Synthesis and self-assembly of amphiphilic semi-brush and dual brush block copolymers in solution and on surfaces†‡
Daniel
Zehm
a,
André
Laschewsky
*a,
Peggy
Heunemann
bc,
Michael
Gradzielski
*b,
Sylvain
Prévost
bd,
Hua
Liang
e,
Jürgen P.
Rabe
e and
Jean-François
Lutz
f
aUniversität Potsdam, Institut für Chemie, Karl-Liebknecht-Str. 24-25, 14476, Potsdam-Golm, Germany. E-mail: laschews@rz.uni-potsdam.de; Fax: +49 331 9775036; Tel: +49 331 9775225
bTechnische Universität Berlin, Stranski-Laboratorium für Physikalische und Theoretische Chemie, Institut für Chemie, Straße des 17. Juni 124, Sekr. TC7, 10623, Berlin, Germany. E-mail: michael.gradzielski@tu-berlin.de; Fax: +49 30 314 26602; Tel: +49 30 314 24934
cInstitut Max von Laue-Paul Langevin (ILL), F-38042, Grenoble Cedex 9, France
dHelmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109, Berlin, Germany. E-mail: prevost.sylvain@gmail.com; Fax: +49 30 80623059; Tel: +49 30 80622339
eHumboldt-Universität zu Berlin, Department of Physics, Newtonstraße 15, 12489, Berlin, Germany. E-mail: rabe@physik.hu-berlin.de; Fax: +49 30 20937632; Tel: +49 30 20937788
fFraunhofer Institute for Applied Polymer Research, Geiselbergstrasse 69, D-14476, Potsdam-Golm, Germany. E-mail: lutz@iap.fraunhofer.de; Fax: +49 331 5683000; Tel: +49 331 5681127
First published on 25th September 2010
Abstract
The combination of two techniques of controlled free radical polymerization, namely the reversible addition fragmentation chain transfer (RAFT) and the atom transfer radical polymerization (ATRP) techniques, together with the use of a macromonomer allowed the synthesis of symmetrical triblock copolymers, designed as amphiphilic dual brushes. One type of brush was made of poly(n-butyl acrylate) as soft hydrophobic block, i.e. characterized by a low glass transition temperature, while the other one was made of hydrophilic poly(ethylene glycol) (PEG). The new triblock polymers represent “giant surfactants” according to their molecular architecture. The hydrophobic and hydrophilic blocks microphase separate in the bulk. In aqueous solution, they aggregate into globular micellar aggregates, their size being determined by the length of the stretched polymer molecules. As determined by the combination of various scattering techniques for the dual brush copolymer, a rather compact structure is formed, which is dominated by the large hydrophobic poly(n-butyl acrylate) block. The aggregation number for the dual brush is about 10 times larger than for the “semi-brush” precursor copolymer, due to the packing requirements for the much bulkier hydrophobic core. On mica surfaces the triblock copolymers adsorb with worm-like backbones and stretched out side chains.
Introduction
Graft polymers characterized by a high density of grafted chains were named “molecular polymer brushes”. They have experimental and theoretical interest due to their extended worm-like conformations in solution as well as on surfaces.1–4Polymer brushes have been considered as supersoft elastomers,5,6 as molecular actuators,7 or as synthetic substitutes for proteoglycans.8 Combined with the concept of amphiphilic block copolymers (often referred to as macrosurfactants),9–14 the resulting amphiphilic polymer brushes have been recently introduced as a new class of polymeric surfactants.15–19 Due to their increased pre-organisational level by virtue of the complex macromolecular structure and to their sheer size, amphiphilic polymer brushes may be expected to exhibit unique aggregation behaviour in bulk, in selective solvents as well as at surfaces, which extends the already well-established concepts of aggregation of amphiphilic block copolymers in aqueous solutions to a larger and more complex dimension.9,20Within the group of amphiphilic polymer brushes, a new subclass are “dual brush” block copolymers, which are characterised by densely grafted hydrophilic and hydrophobic linear side chains attached to dissimilar units of the polymer backbone (Scheme 1). The ability of such “giant surfactants” to self-organise in aqueous solution thereby forming giant micelles and at solid surfaces has been demonstrated recently.17 As their synthesis is complex, amphiphilic dual brushes have been virtually unknown up to now.15–19
 | ||
Scheme 1 Comparison of the architecture of a standard surfactant, a macrosurfactant, and a giant surfactant (from top to bottom). The left ( ) and the right side (○) represent the hydrophobic and hydrophilic elements, respectively. ) and the right side (○) represent the hydrophobic and hydrophilic elements, respectively. | ||
A particularity of the design of such amphiphilic dual brushes is their pronounced response to a selective solvent that makes the solvophilic block swell, while the solvophobic block collapses. Accordingly, a dual brush made of two blocks of equal graft lengths will dissolve in selective solvents for either of the blocks, however, the volume of the swollen blocks will be much larger than the volume of the collapsed blocks,17 implying the formation of very small aggregates.9,21 If, however, a more balanced situation between the volumes occupied by the swollen and the collapsed blocks is aspired, the grafts of the solvophilic block must be much shorter than the grafts of the solvophobic one. This implies, for the use of dual brush polymers in water, that the hydrophilic grafts should be preferentially shorter than the hydrophobic ones (cf. ESI†, Scheme S1).
Recently, we reported on amphiphilic dual brush diblock as well as triblock copolymers,17 made accessible by superposing two methods of controlled free radical polymerisation,22–24 namely by the RAFT (Reversible Addition Fragmentation chain Transfer) and by the NMP (Nitroxyl Mediated Polymerisation) methods. These giant surfactants contained polystyrene brushes as a hydrophobic block and poly(ethylene glycol) (PEG) brushes as a hydrophilic block. When dispersed in water, they assemble into large, well-defined spherical micelles, which were so stable that they could be deposited intactly onto solid surfaces, exhibiting certain similarities to latex particles. Characteristically in these first examples, the hydrophobic block consisted of a polymer brush with a high glass transition temperature (Tg (polystyrene) ≈ 105 °C) such that the hydrophobic domains were glassy at ambient conditions. In fact, amphiphilic polymers with a high Tg micellar core form typically so-called “frozen micelles” when associating in water, which cannot rearrange once formed. Therefore, the structures observed may be far from equilibrium.25–27
To enlarge the scope of the possible self-assembled structures, we present the synthesis and aggregation behaviour of new examples of amphiphilic dual brush triblock copolymers, which contain now a soft hydrophobic block, i.e., a hydrophobic polymer brush with a low glass transition temperature (Fig. 1). This modified design offers that the aggregates formed may be closer to equilibrium and that the aggregates might (at least partially) reorganize in changing environments. Again, the new polymers were prepared by combining two methods of controlled free radical polymerisation, but now combining the RAFT and the ATRP (atom transfer radical polymerisation) techniques. While making use of a PEG-based macromonomer to prepare the hydrophilic blocks, the hydrophobic brush block was grown by the ATRP method. This required the use and incorporation of a different type of hydrophobic inimer than previously,17 now carrying an activated halogen atom in the side chain (Fig. 1).
 | ||
| Fig. 1 Chain transfer agents, monomers and polymers studied. | ||
The change from NMP to ATRP for the grafting step was motivated by the formation of a certain amount of polystyrene homopolymer in our previous strategy,17 due to the self-initiation of styrene at high temperature. The homopolymer could not be fully removed, so that it might affect the self-assembly behaviour of the dual brush block copolymers. We reasoned that the use of ATRP might reduce this problem, in particular when the polymerizations are run at temperatures lower than 130 °C as applied previously. Also, ATRP gives access to the polymerization of other monomer groups than useful in NMP.22,23 Still, the successive combination of RAFT and ATRP polymerizations without the use of protective group chemistry has been hardly explored so far.28 Hence, it was not clear whether a large number of ATRP initiating sites would be compatible with a successful controlled RAFT polymerisation, as needed to construct the skeleton of the block copolymers (explaining our initial preference for the combination of RAFT and NMP).
Experimental
Materials
n-Butyl acrylate (BuA, Aldrich, 99%) was distilled in vacuo. Poly(ethylene glycol monomethyl ether acrylate) (PEGA, Aldrich [32171-39-4], number average degree of ethoxylation 9–10 according to 1H NMR) was passed through a column filled with basic Al2O3 (Merck, activity I, 0.0630–0.200 mm) to remove the inhibitor prior to polymerisation. 2,2′-Azobis(isobutyronitrile) (AIBN, Acros-Organics, 98%) was crystallised from methanol and dried in vacuo. Other solvents used for polymerisation and purification were analytical grade and used as received. Zellu-Trans dialysis tubes (nominal molar mass cutoff of 4000–6000 D) were from Roth. Bisbenzyl trithiocarbonate (CTA1) and 2,6-bis-butylsulfanylthiocarbonyl-sulfanyl-heptanedioic acid dimethyl ester (CTA2) were synthesised as reported before.29 The monomer 2-chloropropionyloxyethyl acrylate (ClPEA) was synthesised following literature procedures.30 The synthesis of inimer 2-(2′-bromo-2-methyl propionyloxy)ethylacrylamide (BrMPAEA) is given in the ESI†.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CTA]:[AIBN] as specified in Table 1.
:[CTA]:[AIBN] as specified in Table 1.
| Entry | Polymer | RAFT agent | Reactant ratios [M]:[CTA]:[AIBN] |
Time/h | Conv.a (%) | Molar mass | |||
|---|---|---|---|---|---|---|---|---|---|
| Theoryb | SEC c | PDI | Spectroscopic analysis Mn × 10−3/g mol−1 | ||||||
| M n,theo × 10−3/g mol−1 | M n,app × 10−3/g mol−1 | ||||||||
|
a Determined by 1H NMR analysis of the crude product.
b Calculated from conversion × [M]/[CTA].
c
Eluent
THF, RI detection, apparent values, calibrated against polystyrene standards.
d Calculated by end group analysis via UV band (λ = 309 nm, εCTA1 = 30930 L mol−1 cm−1 in CH2Cl2).
e Calculated by end group analysis via UV band (λ = 309 nm, εCTA1 = 16900 L mol−1 cm−1 in CH2Cl2).
f Calculated from the compositional data according to 1H NMR, assuming that Mn,theo of the 1st block is preserved in the block copolymer.
|
|||||||||
| 1 | (ClPEA)177 | CTA1 | 200:1:0.1 |
8 | 75 | 31 | 21 | 1.34 | 36d |
| 2 | (ClPEA)680 | CTA1 | 1000:1:0.1 |
48 | 68 | 140 | 57 | 2.13 | 160d |
| 3 | (ClPEA)114 | CTA2 | 200:1:0.1 |
7 | 53 | 22 | 13 | 1.36 | 24e |
| 4 | (PEGA)384 | CTA2 | 500:1:0.1 |
21 | 77 | 175 | 16 | 1.26 | 220e |
| 5 | (PEGA)858 | CTA2 | 1000:1:0.1 |
50 | 90 | 408 | 26 | 1.26 | 590e |
| 6 | (PEGA)192-b-(ClPEA)72-b-(PEGA)192 | (PEGA)384 | 300:1:0.1 |
7 | 38 | 190 | 16 | 1.30 | 189f |
| 7 | (PEGA)429-b-(ClPEA)470-b-(PEGA)429 | (PEGA)858 | 1000:1:0.1 |
88 | 44.5 | 500 | 33 | 1.68 | 505f |
| 8 | (PEGA)375-b-(ClPEA)680-b-(PEGA)375 | (ClPEA)680 | 1000:1:0.1 |
24 | 77 | 489 | 12 | 2.55 | 480f |
:[CTA]:[AIBN] and replacing CTA2 by CTA1, as specified in Table 1.
000), monomer ClPEA (0.177 g, 8.5 × 10−4 mol) and AIBN (0.046 mg, 2.85 × 10−7 mol) in dry ethyl acetate (2.5 mL) was degassed by three freeze–pump–thaw cycles, and placed in an oil bath at 65 °C. The reaction was stopped after 7 h by cooling. The solution was dialyzed first against ethanol and subsequently against deionised water. The aqueous polymer solution was lyophilised to give block copolymer (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 (0.51 g). 1H NMR (300 MHz, CDCl3): δH 1.60 (br s, -CH2- backbone), 1.68 (d, J = 6.91 Hz, ClCCH3), 1.86 (br s, -CH2- backbone), 2.30 (br s, -CH- backbone), 3.36 (s, CH3O-), 3.63 (br s, -CH2-O-CH2-), 4.15 (br s, -COOCH2-), 4.29 (br s, -CH2CH2-O), 4.36 (br s, -CH2CH2-O), 4.45 (q, J = 6.91 Hz, ClCHCOO-). Copolymer (PEGA)429429-bb-(ClPEA)470470-bb-(PEGA)429429 was made analogously (Table 1).
In contrast, the synthesis of (PEGA)375375-bb-(ClPEA)680680-bb-(PEGA)375375 started from the alternative macro-RAFT agent: a mixture of (ClPEA)680680 (1 g, 7.1 × 10−7 mol), PEGA (3.2 g, 7.1 × 10−3 mol) and AIBN (0.1 mg, 7.1 × 10−7 mol) in dry ethyl acetate (21 mL) was degassed by three freeze–pump–thaw cycles, and placed in an oil bath at 65 °C. The reaction was stopped after 24 h by cooling. The solution was dialyzed against deionised water. The aqueous polymer solution was lyophilised to give 3.5 g of (PEGA)375375-bb-(ClPEA)680680-bb-(PEGA)375375.
000, corresponds to 7.6 × 10−5 mol initiating Cl groups) was dissolved in ethyl acetate (0.24 g, 10%) and CuCl (7.5 mg, 7.6 × 10−5 mol). BuA (1.95 g, 15.1 mmol) and toluene (2.4 g) were added. The reaction mixture was purged with argon for 30 min, PMDETA (13.1 mg, 7.6 × 10−5 mol) was added and the mixture was polymerized at 70 °C for 7 h. The reaction was quenched by cooling and precipitated into methanol/water (80 v/20 v). The polymer was isolated and dried in vacuo to yield the dual brush block copolymer TriB-1 (1 g). 1H NMR (300 MHz, CDCl3): δH 0.93 (t, J = 7.25 Hz, -CH3), 1.37 (m, -CH2CH3), 1.59 (m, -CH2-), 1.90 (br s, -CH2- backbone), 2.27 (br s, -CH- backbone), 3.37 (s, CH3O-), 3.64 (br s, -CH2-O-CH2-), 4.03 (br s, -CH2COO-), 4.16 (br s, -CH2COO-). Block copolymer brushes TriB-2, TriB-3 and TriB-4 were prepared analogously, as specified in Table 2.
| Polymer | Macroinitiator | Time/h | Conv.a (%) | Molar mass of brush copolymer | Cleaved BuA grafte | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Theoryb | SEC c | PDIf | NMR d | M n | PDI | f h [%] | |||||
| M n × 10−3/g mol−1 | M n,app f × 10−3/g mol−1 | M n × 10−3/g·mol−1 | M n g of PBuA grafts | ||||||||
| a Conversion determined gravimetrically. b Calculated according to eqn (1). c Eluent THF, RI detection, calibration by polystyrene standards. d Calculated by 1H NMR knowing the molar mass of the precursor. e After cleavage, measured by SEC. f Apparent values of brush copolymer fraction. g Number average molar mass of PBuA grafts assuming 100% grafting efficiency. h Grafting density, f = Mn(PBuA grafts)NMR/Mn(PBuA grafts)cleaved. | |||||||||||
| TriB-1 | (PEGA)192-b-(ClPEA)72-b-(PEGA)192 | 7 | 41 | 745 | 128 | 1.31 | 654 | 6528 | 13290 |
1.27 | 49 |
| TriB-2 | (PEGA)429-b-(ClPEA)470-b-(PEGA)429 | 7 | 55 | 7100 | 301 | 1.41 | 9200 | 18550 |
22600 |
1.34 | 82 |
| TriB-3 | (PEGA)429-b-(ClPEA)470-b-(PEGA)429 | 5 | 32 | 4300 | 308 | 1.42 | 5096 | 9770 | 19000 |
1.44 | 51 |
| TriB-4 | (PEGA)375-b-(ClPEA)680-b-(PEGA)375 | 4 | 37 | 6900 | 394 | 1.48 | 12700 |
18000 |
29500 |
1.30 | 61 |
Methods
SEC of the polymers was run at 25 °C in THF (flow rate: 1.0 mL min−1) using a TSP apparatus (Thermo Separation Products from Thermo-Finnigan GmbH, Dreieich, Germany) equipped with a Shodex RI-71 refractive index detector, a TSP UV detector (260 nm), and a set of PSS SDV columns (styrene/divinyl benzene, 1000 Å and 10000 Å porosity, and 5 µm particle size). Calibration was performed with polystyrene standards (PSS GmbH Mainz, Germany). 1H and 13C NMR spectra were taken with a Bruker Avance 300 apparatus. All spectra are referenced to the solvent residual peak (CHCl3 at 7.26 ppm). Monomer conversions were measured before work-up by comparing the intensity of the vinyl proton signals of the monomers PEGA or ClPEA with the intensity of characteristic signals of the polymers. For the dual brush polymers, monomer conversions were approximated via the polymer yields as determined gravimetrically and the theoretically expected molar mass values were estimated according to eqn (1):| Mn,theor = (conversion × Mr,monomer × [M]/[chlorine]) + Mr,precursor | (1) |
Thermal properties were measured with DSC 822 differential scanning calorimeter (Mettler Toledo) under a nitrogen atmosphere, heating rate 10 K min−1, and cooling rate 5 K min−1. Dynamic light scattering data (DLS) were accumulated at a scattering angle of θ = 173° (backscattering detection) with a high-performance particle sizer (HPPS-ET, Malvern Instruments, UK) equipped with a He–Ne laser (λ = 633 nm) and a thermoelectric Peltier element for temperature control. Autocorrelation functions were analyzed with the CONTIN method. Apparent hydrodynamic diameters Dh were calculated according to the Stokes–Einstein equation, Dh = kT/3πηDapp, with Dapp being the apparent diffusion coefficient and η being the viscosity of the solution. Prior to measurements, the polymer solutions were filtered into a quartz glass cuvette (Suprasil from Hellma, Germany) with an optical path length of 1.9 cm using a WICOM OPTI-Flow 0.45 µm disposable filter. Samples employed for the SANS measurements were characterized by angle dependent dynamic and static light scattering (DLS and SLS) at 25 °C using a setup consisting of an ALV 7004 Correlator, an ALV CGS-3 Goniometer and a He–Ne laser with a wavelength of 632.8 nm in the laboratory PSCM of ILL (Grenoble, France). Cylindrical sample cells were placed in an index matching bath filled with toluene. Autocorrelation functions as well as the mean intensity were recorded under different angles between 30° and 150°.
SANS experiments were done on the instrument D11 of the Institut Laue-Langevin (ILL, Grenoble, France), with scattered neutrons recorded on a 128 × 128 He3 detector of 96 × 96 cm2. A wavelength of 0.6 nm (FWHM 10%) and sample-to-detector distances of 1.2, 8 and 34 m were employed with collimation at 4, 8 and 34 m, respectively, thereby covering a q-range of 0.02–5.2 nm−1, where q is the magnitude of the scattering vector defined as:
 | (2) |
For Scanning Force Microscopy (SFM), a droplet of polymer solution in CHCl3 (0.01 g L−1) was deposited on a freshly cleaved mica surface and spun off after 5 s. The surface was dried under a flux of nitrogen gas and then imaged by SFM in tapping-mode under ambient conditions, employing a Nanoscope 3a (Veeco, USA), using silicon cantilevers (Olympus, Japan) with a typical resonance frequency of 300 kHz and a spring constant of about 42 N m−1. Both height and phase images were recorded.
Results and discussion
Three different strategies may be conceived to synthesise amphiphilic dual brush block copolymers (Scheme 2). The first is sequential polymerisation of hydrophilic and hydrophobic macromonomers.18,32 In this strategy, inherent problems arise from the poor compatibility of the hydrophilic and hydrophobic macromonomers, in particular in the case of high molar masses. In the second strategy, independently prepared hydrophobic and hydrophilic brushes are covalently coupled. This strategy requires highly efficient coupling reactions, as e.g. applied in “click” chemistry.33 So far, this approach has been limited to hydrophilic polymers,34 owing to the incompatibility of hydrophilic and hydrophobic brushes. The third strategy exploits sequential grafting-from polymerisations to create the dual brushes, by using a macroinitiator block copolymer with two different and independently addressable initiating sites. Examples for this strategy are very scarce35 and have not been applied to amphiphilic dual brushes yet. Noteworthy, our approach is based on the polymerisation of inimer 2-chloropropionyloxy-ethyl acrylate (ClPEA) to give directly the macroinitiator blocks, without the need for post-polymerisation modifications, as done before.36–38 | ||
| Scheme 2 Synthetic strategies to amphiphilic dual brush block copolymers. | ||
In this work, we synthesised the amphiphilic dual brush copolymers by combining strategies 1 and 3, i.e. by superposing the macromonomer and the grafting-from strategies. While RAFT polymerisation was applied to synthesise the macroinitiator block copolymer in two steps from a hydrophilic macromonomer and a hydrophobic inimer, ATRP was used to grow the hydrophobic brush from the inimer initiating sites (Scheme 3). The final polymers were made of poly(butyl acrylate) as a hydrophobic brush block and poly(ethylene glycol) (PEG) as a non-ionic hydrophilic brush block. Both polymer brushes represent soft blocks as their glass transition temperatures are well below 0 °C.
 | ||
| Scheme 3 Synthesis of amphiphilic poly(PEGA-bb-(ClPEA-gg-BuA)-bb-PEGA) dual brush block copolymer. | ||
Amphiphilic triblock copolymers that bear reactive C–Cl groups suited for initiating ATRP were made by the consecutive RAFT copolymerisation of the hydrophilic macromonomer PEGA and of inimer ClPEA. Depending on the RAFT agent employed, the R- (in the case of using CTA1) or the Z-groups (in the case of using CTA2) were placed in the center of the growing polymer chains, thus requiring polymerization of the inimer first in the former case or of the hydrophilic macromonomer first in the latter case. Subsequently, the pendant chlorine moieties served as initiating sites for the ATRP polymerisation of BuAvia a grafting-from approach, yielding the dual brush polymers finally.
Synthesis of the linear macrosurfactant precursor block copolymers
RAFT polymerisations were performed with CTA1 and CTA2, respectively, as bifunctional RAFT agents. Both are known to produce well-defined symmetrical ABA or BAB triblock copolymers.29 The polymerisation of ClPEA mediated by both CTA1 and CTA2 provided (ClPEA)177177 and (ClPEA)114114, respectively, with monomodal molar mass distributions (Table 1). The relatively narrow polydispersities of 1.3 indicate good control over the polymerisation process.As the molar masses measured by SEC analysis are at best only approximate because of the calibration by polystyrene standards, molar masses were determined by end-group analysis via UV-spectroscopic determination of the trithiocarbonate content, under the assumption that the polymer bears exactly two trithiocarbonate end groups. The good agreement of the obtained values with the theoretically predicted ones demonstrates the successful synthesis of well-defined polyClPEA macroinitiators. Note that under RAFT polymerisation condition, the pendant chlorine moiety is inert according to the thermogravimetric and 1H NMR analysis (see ESI†, Fig. S1). Thus every repeat unit of polyClPEA bears one pendant chloride site, i.e. functionalisation is quantitative. However, when increasing the ratio of monomer to CTA1, the obtained macroinitiator (ClPEA)680680 showed a broadened molar mass distribution (Table 1), pointing to a gradual loss of control over the polymerisation, as one reaches the limits of the controlled free radical polymerisation concept.22
Note that the finding that the halogen bearing inimer ClPEA undergoes a well controlled RAFT polymerization is not trivial. For instance, inimer 2-(2′-bromo-2-methyl propionyloxy)ethylacrylamide, BrMPAEA, which is also suited to initiate ATRP, can be successfully polymerized under RAFT conditions, too. Though, the control of the molar masses and the molar mass distribution is poor, a considerable amount of thiocarbonyl end groups is lost (see ESI†). This difference is attributed to the more labile C-halogen bond in BrMPAEA (tertiary C–Br bond) compared to ClPEA (secondary C–Cl bond) giving rise to more side reactions.
Analogously, the polymerisation of macromonomer PEGA, mediated by CTA2, provided the hydrophilic brush (PEGA)384384 with monomodal molar mass distributions (see Table 1). Again, the relatively narrow polydispersities of about 1.2 indicate good control over the polymerisation process. Even more pronounced than found for the polymers of ClPEA, the apparent molar mass deduced from SEC using calibration by polystyrene standards differs strongly from the value determined by end-group analysis via UV-spectroscopy (Table 1). The latter value, however, agrees well with the theoretically predicted one, indicating the successful synthesis of a defined polyPEGA macro-RAFT agent. The marked mismatch of the molar masses of the polyPEGA brushes when calibrating the SEC elugrams by linear polystyrene standards is not surprising, as elution speed in SEC depends on the hydrodynamic volume, which differs strongly for linear and branched polymers of identical molar mass. An increased ratio of monomer to CTA2 provided the larger hydrophilic brush (PEGA)858858, which was well defined, too (Table 1). Therefore, we applied these CTA2 based homopolymers for the synthesis of symmetrical ABA triblock copolymers.
Triblock copolymers with a reactive central block made of inimers were prepared by RAFT polymerisation of ClPEA, starting from the bifunctional macro-RAFT agents (PEGA)384384 or (PEGA)858858. The semi-brush triblock copolymer (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 dispersed in water forms aggregates, which are stable over several months. This observation implies that the hydrophobic ClPEA was successfully added onto the hydrophilic (PEGA)384384 or (PEGA)858858 macro-RAFT agents, indicating qualitatively the successful synthesis of the amphiphilic reactive triblock copolymer, as discussed below.
Table 1 lists the data of their characterisation by 1H NMR and SEC. The SEC elugrams of (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 in Fig. 2a exemplify the very small shift and broadening of the molar mass distributions seen after the block copolymerisation step, without the presence of a second, new peak. The macroscopic increase in mass after the chain extension step, however, implies that copolymer is formed. The weak effect of the incorporated central polyClPEA block on the SEC elugram may be explained by brush character of the outer long polyPEGA blocks that dominate the hydrodynamic behaviour in the eluent.
 | ||
| Fig. 2 (a) SEC traces of (i, continuous line) (PEGA)384384, (ii, dashed line) (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192, and (iii, continuous line) TriB-1 in THF as eluent. (b) SEC traces of (i, continuous line) TriB-1 and (ii, continuous line) of corresponding polyBuA graft chains after the cleavage in THF as eluent. | ||
Indeed, Fig. 3ii depicting exemplarily the 1H NMR spectrum of (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 in comparison with the spectrum of the macro-RAFT agent (PEGA)384384 (Fig. 3i) clearly shows the presence of both the hydrophilic polyPEGA block and the polyClPEA block in the reaction products. While SEC analysis alone is not conclusive, the combined SEC and 1H NMR data prove the successful chain extension yielding the amphiphilic triblock copolymers. Accordingly to their overall composition determined from the integrated 1H NMR spectra, the molar masses of the triblock copolymer were calculated, assuming that the molar mass values of the polyPEGA blocks remained unchanged in the copolymerisation steps. Again, the calculated values agreed well with the theoretical molar masses derived from the conversion and the amounts of monomer and RAFT agent used (Table 1).
 | ||
| Fig. 3 1H NMR spectra in CDCl3 of (i) (PEGA)384384, (ii) (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192, and (iii) TriB-1 (from bottom to top). | ||
Following the successful synthesis of (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192, the macro-RAFT agents (PEGA)858858 and (ClPEA)680680 were subsequently employed to prepare even larger amphiphilic triblock copolymer analogues. 1H NMR analysis shows the presence of both polyPEGA and polyClPEA blocks in the reaction product. However, products with much broader molar mass distributions were obtained with both macro-RAFT agents according to SEC (cf. ESI†, Fig. S2). The synthesis sequences starting with CTA1 as well as with CTA2 provided bimodal distributions, with a minor peak at the same elution time as the macro-RAFT agents, suggesting the presence of inactive, residual precursor polymer, and the major peak at considerably longer elution times, attributed to the newly formed triblock intermediates. The delayed elution of the block copolymers compared to their macro-RAFT agent precursors may be caused by attractive interactions with the column material or by attractive interactions of the blocks resulting in a compact conformation. In any case, the combined analytical data show the successful making of the large reactive triblocks (PEGA)375375-bb-(ClPEA)680680-bb-(PEGA)375375 and (PEGA)429429-bb-(ClPEA)470470-bb-(PEGA)429429.
Still, it is evident from the synthesis of the reactive block copolymers with high molar masses, that controlled free radical polymerisation methods, such as RAFT, become increasingly challenging and seem to approach their limits, as to be expected from theory.22 In comparison, RAFT polymerisation mediated by CTA2, where the active group of the growing polymer chain is placed at the outer ends of the polymer, seemed to be more effective in order to get defined block copolymers than the use of CTA1, as judged from the relative importance of the residual precursor peaks.
Synthesis of giant surfactants by “grafting-from” of BuA
The multiple pendant chlorine groups of the polyClPEA block were used to initiate the graft polymerisation of BuA onto the various precursor triblock copolymers in ethyl acetate/toluene (20 v/80 v) by ATRP at 70 °C. The monomer-to-initiator ratio was chosen as about 200 and the grafting was stopped at monomer conversions below 50% (cf.Table 2 for polymerisation conditions and analytical data from 1H NMR and SEC). Again, accurate estimations of molar mass distributions of the amphiphilic copolymer dual brushes were complicated by non-size exclusion effects in SEC analysis, as found for the precursor block copolymers.8,39,40 Nevertheless, for the synthesis of TriB-1, one peak at considerably shorter elution times was observed in the elugrams, corresponding to higher molar mass of the product and indicating successful grafting of BuA to the backbone (Fig. 2a). The polydispersity indices were in the range of 1.3, suggesting controlled grafting of BuA. The combined SEC and 1H NMR analyses (see Fig. 3) show the successful grafting to yield the novel amphiphilic dual brush (PEGA)192192-bb-(ClPEA-gg-BuA5151)7272-bb-(PEGA)192192 and (TriB-1) with 23 vol% PEGA.In addition to the successful controlled synthesis of TriB-1, both ATRP initiators (PEGA)429429-bb-(ClPEA)470470-bb-(PEGA)429429 and (PEGA)375375-bb-(ClPEA)680680-bb-(PEGA)375375 were engaged for the synthesis of the higher homologues TriB-2, TriB-3 and TriB-4 (Table 2). Owing to the reported intra- or even intermolecular radical coupling reaction for macroinitiator systems with multiple initiating sites,41 a catalyst-to-initiator molar ratio of 0.5 for TriB-3 and TriB-4 was chosen to minimize such side reactions, while keeping in mind that a reduced amount of copper catalyst increases the grafting efficiency.40,42,43 Despite the bimodal polymer distribution of the large ATRP macroinitiators, one major peak at considerably shorter elution times, that correspond to products with increased molar mass, was observed in the elugrams after purification, pointing to successful grafting of BuA to the backbone (cf. ESI†, Fig. S2). PDI values in the range of 1.4–1.5 for the major peak indicate satisfactory control over the ATRP process. Furthermore, the combined SEC and 1H NMR analytical data unambiguously demonstrate the successful grafting to yield the amphiphilic dual brushes TriB-2, TriB-3, and TriB-4 with 6, 10 and 5 vol% PEGA, respectively.
Analysis of the grafted side chains
To analyse the grafted side chains of polyBuA in the dual brush copolymers, the side chains were cleaved from the backbone, using acid-catalyzed transesterfication in n-butanol. This reaction medium was selected to ensure the preservation of the butyl ester groups during the cleavage. SEC analysis of the cleaved side chains reveals monomodal molar mass distributions with polydispersities in the range of 1.3–1.45, being in the range of the theoretical value for slow initiation.40,43,44 As shown in Fig. 2b, the peak of the TriB-1 disappeared during cleavage reaction. As the weight fraction of the backbone polymers is very small in comparison to the amount of side chains, the effect of the backbone can be neglected. The apparent grafting efficiencies, as calculated from the ratio of the theoretically calculated molar masses of the grafts to the experimentally found ones, are in the range of 50–80% (see Table 2). They are in the range that can be expected for polyBuA brushes.42Self-organisation in aqueous solution
Differential scanning calorimetry (DSC) revealed that all the dual brush block copolymers exhibit an intense melting peak Tm at about −10 °C as well as a glass transition Tg at about −65 °C, which is characteristic for the polyPEGA blocks. Additionally, a second glass transition at about −40 °C is found, which is attributed to the polyBuA grafts (cf. ESI†, Fig. S3). The intense glass transition signal of the polyBuA grafts in TriB-2, TriB-3, and TriB-4 is due to the substantial weight fraction of BuA in these dual brush polymers. The occurrence of two separate glass transitions in TriB-1, TriB-2, TriB-3 and TriB-4 well below 0 °C indicates, on the one hand, that the copolymers are made of soft blocks. On the other hand, the two glass transitions indicate that the hydrophilic polyPEGA brushes and the hydrophobic polyBuA brushes are incompatible and micro-phase separate in bulk. In addition to their amphiphilic nature, this should favour the tendency of the dual brush copolymers to self-organise in selective solvents, too, for instance in aqueous solutions. In fact, a first qualitative test by 1H NMR in D2O solution reveals missing signals of the hydrophobic polyBuA chains, thus pointing to the aggregation of the hydrophobic blocks (cf. ESI†, Fig. S4). The aggregation behaviour in aqueous solution was studied in more detail by DLS and complementary SANS and SLS measurements. The SANS measurements in D2O (protocol A) and D2O/DMF mixtures (protocol B) are shown in Fig. 4 (for SLS data, see ESI†, Fig. S5). Intensity and shape of the curves show immediately that the molar mass of the aggregates formed by the amphiphilic precursor polymer (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 is much less than that of the ones formed by TriB-1, i.e. after grafting the hydrophobic polyBuA chains.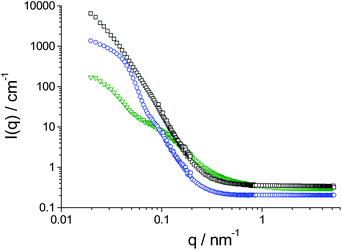 | ||
| Fig. 4
SANS intensity as function of the magnitude of the scattering vector, q, at 25 °C: (▽) 1.0 wt% (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 in D2O; (□) 0.9 wt% TriB-1 in D2O/DMF (10:1); and (○) 0.1 wt% TriB-1 in D2O. | ||
From a model-independent analysis by the Guinier approximation (eqn (S1)†) we derived radius of gyration, Rg, intensity extrapolated to zero scattering angle, I(0),45 and from that the molecular weight Mw of the aggregates by eqn (S2)†. The values obtained from this way are summarized in Table 3. A similar analysis was done for the SLS data (Fig. S5†) but it should be noted that in general the light scattering data have a tendency for yielding somewhat larger values, as here one simply probes a larger length scale than in SANS.
| Polymer | Precursor | TriB-1 | |
|---|---|---|---|
| PEGA (vol%) | 92 | 23 | |
| Solvent | D2O | D2O | D2O/DMF 10/1 |
| Concentration (wt%) | 0.996 | 0.100 | 0.904 |
| R g (SANS)/nm | 60.2 | 48.9 | 73.1 |
| I(0) (SANS)/cm−1 | 280 | 1800 | 11000 |
| M w (SANS)/g mol−1 | 7.4 × 106 | 3.4 × 109 | 2.3 × 109 |
| N agg | 31 | 460 | 390 |
| R comp (I(0))/nm | 13.7 | 50.6 | 44.4 |
| R g (SLS)/nm | 76.1 | 60.9 | 226.3 |
| M w (SLS)/g mol−1 | 3.4 × 106 | 2.5 × 109 | 4.8 × 109 |
| N agg | 75 | 620 | 810 |
| R h (DLS)/nm | 106.6 | 91.2 | 205.7 |
In the case of (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192SANS yields an aggregation number of 31 and a corresponding radius of a compact sphere aggregate of 13.7 nm. In contrast the size deduced from the radius of gyration is 60 nm, thereby making it clear that no compact micellar aggregates can be present, but much more extended structures which would be compatible with a vesicular structure. This picture is in agreement with the light scattering data that would yield a compact radius of 18.4 nm, while the hydrodynamic radius Rh (that probes the real extension of the aggregates) is 107 nm. Furthermore the oscillatory scattering pattern of (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 aggregates in D2O is indicative of a core–shell structure. From the minimum observed at 0.055 nm−1 one can conclude that these shells have a mean radius of 57 nm, in very good agreement with the picture of a vesicle structure.
In the case of the dual brush TriB-1, the analysis of the scattering data is in agreement with a micellar structure consisting of a hydrophobic core made from the polyBuA brush middle block and of a hydrophilic corona made from the polyPEGA brushes. The picture that arises from these data differs notably for the precursor macrosurfactant (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 and the derived giant surfactant TriB-1. These sizes must be discussed keeping in mind that, according to synthetic procedure and experimental characterisation, the maximum length of the stretched molecules is 114 nm.
For the dual-brush TriB-1, all three methods yield a rather consistent picture with radii of gyration deduced from SANS and SLS being 49 nm and 61 nm, respectively, a hydrodynamic radius of 91 nm, and a compact radius deduced from the intensity extrapolated to zero scattering angle, I(0), of 51 nm. The ratio Rg/Rh of 0.67 is significantly lower than the value of 0.778 expected for hard spheres and closer to values typically observed for microgels.45,46 Accordingly the dual-brush polymer forms spherical aggregates with an aggregation number of about 460 and with a maximum extension of 91 nm. This is in reasonable agreement with the size expected from its contour length. Assuming a compact polyBuA core that would be of 45.3 nm radius, we can calculate an area of 28.0 nm2 per hydrophilic brush at the hydrophobic/hydrophilic interface which is in rather good agreement with the 48 nm2 one can calculate as an upper limit based on the fully stretched PEG brush. The experimental value is somewhat larger than the 18–20 nm2 observed before for similar giant surfactants having the same hydrophilic brush but a polystyrene hydrophobic brush. Note that these aggregates are long-time stable as during several months we observed no changes in aggregates size by DLS or SANS.
However, these aggregates depend notably on the way of preparation and of solvent present. When prepared by protocol B, TriB-1 gives much larger particles, for which one mainly observes the q−4 behaviour of large particles by SANS. Actually, the aggregates present are too large to be well-observed within the q-window of the SANS experiment. Therefore the values in Table 3 derived from SANS are lower limits for the true size values. In contrast, SLS and DLS give a picture of rather large particles with radii of about 200–220 nm. This implies that we do not observe simple micellar aggregates, since the radii are much too large for spheres built with molecules of 114 nm contour length. Instead, multiply aggregated particles must be present here and this is also indicated by strong angular dependence of the SLS curves (cf. ESI†, Fig. S5c).
In the case of TriB-1, the samples made in D2O obviously form rather compact micellar aggregates with a polyBuA core and a dense corona of the PEGA brush. It is interesting to note that the aggregates formed by the precursor (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192 possess similar extensions by Rg and Rh (Table 3). This is not surprising as the maximum size of aggregates formed should be determined by the maximum length of the stretched molecules, which is the same for both polymers. The difference between the two types of triblock copolymers is that a bulky hydrophobic core is present for the dual-brush TriB-1, with only a relatively small hydrophilic PEGA brush, while the opposite holds for the precursor semi-brush (PEGA)192192-bb-(ClPEA)7272-bb-(PEGA)192192. Accordingly, a large hydrophobic core is formed in the case of dual brush TriB-1, thereby making Rg smaller than for the precursor polymer, where the scattering arises from a vesicular structure.
In summary, the combined scattering experiments confirm that globular micellar aggregates are formed. These are rather well-defined for the case of protocol A, whereas much larger and lesser defined aggregates are formed by protocol B. The aggregation number of the dual brush copolymers is much larger than the one of the precursor copolymer, due to the much larger hydrophobic block to be covered by the same stabilising hydrophilic block.
Single-molecule visualisation by SFM
Single molecules of the semi-brush precursor (PEGA)375375-bb-(ClPEA)680680-bb-(PEGA)375375 and the corresponding dual brush TriB-4 were deposited from THF and CHCl3 on mica, in order to investigate their shape and size distributions at surfaces. Whereas deposition from THF resulted only in spherical objects, deposition from CHCl3 led to stretched adsorbed polymers. While the flexible semi-brush precursor (PEGA)375375-bb-(ClPEA)680680-bb-(PEGA)375375 exhibits small snake-like conformations (cf. ESI†, Fig. S6), the morphology of the corresponding dual brush differs significantly.Fig. 5a presents SFM images of the long amphiphilic dual brush TriB-4 exhibiting worm-like structures. The distribution of the contour lengths of single worm-like structures exhibits a maximum around 100 nm with a number-averaged length Ln = 107 nm (Fig. 5b). Considering the number average degree of polymerization of the main chain of 1430 (Table 2), the value of Ln is only a third of the contour length of a fully extended chain Lc, with Lc = 1430 × 0.25 nm = 357 nm. This large difference is difficult to explain by the limited grafting density of about 60% of the side chains (see ESI†, Table S2).43,47
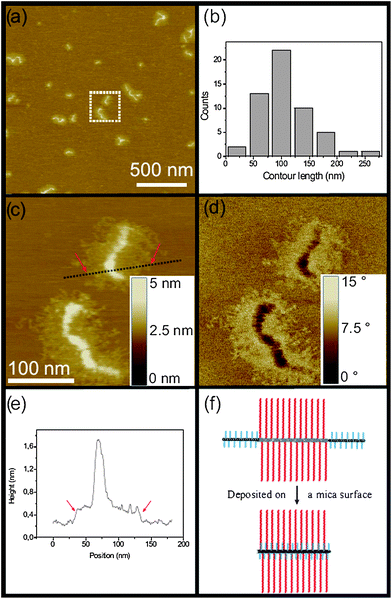 | ||
| Fig. 5 SFM images of dual brush TriB-4 deposited on mica: (a) height image; (b) histogram of measured contour lengths; (c) height image and (d) phase image of zoom in area marked in (a). (e) Cross-section, indicated as dotted line in the height image (c), showing the height difference between the substrate and the graft chains and the backbone, respectively. (f) Backfolding model suggesting that the end blocks fold back on the top of the middle block (top view). | ||
Fig. 5c and d reveal the structure of single polymer brushes of TriB-4 at higher resolution. A cross-sectional analysis shows that the height of the spine of TriB-4 is about 1.6 nm, while the width is approximately 100 nm. The experimentally observed half width of the brushes on the surface of 50 nm compares well to the length of the fully stretched grafted chains of the hydrophobic brushes, LSC. With a molar mass of the cleaved grafted chains of Mn = 29.5 kg mol−1 and thus a DPn,sc,GPC = 230, one obtains LSC = 230 × 0.25 nm = 57 nm. This agreement suggests that the soft polyBuA side chains are highly extended, resulting in the well resolved corona in the images. On the other hand, there is no indication of the stretched hydrophilic blocks, which if extended would be each 94 nm long with side chains that are only 4 nm long if fully extended. Also there is no indication of coiled end blocks, which should exhibit a diameter of 8 nm, assuming a PEO density of 1.1 g cm−3. Moreover, the spine is surprisingly thick.
This puzzle is solved, if one assumes a backfolding of the main chain, which reduces the contour length to less than half the fully extended contour, and also explains the thickened spine. Similar backfolding has been observed for charged dendronized polymers in a polar solvent, driven by the hydrophobic effect.48 Here, the driving force is not clear. Possibly, the stretched two-dimensional arrangement induces an orientation of the ester moieties at the polymer backbone, the resulting dipole moment being compensated by the ether moieties of the backfolded polyPEGA chains.
Conclusions
The combination of two techniques of controlled free radical polymerization, namely the RAFT and the ATRP techniques, together with the use of a macromonomer is a powerful strategy to prepare novel amphiphilic dual brush copolymers in form of symmetrical triblock copolymers. Both the hydrophilic and the hydrophobic blocks are soft blocks with low glass transitions, which micro-phase separate in the bulk. In aqueous solution, they behave as amphiphiles and, thus represent by virtue of their unusual large size giant surfactants. These form long-time stable micellar or vesicular aggregates depending on their molecular architecture. On mica surfaces, the triblock copolymers can be adsorbed as single molecules with worm-like backbones and stretched out side chains. The SFM images indicate that end blocks may back-fold onto the middle block.Acknowledgements
We thank M. Gräwert and H. Schlaad (Max-Planck-Institut for Colloids and Interfaces, Potsdam-Golm) for help with SEC measurements, and M. Heydenreich and A. Krtitschka for NMR measurements (Universität Potsdam). Furthermore we thank N. Severin (Humboldt-Universität zu Berlin) for fruitful discussions. Financial support was given by Sfb 448 of Deutsche Forschungsgemeinschaft DFG and by Fonds der Chemischen Industrie.Notes and references
- M. Zhang and A. H. E. Müller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3461–3481 CrossRef CAS.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS.
- S. Bolisetty, S. Rosenfeldt, C. N. Rochette, L. Harnau, P. Lindner, Y. Xu, A. H. E. Müller and M. Ballauff, Colloid Polym. Sci., 2009, 287, 129–138 CrossRef CAS.
- A. Zhang, J. Barner, I. Gössl, J. P. Rabe and A. D. Schlüter, Angew. Chem., Int. Ed., 2004, 43, 5185–5188 CrossRef CAS.
- D. Neugebauer, Y. Zhang, T. Pakula, S. S. Sheiko and K. Matyjaszewski, Macromolecules, 2003, 36, 6746–6755 CrossRef CAS.
- T. Pakula, Y. Zhang, K. Matyjaszewski, H. Lee, H. Börner, S. Qin and G. C. Berry, Polymer, 2006, 47, 7198–7206 CrossRef CAS.
- C. Li, N. Gunari, K. Fischer, A. Janshoff and M. Schmidt, Angew. Chem., Int. Ed., 2004, 43, 1101–1104 CrossRef CAS.
- H. Lee, J. R. Boyce, A. Nese, S. S. Sheiko and K. Matyjaszewski, Polymer, 2008, 49, 5490–5496 CrossRef CAS.
- S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688–714 CrossRef CAS.
- S. Garnier, A. Laschewsky and J. Storsberg, Tenside, Surfactants Deterg., 2006, 43, 88–102 Search PubMed.
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3(3–4), 38–46 CrossRef CAS.
- M. A. Cohen Stuart, Colloid Polym. Sci., 2008, 286, 855–864 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC.
- K. Ishizu, J. Satoh, K. Toyoda and A. Sogabe, J. Mater. Sci., 2004, 39, 4295–4300 CrossRef CAS.
- K. Huang and J. Rzayev, J. Am. Chem. Soc., 2009, 131, 6880–6885 CrossRef CAS.
- D. Zehm, A. Laschewsky, M. Gradzielski, S. Prévost, H. Liang, J. P. Rabe, R. Schweins and J. Gummel, Langmuir, 2010, 26, 3145–3155 CrossRef CAS.
- Z. Li, J. Ma, C. Cheng, K. Zhang and K. L. Wooley, Macromolecules, 2010, 43, 1182–1184 CrossRef CAS.
- A. Laschewsky, M. Mertoglu, S. Kubowicz and A. F. Thünemann, Macromolecules, 2006, 39, 9337–9345 CrossRef CAS.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, Acc. Chem. Res., 2009, 42, 681–692 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1569 RSC.
- Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. P. Davis, John Wiley and Sons, Inc., Hoboken, 2002 Search PubMed.
- Controlled and Living Polymerizations. From Mechanisms to Applications, ed. K. Matyjaszewski and A. H. E. Müller, Wiley-VCH, Weinheim, 2009 Search PubMed.
- B. Le Droumaget and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- N. S. Cameron, M. K. Corbierre and A. Eisenberg, Can. J. Chem., 1999, 77, 1311–1326 CrossRef.
- I. W. Hamley, Block Copolymers in Solution: Fundamentals and Applications, John Wiley & Sons Ltd, Chichester, England, 2005 Search PubMed.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- Y. Zhang, Z. Shen, D. Yang, C. Feng, J. Hu, G. Lu and X. Huang, Macromolecules, 2010, 43, 117–125 CrossRef CAS.
- A. M. Bivigou-Koumba, J. Kristen, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Macromol. Chem. Phys., 2009, 210, 565–578 CrossRef CAS.
- M. Claes, S. Voccia, C. Detrembleur, C. Jérôme, B. Gilbert, P. Leclère, V. M. Geskin, R. Gouttebaron, M. Hecq, R. Lazzaroni and R. Jérôme, Macromolecules, 2003, 36, 5926–5933 CrossRef CAS.
- U. Keiderling, Appl. Phys. A: Mater. Sci. Process., 2002, 74, s1455–s1457 CrossRef CAS.
- Y. Xia, B. D. Olsen, J. A. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 18525–18532 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- Z. Zarafshani, Ö. Akdemir and J.-F. Lutz, Macromol. Rapid Commun., 2008, 29, 1161–1166 CrossRef CAS.
- J. Rzayev, Macromolecules, 2009, 42, 2135–2141 CrossRef CAS.
- G. Cheng, A. Böker, M. Zhang, G. Krausch and A. H. E. Müller, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- N. Khelfallah, N. Gunari, K. Fischer, G. Gkogkas, N. Hadjichristidis and M. Schmidt, Macromol. Rapid Commun., 2005, 26, 1693–1697 CrossRef.
- C. Tang, B. Dufour, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2007, 40, 6199–6205 CrossRef CAS.
- S. Podzimek and T. Vlcek, J. Appl. Polym. Sci., 2001, 82, 454–460 CrossRef CAS.
- B. S. Sumerlin, D. Neugebauer and K. Matyjaszewski, Macromolecules, 2005, 38, 702–708 CrossRef CAS.
- K. L. Beers, S. G. Gaynor, K. Matyjaszewski, S. S. Sheiko and M. Möller, Macromolecules, 1998, 31, 9413–9415 CrossRef CAS.
- D. Neugebauer, B. S. Sumerlin, K. Matyjaszewski, B. Goodhart and S. S. Sheiko, Polymer, 2004, 45, 8173–8179 CrossRef CAS.
- Y. Xu, H. Becker, J. Yuan, M. Burkhardt, Y. Zhang, A. Walther, S. Bolisetty, M. Ballauff and A. H. E. Müller, Macromol. Chem. Phys., 2007, 208, 1666–1675 CrossRef CAS.
- L. Gold, J. Chem. Phys., 1958, 28, 91–99 CrossRef CAS.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-rays, Wiley, New York, 1955 Search PubMed.
- M. Gradzielski, D. Langevin and B. Farago, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 53, 3900–3919 CrossRef CAS.
- Y. Xu, S. Bolisetty, M. Drechsler, B. Fang, J. Yuan, M. Ballauff and A. H. E. Müller, Polymer, 2008, 49, 3957–3964 CrossRef CAS.
- W. Zhuang, E. Kasëmi, Y. Ding, M. Kröger, A. D. Schlüter and J. P. Rabe, Adv. Mater., 2008, 20, 3204–3210 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Scheme of dual brush block copolymers in selective and non-selective solvents. Details on the synthesis and molecular characterization of the polymers made, including 1H NMR spectra, SEC elugrams, and thermal characterisations, as well as additional DLS, SLS and SANS data of polymer solutions, and SFM image of reactive triblock copolymer precursor adsorbed on mica. See DOI: 10.1039/c0py00200c |
| ‡ This paper contains work as a result of a collaborative research project of the German Science Foundation (DFG Sonderforschungsbereich 448) on “Mesoscopically organized composites”. |
| This journal is © The Royal Society of Chemistry 2011 |