 Open Access Article
Open Access ArticleSynthesis of cyclic adenosine 5′-diphosphate ribose analogues: a C2′ endo/syn “southern” ribose conformation underlies activity at the sea urchin cADPR receptor†
Christelle
Moreau
a,
Gloria A.
Ashamu
a,
Victoria C.
Bailey
a,
Antony
Galione
b,
Andreas H.
Guse
c and
Barry V. L.
Potter
*a
aWolfson Laboratory of Medicinal Chemistry, Department of Pharmacy and Pharmacology, University of Bath, Bath, UK BA2 7AY. E-mail: b.v.l.potter@bath.ac.uk; Fax: +44 1225 386114; Tel: +44 1225 386639
bDepartment of Pharmacology, University of Oxford, Mansfield Road, Oxford, UK OX1 3QT
cInstitute of Biochemistry and Molecular Biology I: Cellular Signal Transduction, University Medical Center Hamburg-Eppendorf, Germany
First published on 25th October 2010
Abstract
Novel 8-substituted base and sugar-modified analogues of the Ca2+ mobilizing second messenger cyclic adenosine 5′-diphosphate ribose (cADPR) were synthesized using a chemoenzymatic approach and evaluated for activity in sea urchin egg homogenate (SUH) and in Jurkat T-lymphocytes; conformational analysis investigated by 1H NMR spectroscopy revealed that a C2′ endo/syn conformation of the “southern” ribose is crucial for agonist or antagonist activity at the SUH-, but not at the T cell-cADPR receptor.
Introduction
Cyclic adenosine 5′-diphosphoribose (cADPR, 1, Fig. 1), a metabolite of nicotinamide adenine dinucleotide (NAD+), was first discovered in 1987 by Lee and co-workers as a potent Ca2+ releasing second messenger.1 Based on NMR and mass spectroscopy this dinucleotide was suggested to possess a cyclic structure with a glycosidic bond between N6 of the adenine ring and the anomeric carbon C1′′ of the ribose linked to nicotinamide.2 Later, the structure of cADPR was finally revealed by X-ray analysis to be a unique cyclic dinucleotide bearing two glycosidic bonds between N9 and C1′ of the ribose ring (or “southern” ribose) and N1 and C1′′ of the second ribose moiety (or “northern” ribose). The structure also revealed both N-glycosidic bonds to be in the β-configuration and the “southern” ribose to be predominantly in the C2′-endo conformation.3 The cADPR/Ca2+ signalling system is active in diverse cellular systems such as animal cellse.g. smooth, skeletal and cardiac muscle, acinar cells, protozoa and plant cells.4 Pharmacological studies indicate that ryanodine receptors are the intracellular Ca2+ channels involved in cADPR-induced Ca2+ release.5–7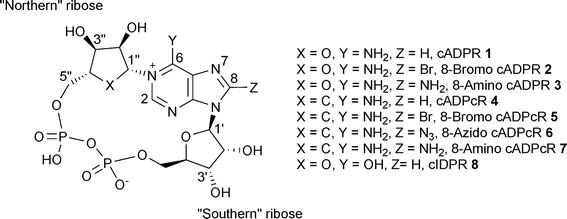 | ||
| Fig. 1 cADPR analogue structures and numbering system. | ||
Many cADPR analogues have been synthesized since and their Ca2+ mobilising activities examined in various systems, but mainly in sea urchin egg homogenates (SUH) and Jurkat T cells (JTC).8–12 Despite these efforts and although many useful synthetic tools have been developed, the structural features needed for both agonist/antagonist activities at the receptor still remain somewhat unclear. Early findings seemed to reveal that substitution at the 8-position of the adenine ring of cADPR (2 and 3, Fig. 1) converts a cADPR agonist into an antagonist in both SUH and JTC.13,14 However, it was later discovered that some 8-substituted cyclic adenosine diphosphocarbocyclic ribose (cADPcR, 4–7, Fig. 1) analogues are agonists rather than antagonists in SUH, therefore suggesting that the oxygen atom of the “northern” ribose could be a crucial feature for antagonistic activity.15 A small number of 8-substituted cyclic inosine diphosphoribose (cIDPR, 8, Fig. 1) analogues have been synthesised in our laboratory. Some of these acted as agonists in T cells, suggesting the 6-amino group to be an important structural feature for antagonistic activity.16–18 Use of this template has lead to structural biology insight on the cADPR hydrolase CD38.19 Modification of the base moiety of cADPR has produced an agonist analogue in 3-deaza cADPR, 70 times more potent than cADPR in SUH.20 More radical structural modifications of the “northern” ribose led to agonist analogues.21 Agonistic activity was also observed when the pyrophosphate linkage was extended to a triphosphate.22 Further modifications of the “southern” ribose revealed that the 2′-OH has little effect on agonist activity in SUH, but that 3′-O-alkylation could generate an antagonist.23 A recent NMR conformational study of this compound related the antagonism observed to an altered “backbone” conformation, but the evidence was not sufficient to establish this idea.24
Recently, we successfully synthesized a series of 8-substituted 2′-deoxy analogues of cADPR.25 Structure–activity relationship studies revealed that deletion of the 2′-OH group decreases antagonistic activity but, more importantly, some classical antagonist analogues unexpectedly showed agonistic activity at high concentrations in SUH. While some parallel trends were observed between analogues acting at both SUH and JTC cADPR receptors it is becoming clear, not surprisingly, that the receptors in these invertebrate and mammalian systems are different. In any case, these results illustrate that the global structural features required for cADPR agonism/antagonism are far from clear and are also different from system to system. There is clearly a need to rationalise the considerable range of activities now observed and deduce trends to underpin future design strategies in this area for chemical biological intervention.
Here, we report the synthesis of several new cADPR analogues (9–18) modified at the “southern” ribose and in the purine ring, as well as their biological evaluation in both SUH and JTC. Also, we report, for the first time, how agonism/antagonism in SUH may be linked to partial conformational preference in cADPR. A preliminary account of some of the synthetic work has appeared.14
Results and discussion
Chemistry
 | ||
Scheme 1 Synthesis of 8-modified cADPR analogues. Reagents and conditions: (a) PdCl2, PPh3, AlMe3, THF, reflux, 2.5 h. (b) POCl3, TEP, 0 °C. (c) β-NMN+, DCC, pyridine:water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 7 days, rt. (d) Aplysiacyclase, 25 mM HEPES (pH 6.8), 20 min, rt. :1), 7 days, rt. (d) Aplysiacyclase, 25 mM HEPES (pH 6.8), 20 min, rt. | ||
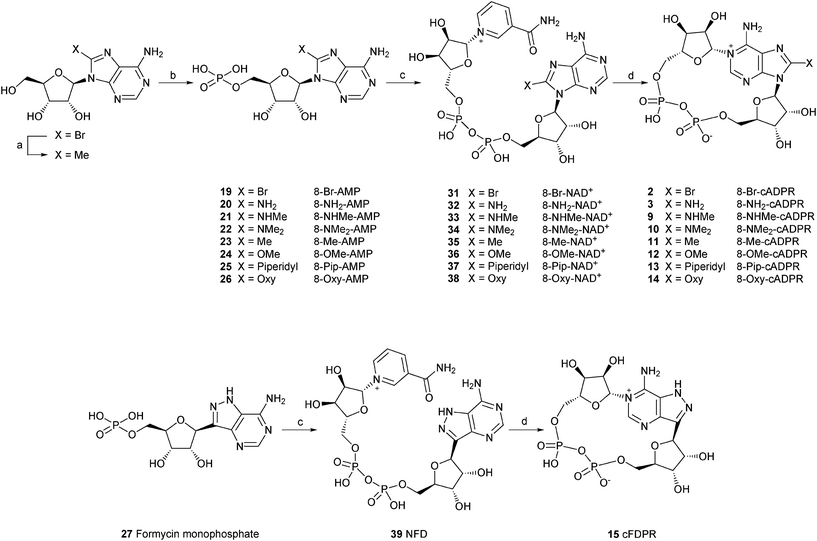
8-Bromo adenosine 5′-monophosphate (19) was prepared using established methodology developed by Yoshikawa et al.26 Crude product was purified on an ion-exchange Q-Sepharose column eluted with a gradient of 1 M TEAB followed by a second column of activated charcoal to removed the inorganic phosphate contaminant. 8-Methyl adenosine 5′-monophosphate (8-Me-AMP, 23) was prepared in 2 steps by a palladium-catalysed coupling reaction of 8-Br-adenosine with a methylating agent, followed by phosphorylation. Other AMP derivatives (20–26) were prepared by nucleophilic displacement on 8-bromo AMP by the appropriate agent. 8-modified NAD+ analogues (31–39) were synthesised by a method developed by Hughes et al. which consists of the coupling of β-nicotinamide mononucleotide (β-NMN+) with the corresponding monophosphate in the presence of dicyclohexylcarbodiimide (DCC) as a coupling reagent.27 This method, however, was low yielding (7–61%) and lately we have used a better approach involving the coupling of a monophosphate with a nucleotide phosphoromorpholidate in the presence of a Lewis acid.16,25
All of the final nucleotide pyrophosphates were carefully separated from any monophosphates and purified to homogeneity by ion-exchange chromatography. Subsequent incubation with the Aplysiaenzyme, followed by purification by ion-exchange produced the desired 8-modified cADPR analogues. These were: 8-bromo cyclic adenosine diphosphoribose (8-Br-cADPR, 2), 8-amino cyclic adenosine diphosphoribose (8-NH2-cADPR, 3), 8-methylamino cyclic adenosine diphosphoribose (8-NHMe-cADPR, 9), 8-dimethylamino cyclic adenosine diphosphoribose (8-NMe2-cADPR, 10), 8-methyl cyclic adenosine diphosphoribose (8-Me-cADPR, 11), 8-methoxy cyclic adenosine diphosphoribose (8-OMe-cADPR, 12), 8-piperidyl cyclic adenosine diphosphoribose (8-Pip-cADPR, 13), 8-oxy cyclic adenosine diphosphoribose (8-Oxy-cADPR, 14) and 8-aza-9-deaza cyclic adenosine diphosphoribose (cFDPR, 15).
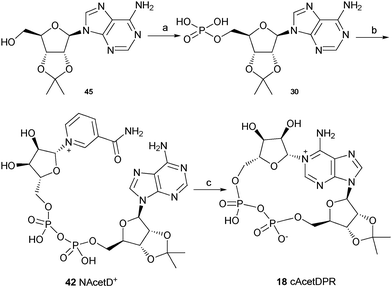
The synthesis of these three analogues 16, 17 and 18 is summarised in Schemes 2, 3 and 4 respectively. Acyclic adenosine28,29 (43), arabinofuranoside (44) and isopropylidene protected adenosine (45) were selectively phosphorylated at the 5′-hydroxyl using a POCl3, triethylphosphate and water mixture to give their respective monophosphates 28, 29 and 30. Subsequent pyrophosphate bond formation followed by cyclase incubation under the general conditions described for the 8-modified analogues, generated the cAcycloDPR (16), cAraDPR (17), cAcetDPR (18) analogues respectively.
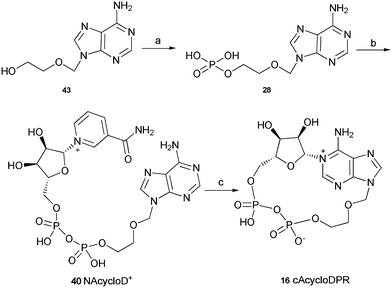 | ||
| Scheme 2 Synthesis of cAcycloDPR. Reagents and conditions: (a) POCl3, TEP, 0 °C, 1.5 h then ice/water. (b) β-NMN+, DCC, 7 days, rt. (c) Aplysiacyclase, 25 mM HEPES (pH 6.8), rt. | ||
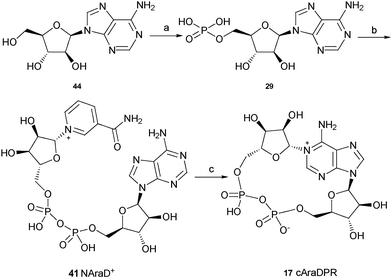 | ||
| Scheme 3 Synthesis of cAraDPR. Reagents and conditions: (a) POCl3, TEP, 0 °C, 1.5 h then ice/water. (b) β-NMN+, DCC, 7 days, rt. (c) Aplysiacyclase, 25 mM HEPES (pH 6.8), rt. | ||
Biology
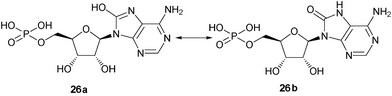
8-Amino-cADPR (3) was also synthesized as a control to enable us to carry out comparative studies. 8-Amino-cADPR was confirmed as a potent antagonist in SUH with an IC50 value of 0.01 μM. Replacement of the amino group with a methyl group as in 8-methyl-cADPR (11) gave an antagonist with an IC50 value of 0.53 μM. The –CH3group is similar in atomic mass to –NH2 and yet 8-Me-cADPR was 53 times less potent as an antagonist compared to 8-NH2-cADPR (3). Substitution with an oxy group (with an atomic mass of 17) was attempted. The analogue obtained (14) showed still weaker activity as an antagonist with an approximate IC50 of 2 μM. 8-“Hydroxy” AMP (26a) is known to exist predominantly in the keto form (26b) at physiological range 5 < pH < 9,30,31 hence the nitrogen N7 is protonated (Fig. 2). It is reasonable to assume that 8-oxy cADPR is also predominantly in the keto form at the same pH range. The reduction in activity could be due to the protonation at N7 which may affect receptor binding. Substitution with groups of varying size such as -NHMe, -NMe2 and -piperidyl produced novel compounds (9, 10 and 13 respectively) with perturbations to the 8-amino motif, which were investigated for antagonist activity (Table 1). 8-NHMe-cADPR (9) was a much weaker antagonist compared to 8-NH2-cADPR (3) with an IC50 of ∼40 μM, 8-NMe2 cADPR (10) was weaker than 8-NHMe-cADPR (9) as an antagonist, but 8-piperidyl (13) was not active at all as an antagonist up to 50 μM. Other analogues such as 8-OCH3-cADPR (12) and 8-Br-cADPR (2) were also synthesised for comparative study. These analogues were antagonists with IC50 values of 4.8 and 0.97 μM respectively. It appears, therefore, from this study, that the ability of the molecule to hydrogen bond at the 8-position with its target protein in SUH is not critical for activity as an antagonist as 8-CH3-cADPR (11) is a better antagonist compared to those that can form hydrogen bonds such as 8-OCH3-cADPR (12) and 8-NHMe-cADPR (9).
| 8-X-cADPR | IC 50 (μM) in SUH | IC 50 (μM) in JTC |
|---|---|---|
| a Ca2+ mobilisation from sea urchin egg microsomes was evaluated fluorimetrically using 2.5% egg homogenates containing fluo-3 (3 μM) as previously described.25,33Ca2+ release assay in Jurkat T cells was carried out as reported previously.34,35 b IC50 was not reached (partial antagonist).nd = active compound, IC50 was not determined. | ||
| 8-NH2-cADPR (3) | 0.01 | 1 |
| 8-Me-cADPR (11) | 0.53 | n.d |
| 8-Br-cADPR (2) | 0.97 | b |
| 8-Oxy-cADPR (14) | 2 | n.d |
| 8-OMe-cADPR (12) | 4.8 | 0.2 |
| 8-NHMe-cADPR (9) | <40 | n.d |
| 8-NMe2-cADPR (10) | >40 | n.d |
| 8-Pip-cADPR (13) | No effect | n.d |
Modification in the purine ring as in 8-aza-9-deaza cADPR (15), resulted in an analogue that is 10 times less potent as an agonist compared to cADPR (EC50 = 0.31 μM). The C8 carbon in the purine ring was replaced by a nitrogen atom and the N9 nitrogen was replaced by a carbon atom. Modification in the purine ring hence did not produce an antagonist compared to exocyclic modification as discussed above. The reduction in activity could be due to protonation of the N7 nitrogen which may affect receptor binding. A small modification at this position as in 7-deaza cADPR (51), has been reported to result in partial agonist activity 32
In order to further study these interactions three compounds (16, 17 and 18) were designed, synthesised and evaluated for their Ca2+ mobilising activities in SUH. Both cAcycloDPR (16) and cAraDPR (17) were inactive whilst cAcetDPR (18) was a poor agonist (EC50 = 12 μM). A careful study of the biological data obtained for these compounds suggested that instead of there being a relationship between the groups attached to the ribose ring and activity, the conformation of the ribose ring is of importance and will be discussed later.
Modification in the purine ring in analogue (15) did not affect the Ca2+ release property of the analogue. This analogue showed a similar Ca2+ release profile compared to cADPR in JTC even though the 8-position is altered (although not outside the ring). These alterations appear to be unimportant for Ca2+ releasing activity in this system.
Conformational analysis
Despite significant past work by several groups, the structural features required for cADPR-mediated agonism/antagonism remain unclear. Recently, Shuto et al. reported that 2′′,3′′-dideoxydidehydro cADPcR (62), an inactive compound, adopted a major C3′ endo and a high anti conformation in aqueous solution,38 therefore indicating that both the N9-ribose moiety and the N9-glycosidic bond conformations may be of crucial importance for the Ca2+ release activities of cADPR analogues.In solution, nucleosides and nucleotides exist in conformational equilibrium between C2′-endo and C3′-endo forms (Fig. 3). In addition, the nucleobase can be oriented towards (syn) or away (anti) from the ribose ring. These local changes will affect the overall conformation of the cyclic dinucleotides. Extensive studies developed by Altona and Sundaralingam have established that the C2′-endo/C3′-endo ratio can be mathematically calculated from a 1H NMR spectrum by adaptation of the equation C2′-endo = [J1′,2′/(J1′,2′ + J3′,4′)] × 100. Moreover, they have also shown that the sum J1′,2′ + J3′,4′ is close to 10 and therefore the ratio C2′-endo/C3′-endo can be estimated from the equation 10J1′,2′.39,40
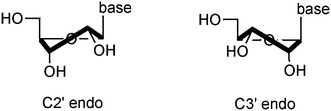 | ||
| Fig. 3 Schematic representation of the ribofuranose ring in both C2′endo and C3′ endo conformation. | ||
In cADPR, the adenine base is oriented in the syn conformation about the glycosidic bond both in the crystal structure3 and in solution.24,38 Information on the conformation about the glycosidic bond has been well documented, and has revealed that the chemical shift of the H-2′ proton could be used very effectively as a good indicator for the syn/anti equilibrium in nucleosides and nucleotides.41,42 Typically, purine nucleosides and nucleotides with a bulky substituent at C8 display a characteristic downfield shift of H-2′ upon 8-substitution. The chemical shift values for the protons common to some AMP, NAD+ and cADPR analogues prepared during the course of this study are listed in Table 2 and indeed, in agreement with early reports, we have found that the NMR resonance for H-2′ can be used very effectively to assign the favoured glycosyl bond conformation. Therefore, AMP, 8-amino- and 8-aminomethyl AMP appear to be predominantly anti as previously reported.43,44 The H-2′ resonance of 7-deaza AMP suggests that this nucleotide has significant antiglycosyl bond character, a result that is not surprising due to its resemblance to AMP. Our data show a similar trend for the H-2′ resonance values in the NAD+ series as in the AMP series. We had previously observed in the hypoxanthine series that the glycosyl bond conformation is unaffected by pyrophosphate bond formation.16 However, during the cyclization reaction, the NAD+ analogues in the anti conformation e.gNAD+, 8-amino-, 8-methylamino- and 7-deaza NAD+ have their cyclic counterpart predominantly in the syn configuration.
| X | 8-X-AMP | 8-X-NAD + | 8-X-cADPR | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H-1′ | H-2 ′ | Δ 1′-2′ a | Conf b | H-1′ | H-2 ′ | Δ 1′-2′ | Conf | H-1′ | H-2 ′ | Δ 1′-2′ | Conf | |
| a Difference in chemical shifts between H-1′ and H-2′. b Favoured glycosidic bond conformation. | ||||||||||||
| H | 6.1 | 4.8 | 1.3 | anti | 6.0 | 4.7 | 1.3 | anti | 5.8 | 5.2 | 0.6 | syn |
| Br | 5.8 | 5.1 | 0.7 | syn | 5.7 | 5.0 | 0.7 | syn | 6.3 | 5.6 | 0.7 | syn |
| Me | 5.8 | 4.8 | 1.0 | syn | 5.7 | 4.7 | 1.0 | syn | 5.8 | 5.3 | 0.5 | syn |
| NH2 | 5.8 | 4.6 | 1.2 | anti | 5.9 | 4.6 | 1.2 | anti | 5.8 | 5.4 | 0.4 | syn |
| NHMe | 5.8 | 4.6 | 1.2 | anti | 5.7 | 4.5 | 1.2 | anti | 5.7 | 5.6 | 0.1 | syn |
| NMe2 | 5.6 | 5.1 | 0.5 | syn | 5.9 | 5.4 | 0.5 | syn | 6.2 | 5.6 | 0.6 | syn |
| Pip | 5.6 | 5.1 | 0.5 | syn | 5.5 | 5.0 | 0.5 | syn | 5.9 | 5.6 | 0.3 | syn |
| OMe | 5.7 | 4.8 | 0.9 | syn | 5.7 | 4.8 | 0.9 | syn | 5.9 | 5.6 | 0.3 | syn |
| Oxy | 5.6 | 4.9 | 0.7 | syn | 5.5 | 4.9 | 0.6 | syn | 5.8 | 5.6 | 0.2 | syn |
| 7-Deaza | 6.1 | 4.5 | 1.6 | anti | 6.1 | 4.4 | 1.7 | anti | 5.7 | 5.3 | 0.4 | syn |
| 7-Deaza-8-Br | 6.0 | 5.1 | 0.9 | syn | 6.0 | 5.1 | 0.9 | syn | 6.0 | 5.4 | 0.6 | syn |
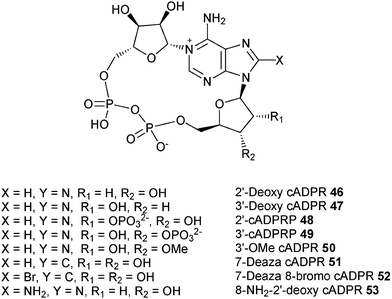
We have therefore extracted information on the preferred conformation about the glycosidic bond based on NMR results, and have determined the conformation of the “southern” ribose using Altona's approach for all the cADPR analogues synthesized in our laboratory as well as those from other groups. The results of our analysis are summarized in Table 3. Structures of analogues 46–62 are shown in Fig. 4 and 5.
| Compounds | J 1′,2′ | J 3′,4′ | C2′ endo | H-1′ | H-2′ | Δ1′-2′ | Syn/Anti | Activity in SUH |
|---|---|---|---|---|---|---|---|---|
| a Not determined.The compounds in italics do not display a C2′ endo/syn conformation. | ||||||||
| Compounds synthesized by our group 14,23,25,32,45–50 | ||||||||
| cADPR (1) | 5.6 | 3.2 | 64% | 5.8 | 5.2 | 0.6 | Syn | Agonist EC50 = 32 nM |
| 8-Bromo-cADPR (2) | 5.3 | n.da | 53% | 6.3 | 5.6 | 0.7 | Syn | Antagonist IC50 = 0.97 μM |
| 8-NH2 cADPR (3) | 5.6 | n.d | 56% | 5.8 | 5.3 | 0.5 | Syn | Antagonist IC50 = 0.01 μM |
| 8-NHMe cADPR (9) | 5.5 | n.d | 55% | 5.7 | 5.6 | 0.1 | Syn | Weak antagonist IC50 = 40 μM |
| 8-NMe2 cADPR (10) | 6.4 | n.d | 64% | 6.2 | 5.6 | 0.6 | Syn | Weak antagonist IC50>40 μM |
| 8-Me cADPR (11) | 5.3 | n.d | 53% | 5.8 | 5.3 | 0.5 | Syn | Antagonist IC50 = 0.53 μM |
| 8-OMe cADPR (12) | 5.9 | n.d | 59% | 5.9 | 5.6 | 0.3 | Syn | Antagonist IC50 = 4.8 μM |
| 8-Aza-9-deaza cADPR (15) | 5.3 | n.d | 53% | 5.9 | 5.0 | 0.9 | Syn | Agonist EC50 = 0.3 μM |
| cAcycloDPR (16) | — | — | — | — | — | — | — | Inactive |
| cAraDPR (17) | 7.3 | 8.1 | 47% | 6.2 | 5.1 | 1.1 | Anti | Inactive |
| cAcetDPR (18) | 2.0 | 3.1 | 22% | 6.2 | 5.1 | 1.1 | Anti | Poor agonist EC 50 = 12 μM |
| 2′-Deoxy cADPR (46) | 7.0 | n.d* | 70% | Syn | Agonist EC50 = 58 nM | |||
| 3′-Deoxy cADPR (47) | 3.0 | n.d | 30% | 5.9 | 5.2 | 0.7 | Syn | Poor agonist EC 50 = 5 μM |
| 2′-cADPRP (48) | 4.0 | n.d | 40% | 6.2 | 5.7 | 0.5 | Syn | Inactive |
| 3′-cADPRP (49) | 3.7 | n.d | 37% | 6.0 | 5.3 | 0.7 | Syn | Inactive |
| 3′-OMe cADPR (50) | 5.6 | 2.7 | 67% | 5.9 | 5.3 | 0.6 | Syn | Antagonist IC50 = 4.8 μM |
| 7-Deaza cADPR (51) | 6.4 | n.d | 64% | 5.7 | 5.3 | 0.4 | Syn | Agonist EC50 = 90 nM |
| 7-Deaza-8-Br-cADPR (52) | 5.9 | 3.5 | 63% | 6.0 | 5.4 | 0.6 | Syn | Antagonist IC50 = 0.73 μM |
| 8-Amino-2′-deoxy cADPR (53) | 6.9-6.6 | n.d | 66-69% | 6.06 | — | — | Syn | Antagonist IC50 = 0.22 μM |
| Compounds synthesized by Shuto's group 15,38,51,52 | ||||||||
| cADPcR (4) | 6.1 | 2.6 | 70% | 6.0 | 5.1 | 0.9 | Syn | Agonist EC50 = 79 nM |
| 8-Cl-cADPcR (5) | 6.3 | 2.4 | 72% | 6.1 | 5.2 | 0.9 | Syn | Agonist EC50 = 19 μM |
| 8-NH2-cADPcR (6) | 6.3 | n.d | 63% | 5.9 | 5.2 | 0.7 | Syn | Agonist EC50 = 80 nM |
| 8-N3-cADPcR (7) | 6.2 | n.d | 62% | 5.9 | 5.2 | 0.7 | Syn | Agonist EC50 = 3.9 μM |
| 3′′-Deoxy cADPcR (54) | 6.1 | 2.6 | 70% | 6.1 | 5.2 | 0.9 | Syn | Agonist EC50 = 14 nM |
| 8-Cl-3′′-deoxy cADPcR (55) | 6.2 | n.d | 62% | 6.1 | 5.2 | 0.9 | Syn | Partial agonist EC50 = 0.19 μM |
| 8-NH2-3′′-deoxy cADPcR (56) | 6.3 | 2.3 | 73% | 5.9 | 5.3 | 0.6 | Syn | Partial agonist EC50 = 17 nM |
| 8-N3-3′′-deoxy cADPcR (57) | 6.2 | n.d | 62% | 5.9 | 5.2 | 0.7 | Syn | Partial agonist EC50 = 0.49 μM |
| 2′′-Deoxy cADPcR (58) | 5.9 | 3.0 | 66% | 6.1 | 5.2 | 0.9 | Syn | Agonist EC50 = 0.61 μM |
| 2′′,3′′-Dideoxy cADPcR (59) | 5.4 | 2.4 | 69% | 6.1 | 5.2 | 0.9 | Syn | Agonist EC50 = 0.73 μM |
| 2′′-OMOM-3′′-OMe cADPcR (60) | 6.3 | 2.4 | 72% | 6.0 | 5.1 | 0.9 | Syn | Agonist EC50 = 0.88 μM |
| 2′′-OMOM-3′′-deoxy cADPcR (61) | 5.3 | 2.2 | 70% | 6.1 | 5.2 | 0.9 | Syn | Agonist EC50 = 0.88 μM |
| 2′′,3′′-Dideoxy- didehydro cADPcR (62) | 1.5 | 6.3 | 19% | 6.1 | 5.0 | 1.1 | Anti | Poor agonist EC 50 > 20 μM |
 | ||
| Fig. 4 Structures of the cADPR analogues mentioned herein – our group. | ||
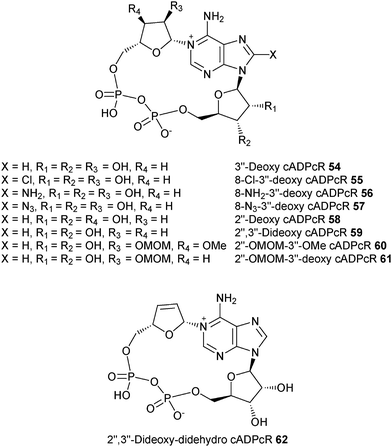 | ||
| Fig. 5 Structures of the cADPR analogues mentioned herein – Shuto's group. | ||
At first glance, it can be seen that most cADPR analogues adopt a syn conformation about the N9-glycosyl linkage except cAcetDPR (18), cAraDPR (17) and 2′′,3′′-dideoxydidehydro cADPcR (62). These same compounds also adopt a C3′ endo form in the N9-ribose moiety.
Additionally, 3′-deoxy cADPR, 2′-cADPRP and 3′-cADPRP also display a C3′ endo form, but with a predominantly syn glycosidic bond. It appears that these compounds are either inactive or are really poor agonists in SUH.23 Conversely, all the other analogues in this table displaying a C2′ endo/syn conformation are either agonists or antagonists in SUH.
Whilst this analysis does not provide structural clues about relative agonistic/antagonistic activity or potency in either case, it does seem that the C2′ endo/syn conformation may be a key requirement for activity in SUH only. Indeed, it has been demonstrated earlier that there are differences in ligand recognition by the protein between the sea urchin and T cell receptor. For example, 2′-deoxy cADPR (46) is inactive in T cells, but is a potent agonist in SUH, whereas 2′-cADPRP (48) is active in T cells but not in SUH (Table 4). 3′-OMe cADPR (50) was designed to investigate further the role of the hydroxyl group. 3′-Deoxy cADPR (47) can neither donate or accept a hydrogen bond at the 3′ position, whilst 3′-OMe cADPR can only act as a hydrogen bond acceptor. This compound is an antagonist in SUH, thus showing that the oxygen atom must interact with the receptor in order to inhibit Ca2+ release. However, the same compound is an agonist in JTC, thus showing that the OMe group has little effect on the activity of cADPR. These results further highlight that there may be differences in the cADPR-Ca2+ release mechanism for the ryanodine receptor of SUH and JTC. This trend appears to be valid only with cADPR analogues and not with the cIDPR series. Indeed, some cIDPR derivatives (e.g 8-Br cIDPR) show agonist activity in T cells but are apparently inactive in SUH.
| Compounds | Activity in SUH | Activity in JTC | Ref. |
|---|---|---|---|
| cADPR (1) | Agonist | Agonist | 1,34 |
| 8-Bromo cADPR (2) | Antagonist | Antagonist | 14,25 |
| 8-NH2 cADPR (3) | Antagonist | Antagonist | 13,34 |
| 8-NHMe cADPR (9) | Weak antagonist | Antagonist | 50 |
| 8-NMe2 cADPR (10) | Weak antagonist | Antagonist | 50 |
| 8-Me cADPR (11) | Antagonist | Antagonist | 46 |
| 8-OMe cADPR (12) | Antagonist | Antagonist | 37 |
| 8-Piperidyl cADPR (13) | Inactive | Antagonist | 14 |
| 8-Aza-9-deaza cADPR (15) | Active | Active | 50 |
| cAcycloDPR (16) | Inactive | Not tested | 49 |
| cAraDPR (17) | Inactive | Not tested | 49 |
| cAcetDPR (18) | Poor agonist | Not tested | 49 |
| 2′-Deoxy cADPR (46) | Agonist | Inactive | 23 |
| 3′-Deoxy cADPR (47) | Poor agonist | Inactive | 23 |
| 2′-cADPRP (48) | Inactive | Agonist | 23,47 |
| 3′-cADPRP (49) | Inactive | Inactive | 23,47 |
| 3′-OMe cADPR (50) | Antagonist | Not tested | 23 |
| 7-Deaza-8-bromo cADPR (52) | Antagonist | Antagonist | 37,48,49 |
| 8-Bromo cIDPR | Inactive | Agonist | 17 |
We naturally need to invoke the caveat for all work of this nature that, while our study focuses upon cADPR analogue conformation in solution, we can draw no firm conclusions regarding actual ligand conformation as bound to the cADPR receptor. A recent trend in nucleoside/nucleotide work in general has been to employ the use of conformationally locked rigid ribose motifs using a variety of strategies.53–56 Taking our observations reported here into account it could be productive to apply such approaches also to the cADPR field and these could hopefully extend the work reported here to encompass receptor bound conformations.
Conclusion
A series of novel cADPR derivatives has been synthesized in order to investigate the determinants for both agonist and antagonist activity. These compounds were tested for their Ca2+ releasing activity in both SUH and JTC. A careful analysis of all the cADPR analogues synthesized over the past decade reveals that a C2′ endo/syn conformation (Fig. 6) is crucial for activity (agonistic or antagonistic), whereas compounds in their C3′ endo conformation are either inactive or are poor agonists. This trend appears to be valid in sea urchin egg homogenate only (not in T cells) and with cADPR analogues only (not cIDPR).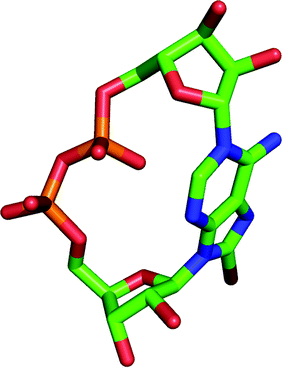 | ||
| Fig. 6 Model of 8-Br cADPR (2) with its “southern” ribose in the C2′ endo/syn conformation. Hydrogen atoms are not shown for clarity. | ||
Experimental
General Procedures
All reagents and solvents were of commercial quality and were used directly unless otherwise described. Pyridine was dried overnight with calcium hydride, distilled and stored over potassium hydroxide pellets. Aplysia californica ovotestis homogenate containing ADP-ribosyl cyclase was prepared as previously described.57 The protein concentration of the Aplysiacyclase used in all cases was estimated as 10 mg ml−1 using a Bio-rad protein estimation assay. Ion-exchange chromatography was performed on an LKB-Pharmacia medium pressure ion-exchange chromatograph using a Sepharose Q fast flow column with gradients of triethylammonium bicarbonate buffer (TEAB) pH 7.6 as eluent. 1 M TEAB was prepared by bubbling carbon dioxide gas into 1 M triethylamine solution. HPLC was performed on a Shimadzu LC-6A chromatograph with the UV detector operating at 259 nm using a combination of a Partisil 10 μ SAX guard column (10 × 0.46 cm) and a Technicol (10 × 0.46 cm) 10 μ SAX HPLC column, or using a Spherisorb 10 μ SAX (25 × 0.46 cm) column with an isocratic elution using phosphate buffer (KH2PO4), pH 3.0 at a flow rate of 1 ml min−1. 1H NMR and 31P NMR spectra were recorded on either Jeol JNM GX-270 FT NMR or Jeol EX-400 FT NMR spectrometers. Chemical shifts were measured in ppm relative to deuterated water (D2O) for 1H NMR and to external 85% H3PO4 for 31P NMR. 31P NMR spectra were measured at 162 MHz or 109 MHz in D2O. J values are given in Hertz (Hz). Mass spectra were recorded at the EPSRC Mass Spectrometry Service Center at the University of Swansea and at the University of Bath. Ultraviolet (UV) absorbance was measured with a Perkin-Elmer Lambda 3 UV/VIS spectrophotometer. Melting points were determined with a Reichert-Jung Thermo Galen Kofler block and are uncorrected.Biology
Chemistry
Synthesis of nucleoside analogues
:3, v/v) and left stirring for a further 30 min. The sample was dried in vacuo and 31P NMR spectroscopy showed the presence of inorganic phosphate and 8-Br-AMP at δ −0.1 and 0.4 ppm respectively. Inorganic phosphate was removed by passing the nucleotide mixture dissolved in 50 mL of water through a charcoal column. This column (25 × 4 cm) was prepared by using ca. 1 cm of celite as a bed on top of which 7 cm of activated charcoal (NoritB) were added. The nucleotide solution in water was poured unto the column and the eluent was collected. Water was used to flush inorganic phosphate off the column and a total of 75 mL of eluent was collected. The inorganic phosphate-free nucleotide was eluted off the column using ethanol:water:conc. ammonia (25:24:1, v/v, 2 × 500 mL). δH (D2O, 400 MHz) 3.8–3.9 (2H, m, H-5′a, H-5′b), 4.25 (1H, m, H-4′), 4.56 (1H, dd, J3′,2′ 5.8, J3′,4′ 4.2, H-3′), 5.19 (1H, dd, J2′,3′ 5.8, J2′,1′ 5.7, H-2′), 6.07 (1H, d, J1′,2′ 5.8, H-1′), 8.14 (1H, s, H-2). δC (D2O, 100 MHz) 65.3 (d, JCP 3.7, C-5′), 70.5 (C-3′), 71.8 (C-2′), 84.4 (d, JCP 9.2, C-4′), 89.2 (C-1′), 118.3 (C-5), 139.9 (C-8), 150.7 (C-4), 153.6 (C-2), 154.9 (C-6). δP (D2O, 162 MHz, 1H-decoupled) 3.4 (s, 1P) and δP (D2O, 162 MHz, 1H-coupled) 3.7 (s, J 5.7, 1P). λmax 264 nm (ε 15100, pH 7.0).
δ H (D2O, 270 MHz) 3.8-3.9 (2H, m, H-5′a, H-5′b), 4.2-4.1 (1H, m, H-4′), 4.3-4.4 (m, 1H, H-3′), 4.7 (1H, app.t, J2′,1′ = J2′,3′ 7.6, H-2′), 5.9 (1H, d, J1′,2′ 7.6, H-1′), 7.8 (1H, s, H-2). δP (D2O, 162 MHz, 1H-decoupled) 0.35 (s, 1P). λmax 274 nm (ε 16000, pH 8.3).
:1). The crude mixture was dried in vacuo to give a green residue which was dissolved in methanol (50 mL) and refluxed for 4 h with a small amount of ammonium chloride (Rf 0.05, DCM–MeOH 9:1). The crude sample was purified on a short silica gel column eluted with CHCl3–EtOH (3:2) to give a white solid which was further recrystallised from water (0.39 g, 1.4 mmol, 48%). δH (D2O, 270 MHz) 3.5–3.7 (2H, m, H-5′a, H-5′b), 4.0 (m, 1H, H-4′), 4.1 (m, 1H, H-3′), 4.8 (dd, J2′,1′ = 7.3 and J2′,3′ = 5.1 Hz, 1H, H-2′), 5.8 (1H, d, J1′,2′ 7.3, H-1′), 8.0 (1H, s, H-2). m/z (FAB+) 282 [100%, (M + H)+]. HRMS (FAB+) calcd for C11H16N4O4 282.1207 (M + H)+ found 282.1202. m.p. 208 °C.
Phosphorus oxychloride (100 μL, 1.08 mmol) was added dropwise at 0 °C to a solution of 8-methyl adenosine (170 mg, 0.6 mmol) in triethylphosphate (4 mL) under N2. The reaction was left stirring for 3 h at rt after which HPLC analysis showed the presence of a new product at 2 min. The reaction mixture was quenched by stirring in 8 mL of pyridine:water (1:3) for 30 min and the solvents were removed in vacuo. The residue was dissolved in milliQ water (350 mL) and applied to an ion-exchange Q-sepharose column eluted with 1 M TEAB. The product eluted off between 110–160 mM TEAB, and the inorganic phosphate impurity was removed by passing the solution through a charcoal column as previously described to afford the nucleotide as a white solid (0.38 mmol, 60.3%). δH (D2O, 400 MHz) δH (D2O, 400 MHz) 2.46 (3H, s, Me), 3.8-3.9 (2H, m, H-5′a, H-5′b), 4.1-4.21 (1H, m, H-4′), 4.3 (app.t, J3′,2′ = J3′,4′ = 6.2 Hz, 1H, H-3′), 4.8 (1H, app.t, J2′,1′ = J2′,3′ 6.2, H-2′), 5.8 (1H, d, J1′,2′ 6.8, H-1′), 7.9 (1H, s, H-2). δP (D2O, 162 MHz, 1H-decoupled) 4.0 (s, 1P) and δP (D2O, 162 MHz, 1H-coupled) 3.7 (s, J 5.6, 1P). λmax 260 nm (ε 15300, pH 8.3).
:10 (2 mL) to give the pure product 21. δH (400 MHz, D2O) 3.57–3.60 (2H, m, POCH2CH2), 3.74–3.78 (2H, m, POCH2), 5.66 (2H, s, OCH2base), 8.16 (1H, s, H-2), 8.26 (1H, s, H-8), and. δC (D2O, 100 MHz) 64.5 (d, JCP 5.5, POCH2), 69.8 (d, JCP 9.2, POCH2), 73.9 (OCH2base), 119.2 (C-5), 143.4 (C-8), 149.7 (C-4), 153.6 (C-2), 156.3 (C-6). δP (D2O, 161 MHz) 1.48. m/z (FAB+) 290.1 [100%, (M + H)+]. λmax 259 nm (ε 15300). HPLC Rt = 2.5 mins.
Synthesis of NAD+ analogues
β-NMN+ and the appropriate AMP analogue were dissolved in water in a round bottom flask, dry pyridine was added to make a 4:1 pyridine:water mixture and excess DCC was subsequently added to the nucleotide mixture. The solution was left stirring at room temperature for 7 days. The resulting mixture was poured into 100 mL of cold distilled water and left at 4 °C for 2 h to precipitate the DCU formed in the reaction. DCU was filtered off and the filtrate was extracted with 3 × 50 mL aliquots of chloroform to remove other water insoluble organic impurities. The aqueous layer was collected and dried in vacuo. The residue was dissolved in milliQ water and purified by ion-exchange chromatography on Q-sepharose eluted with a gradient of 1 M TEAB.
:1) as described. The pure dinucleotide was obtained in 16% yield. δH (D2O, 400 MHz) 4.2-4.6 (9H, m, ribose-H), 4.7-4.8 (1H, m, H-2′), 5.86 (1H, d, J1′,2′ 7, H-1′), 6.1 (1H, d, J1′′,2′′ 4.7, H-1′′), 8.0 (1H, s, HA2), 8.2 (1H, t, J5,6 = J5,4 6.6, HN5), 8.7 (1H, d, J4,5 6.6, HN4), 9.1 (1H, d, J6,5 6.6, HN6), 9.3 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −11.83 and −11.97 (AB system, JAB 19.4, 2P). m/z (ES−) 677 [10% (M − H)−]. HPLC Rt = 4.1 mins.
:1) as described. The pure product was obtained in 11% (17.1 μmol) yield. δH (D2O, 400 MHz) 3.1 (3H, s, NMe2), 4.4-5.0 (9H, m, ribose-H), 5.0 (1H, app.t, J2′,1′ = J2′,3′ 6.3, H-2′), 5.9 (1H, d, J1′,2′ 6.3, H-1′), 6.3 (1H, d, J1′′,2′′ 5.4, H-1′′), 8.2 (1H, s, HA2), 8.4 (1H, t, J 7, HN5), 8.9 (1H, d, J 8.0, HN4), 9.3 (1H, d, J 6.3, HN6), 9.5 (1H, s, HN2). δP (D2O, 109 MHz, 1H-decoupled) −11.42 and −11.82 (AB system, JAB 21, 2P). m/z (FAB+) 707 [70% (M + H)+]. λmax 273 nm (ε 16100, pH 8.3). HPLC Rt = 5.8 mins.
:1) as described. The pure dinucleotide was obtained in 12% yield after 2 purifications. δH (D2O, 400 MHz) 2.5 (3H, s, Me), 4.2–4.6 (9H, m, ribose-H), 4.7 (1H, d, J2′,3′ = J2′,1′ 6.4, H-2′), 5.7 (1H, d, J1′,2′ 6.4, H-1′), 5.8 (1H, d, J1′′,2′′ 5.2, H-1′′), 7.9 (1H, s, HA2), 8.1 (1H, t, J 7.0, HN5), 8.6 (1H, d, J4,5 6.3, HN4), 8.9 (1H, d, J6,5 6.1, HN6), 9.3 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −10.96 and −11.34 (AB system, JAB 20.5, 2P). m/z (ES−) 676 [100% (M − H)−]. λmax 259 nm (ε 13755, pH 8.3). HPLC Rt = 2.46 mins.
:1) as described. The pure dinucleotide was obtained in 16% yield. δH (D2O, 400 MHz) 4.0 (3H, s, OMe), 4.6-4.0 (9H, m, ribose-H), 4.8 (1H, dd, J2′,3′ 5.8, J2′,1′ 5.2, H-2′), 5.7 (1H, d, J1′,2′ 5.2, H-1′), 5.8 (1H, d, J1′′,2′′ 4.9, H-1′′), 7.9 (1H, s, HA2), 8.1 (1H, dd, J5,4 7.9, J5,6 6.1, HN5), 8.6 (1H, d, J4,5 7.9, HN4), 9.0 (1H, d, J6,5 6.1, HN6), 9.1 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −11.38 and −11.85 (AB system, JAB = 20.9 Hz, 2P). m/z (ES+) 694 [100%, M+H)+]. λmax 259 nm (ε 13000, pH 8.3). HPLC Rt = 2.4 mins.
:1) as described. The pure dinucleotide was obtained in 7% yield after 2 purifications. δH (D2O, 400 MHz) 1.6 (6H, m, 3 × CH2), 3.2 (4H, m, 2 × CH2), 4.0–4.5 (9H, m, ribose-H), 5.0 (1H, d, J2′,3′ 6.4, J2′,1′ 6.1, 1H, H-2′), 5.5 (1H, d, J1′,2′ 6.1, H-1′), 5.8 (1H, d, J1′′,2′′ 5.5, H-1′′), 7.9 (1H, s, HA2), 8.1 (1H, dd, J5,4 7.0, J5,4 6.1, HN5), 8.6 (1H, d, J4,5 7.0, HN4), 8.9 (1H, d, J6,5 6.1, HN6), 9.1 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −11.44 and −11.84 (AB system, JAB 20.8, 2P). m/z (ES−) 746 [50%, (M − H)−]. λmax 274 nm (ε 12800, pH 8.3). HPLC Rt = 6.3 mins.
:1) as described. The pure dinucleotide was obtained in 9% yield after 2 purifications. δH (D2O, 400 MHz) 4.4-4.0 (9H, m, ribose), 4.9 (1H, app.t, J2′,3′ = J2′,1′ 5.6, H-2′), 5.5 (1H, d, J1′,2′ 5.6, H-1′), 5.85 (1H, d, J1′′,2′′ 4.5, H-1′′), 7.7 (1H, s, HA2), 8.1 (1H, t, J 7.0, HN5), 8.5 (1H, d, J4,5 6.5, HN4), 8.9 (1H, d, J6,5 6.0, HN6), 9.1 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −11.44 and −11.92 (AB system, JAB 21.0, 2P). m/z (ES−) 678 [100%, (M − H)−]. λmax 266 nm (ε 13100, pH 8.3). HPLC Rt = 3.4 mins.
:1) as described. The pure dinucleotide was obtained in 9% yield. δH (D2O, 400 MHz) 4.0-5.0 (10H, m, ribose-H), 5.2 (1H, d, J1′,2′ 7.1, H-1′), 6.0 (1H, d, J1′′,2′′ 5.3, H-1′′), 8.0 (1H, s, HA2), 8.1 (1H, m, HN5), 8.6 (1H, d, J4,5 8.0, HN4), 9.05 (1H, d, J6,5 5.3, HN6), 9.2 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −11.88 and −11.78 (AB system, JAB = 21.6 Hz, 2P). m/z (ES−) 662 [30%, (M − H)−]. λmax 275 nm (ε 4600, pH 8.3). HPLC Rt = 2.2 mins.
:1) as described. The pure dinucleotide was obtained in 12% yield. δH (D2O, 400 MHz) 3.81 (2H, m, POCH2CH2), 4.07 (2H, m, POCH2), 4.21-4.75 (5H, m, H-2′′, H-3′′, H-4′′ and H-5′′), 5.66 (2H, s, OCH2base), 6.11 (1H, d, J1′′,2′′ 5.5, H-1′′), 8.18-8.26 (3H, m, HN5, HA2 and HA8), 8.89 (1H, d, J4,5 6.9, HN4), 9.20 (1H, d, J6,5 6.2, HN6), 9.36 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −10.9 and −10.6 (AB system, JAB 18.5, 2P). λmax 259 nm (ε 17300, pH 8.3). HPLC Rt = 3.1 mins.
:1) as described. The pure dinucleotide was obtained in 10% yield. δH (D2O, 400 MHz) 4.57-4.13 (10H, m, ribose-H), 6.05 (1H, d, J1′,2′ 4.2, H-1′), 6.27 (1H, d, J1′′,2′′ 5.7, H-1′′), 8.13 (1H, s, HA2), 8.19 (1H, m, HN5), 8.34 (s1H,, HA8), 8.83 (1H, d, J4,5 8.1, HN4), 9.17 (1H, d, J6,5 5.7, HN6), 9.31 (1H, s, HN2). δP (D2O, 162 MHz, 1H-decoupled) −11.44 and −11.82 (AB system, JAB 20.8, 2P). m/z (FAB−) 663 [100%, (M − H)−]. λmax 259 nm (ε 17300, pH 8.3). HPLC Rt = 2.32 mins.
:1) as described. The pure dinucleotide was obtained in 10% yield. δH (D2O, 400 MHz) 1.31(3H, s, CH3), 1.52 (3H, s, CH3), 4.53-4.03 (8H, m, ribose-H), 5.04 (1H, dd, J3′,2′ 5.8, J3′,4′ 2.6, H-3′), 5.32 (1H, dd, J2′,3′ 5.8, J2′,1′ 2.8, H-2′), 6.04 (1H, d, J1′′,2′′ 5.8, H-1′′), 6.12 (1H, d, J1′,2′ 2.8, H-1′), 8.07 (1H, s, HA2), 8.15 (1H, dd, J5,4 7.9, J5,6 5.8, HN5), 8.20 (1H, s, HA8), 8.83 (1H, d, J4,5 7.9, HN4), 9.16 (1H, d, J6,5 5.8, HN6). 9.38 (1H, s, HN2), δP (D2O, 162 MHz, 1H-decoupled) −11.8 (m, 2P). m/z (FAB−) 580 [M+ − nicotinamide]. λmax 259 nm (ε 17300, pH 8.3). HPLC Rt = 4.18 mins.
Synthesis of analogues of cyclic-adenosine diphosphate ribose
Acknowledgements
We thank the Wellcome Trust for Project Grant no. 084068 [to BVLP and AHG] and MRC for a studentship (VCB). Work in the Guse lab has been supported by the Deutsche Forschungsgemeinschaft, the Gemeinnützige Hertie-Stiftung and the Deutsche Akademische Austauschdienst. We thank Dr M Thomas for providing Fig. 6.References
- D. L. Clapper, T. F. Walseth, P. J. Dargie and H. C. Lee, J. Biol. Chem., 1987, 262, 9561–9568 CAS
.
- H. C. Lee, T. F. Walseth, G. T. Bratt, R. N. Hayes and D. L. Clapper, J. Biol. Chem., 1989, 264, 1608–1615 CAS
- H. C. Lee, R. Aarhus and D. Levitt, Nat. Struct. Biol., 1994, 1, 143–144 CAS
- A. H. Guse, Curr. Mol. Med., 2004, 4, 239–248 Search PubMed
- A. Galione and G. C. Churchill, Sci STKE, 2000, 2000, pe1 Search PubMed
- A. H. Guse, J. Mol. Med., 2000, 78, 26–35 CrossRef CAS
- H. C. Lee, Physiol. Rev., 1997, 77, 1133–1164 CAS
- F. J. Zhang, Q. M. Gu and C. J. Sih, Bioorg. Med. Chem., 1999, 7, 653–664 CrossRef CAS
- B. V. L. Potter and T. F. Walseth, Curr. Mol. Med., 2004, 4, 303–311 Search PubMed
- S. Shuto and A. Matsuda, Curr. Med. Chem., 2004, 11, 827–845 CrossRef CAS
- A. H. Guse, FEBS J., 2005, 272, 4590–4597 CrossRef CAS
- A. H. Guse, Curr. Med. Chem., 2004, 11, 847–855 CrossRef CAS
- T. F. Walseth and H. C. Lee, Biochim. Biophys. Acta, 1993, 1178, 235–242 CrossRef CAS
- G. A. Ashamu, A. Galione and B. V. L. Potter, Chem. Commun., 1995, 1359–1360 RSC
- S. Shuto, M. Fukuoka, T. Kudoh, C. Garnham, A. Galione, B. V. L. Potter and A. Matsuda, J. Med. Chem., 2003, 46, 4741–4749 CrossRef CAS
- C. Moreau, G. K. Wagner, K. Weber, A. H. Guse and B. V. L. Potter, J. Med. Chem., 2006, 49, 5162–5176 CrossRef CAS
- G. K. Wagner, S. Black, A. H. Guse and B. V. L. Potter, Chem. Commun., 2003, 1944–1945 RSC
- G. K. Wagner, A. H. Guse and B. V. L. Potter, J. Org. Chem., 2005, 70, 4810–4819 CrossRef CAS
- Q. Liu, I. A. Kriksunov, C. Moreau, R. Graeff, B. V. L. Potter, H. C. Lee and Q. Hao, J. Biol. Chem., 2007, 282, 24825–24832 CrossRef CAS
- L. Wong, R. Aarhus, H. C. Lee and T. F. Walseth, Biochim. Biophys. Acta., 1999, 1472, 555–564 CrossRef CAS
- L. J. Huang, Y. Y. Zhao, L. Yuan, J. M. Min and L. H. Zhang, Bioorg. Med. Chem. Lett., 2002, 12, 887–889 CrossRef CAS
- F. J. Zhang, S. Yamada, Q. M. Gu and C. J. Sih, Bioorg. Med. Chem. Lett., 1996, 6, 1203–1208 CrossRef
- G. A. Ashamu, J. K. Sethi, A. Galione and B. V. L. Potter, Biochemistry, 1997, 36, 9509–9517 CrossRef CAS
- S. M. Graham, D. J. Macaya, R. N. Sengupta and K. B. Turner, Org. Lett., 2004, 6, 233–236 CrossRef CAS
- B. Zhang, G. K. Wagner, K. Weber, C. Garnham, A. J. Morgan, A. Galione, A. H. Guse and B. V. L. Potter, J. Med. Chem., 2008, 51, 1623–1636 CrossRef CAS
- M. Yoshikawa, T. Kato and T. Takenishi, Bull. Chem. Soc. Japan, 1969, 42, 3505–3508 CAS
- N. A. Hughes, G. W. Kenner and A. Todd, J. Chem. Soc., 1957, 3733–3736 RSC
- M. Senkus, J. Am. Chem. Soc., 1946, 68, 734–736 CrossRef CAS
- M. J. Robins and P. W. Hatfield, Can. J. Chem., 1982, 60, 547–553 CAS
- R. E. Holmes and R. K. Robins, J. Am. Chem. Soc., 1965, 87, 1772–1776 CrossRef CAS
- J. O. Folayan and D. W. Hutchinson, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1977, 474, 329–333 Search PubMed
- V. C. Bailey, J. K. Sethi, S. M. Fortt, A. Galione and B. V. L. Potter, Chem. Biol., 1997, 4, 51–61 CrossRef CAS
- P. J. Dargie, M. C. Agre and H. C. Lee, Cell Regul., 1990, 1, 279–290 Search PubMed
- A. H. Guse, C. P. daSilva, F. Emmrich, G. A. Ashamu, B. V. L. Potter and G. W. Mayr, J. Immunology, 1995, 155, 3353–3359 CAS
- A. H. Guse, E. Roth and F. Emmrich, Biochem. J., 1993, 291(Pt 2), 447–451 CAS
- R. Aarhus, R. M. Graeff, D. M. Dickey, T. F. Walseth and H. C. Lee, J. Biol. Chem., 1995, 270, 30327–30333 CrossRef CAS
- A. H. Guse, C. P. da Silva, I. Berg, A. L. Skapenko, K. Weber, P. Heyer, M. Hohenegger, G. A. Ashamu, H. Schulze-Koops, B. V. L. Potter and G. W. Mayr, Nature, 1999, 398, 70–73 CrossRef CAS
- T. Kudoh, M. Fukuoka, S. Ichikawa, T. Murayama, Y. Ogawa, M. Hashii, H. Higashida, S. Kunerth, K. Weber, A. H. Guse, B. V. L. Potter, A. Matsuda and S. Shuto, J. Am. Chem. Soc., 2005, 127, 8846–8855 CrossRef CAS
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333–2344 CrossRef CAS
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 94, 8205–8212 CrossRef CAS
- R. Stolarski, L. Dudycz and D. Shugar, Eur. J. Biochem., 1980, 108, 111–121 CAS
- H. Rosemeyer, G. Toth, B. Golankiewicz, Z. Kazimierczuk, W. Bourgeois, U. Kretschmer, H. P. Muth and F. Seela, J. Org. Chem., 1990, 55, 5784–5790 CrossRef CAS
- F. Jordan and H. Niv, Biochim. Biophys. Acta, 1977, 476, 265–271 CrossRef CAS
- F. E. Evans and N. O. Kaplan, J. Biol. Chem., 1976, 251, 6791–6797 CAS
- V. C. Bailey, J. K. Sethi, A. Galione and B. V. L. Potter, Chem. Commun., 1997, 695–696 RSC
- B. Zhang, V. C. Bailey and B. V. L. Potter, J. Org. Chem., 2007, 73, 1693–1703
- A. H. Guse, C. P. daSilva, K. Weber, C. N. Armah, G. A. Ashamu, C. Schulze, B. V. L. Potter, G. W. Mayr and H. Hilz, Eur. J. Biochem., 1997, 245, 411–417 CrossRef CAS
- J. K. Sethi, R. M. Empson, V. C. Bailey, B. V. L. Potter and A. Galione, J. Biol. Chem., 1997, 272, 16358–16363 CrossRef CAS
-
V. C. Bailey, PhD Thesis, University of Bath, 1997
-
G. A. Ashamu, PhD Thesis, University of Bath, 1997
- S. Shuto, M. Fukuoka, A. Manikowsky, Y. Ueno, T. Nakano, R. Kuroda, H. Kuroda and A. Matsuda, J. Am. Chem. Soc., 2001, 123, 8750–8759 CrossRef CAS
- T. Kudoh, T. Murayama, A. Matsuda and S. Shuto, Bioorg. Med. Chem., 2007, 15, 3032–3040 CrossRef CAS
- E. Nandanan, S. Y. Jang, S. Moro, H. O. Kim, M. A. Siddiqui, P. Russ, V. E. Marquez, R. Busson, P. Herdewijn, T. K. Harden, J. L. Boyer and K. A. Jacobson, J. Med. Chem., 2000, 43, 829–842 CrossRef CAS
- P. A. Evans, K. W. Lai, H. R. Zhang and J. C. Huffman, Chem. Commun., 2006, 844–846 RSC
- S. K. Singh, R. Kumar and J. Wengel, J. Org. Chem., 1998, 63, 10035–10039 CrossRef CAS
- P. Nielsen and J. Wengel, Chem. Commun., 1998, 2645–2646 RSC
- M. R. Hellmich and F. Strumwasser, Cell Regul., 1991, 2, 193–202 Search PubMed
- I. L. Cartwright, D. W. Hutchinson and V. W. Armstrong, Nucleic Acids Res., 1976, 3, 2331–2339 CAS
Footnote |
| † This paper is part of an Organic & Biomolecular Chemistry web theme issue on chemical biology. |
| This journal is © The Royal Society of Chemistry 2011 |