T dye (see Fig. 5b). Since no obvious difference is found in the shape and size of two nickel phosphides, the difference on their photocatalytic degradation ability to dyes should be mainly attributed to their different phases. Furthermore, as shown in Fig. 5, Ni2P nanoparticles possess similar photocatalytic capacities for the degradation of the two organic dyes, but Ni12P5 nanoparticles have different capacities. Ni12P5 nanoparticles can slightly promote the degradation of Pyronin B, but strongly for Safranin T within the same time under 365 nm UV light, which is in good agreement with our previous work.19 This experiment indicates that the photocatalytic degradation capacity of Ni12P5 is affected by the structures of organic molecules. Safranin T molecules contain –NH2groups, which can coordinate with Ni atom in Ni12P5. Pyronin B is an ionic compound dye, it cannot bind with Ni atom in Ni12P5. As a result, Safranin T dyes are degraded more easily than Pyronin B in the presence of Ni12P5.
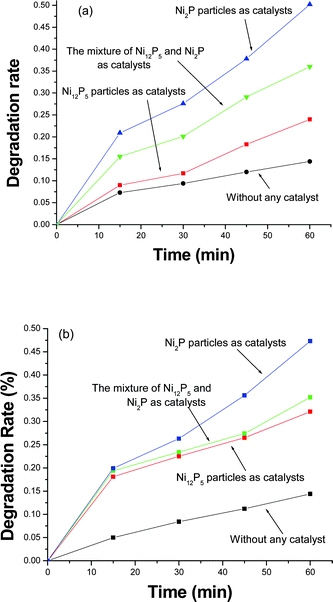 | ||
| Fig. 5 Degradation rates of organic dyes in the absence/presence of nickel phosphides under the irradiation of 365 nm light for different times: (a) Pyronin B and (b) Safranin T. | ||
In summary, phase-controllable synthesis of nickel phosphide nanocrystals has been successfully realized by a simple and reliable hydrothermal route, employing nontoxic red phosphorus and nickel dichloride as the starting materials. Appropriate surfactant, reaction temperature and time are important factors realizing phase-controllable synthesis of nickel phosphide nanocrystals. Experiments indicate that the properties of materials can be affected by their phases. The as-synthesized hexagonal Ni2P nanocrystals are better photocatalysts than tetragonal Ni12P5 nanoparticles for the degradation of organic dyes such as Pyronin B and Safranin T.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (20771005), Key Foundation of Chinese Ministry of Education (210098) and the Education Department of Anhui Province (No. 2006KJ006TD).Notes and references
- S. L. Brock, S. C. Perera and K. L. Stamm, Chem.–Eur. J., 2004, 10, 3364 CrossRef CAS.
- Y. Li, M. A. Malik and P. O. Brien, J. Am. Chem. Soc., 2005, 127, 16020 CrossRef CAS.
- L. Sun, Y. Hao, C. L. Chien and P. C. Searson, IBM J. Res. Dev., 2005, 49, 79 CAS.
- Y. Cui and C. M. Lieber, Science, 2001, 291, 851 CrossRef CAS.
- T. Hyeon, Chem. Commun., 2003, 927 RSC.
- S. T. Oyama, P. Clark, X. Wang, T. Shido, Y. Iwasawa, S. Hayashi, J. M. Ramallo-Lopez and F. G. Requejo, J. Phys. Chem. B, 2002, 106, 1913 CrossRef CAS.
- S. F. Yang, C. H. Liang and R. Prins, J. Catal., 2006, 241, 465 CrossRef CAS.
- J. A. Cecilia, A. Infantes-Molina, E. Rodriguez-Castellon and A. Jimenez-Lopez, J. Catal., 2009, 263, 4 CrossRef CAS.
- S. T. Oyama, X. Wang, Y. K. Lee and W. J. Chun, J. Catal., 2004, 221, 263 CrossRef CAS.
- X. Wang, P. Clark and S. T. Oyama, J. Catal., 2002, 208, 321 CrossRef CAS.
- J. Park, B. Koo, K. Y. Yoon, Y. Hwang, M. Kang, J. G. Park and T. Hyeon, J. Am. Chem. Soc., 2005, 127, 8433 CrossRef CAS.
- A. E. Henkes, Y. Vasquez and R. E. Schaak, J. Am. Chem. Soc., 2007, 129, 1896 CrossRef CAS.
- R. K. Chiang and R. T. Chiang, Inorg. Chem., 2007, 46, 369 CrossRef.
- Y. Chen, H. She, X. Luo, G. H. Yue and D. L. Peng, J. Cryst. Growth, 2009, 311, 1229 CrossRef CAS.
- X. Wang, F. Wan, Y. Gao, J. Liu and K. Jiang, J. Cryst. Growth, 2008, 310, 2569 CrossRef CAS.
- S. Liu, X. Liu, L. Xu, Y. Qian and X. Ma, J. Cryst. Growth, 2007, 304, 430 CrossRef CAS.
- J. Liu, X. Chen, M. Shao, C. An, W. Yu and Y. Qian, J. Cryst. Growth, 2003, 252, 297 CrossRef CAS.
- G. Shi and J. Shen, J. Mater. Chem., 2009, 19, 2295 RSC.
- J. Li, Y. H. Ni, K. M. Liao and J. M. Hong, J. Colloid Interface Sci., 2009, 332, 231 CrossRef CAS.
- Y. H. Ni, A. L. Tao, G. Z. Hu, X. F. Cao, X. W. Wei and Z. S. Yang, Nanotechnology, 2006, 17, 5013 CrossRef CAS.
- Y. H. Ni, J. Li, L. Zhang, S. Yang and X. W. Wei, Mater. Res. Bull., 2009, 44, 1166 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |