Optimised ‘click’ synthesis of glycopolymers with mono/di- and trisaccharides†
Nicola
Vinson
,
Yanzi
Gou
,
C. Remzi
Becer
,
David M.
Haddleton
and
Matthew I.
Gibson
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: m.i.gibson@warwick.ac.uk; Fax: +44 (0)2476 524112
First published on 4th October 2010
Abstract
In this paper we investigate the optimum procedure for the post-polymerisation modification of alkyne-bearing polymer scaffolds with glycosyl azides. We first elaborate the one-pot synthesis of glycosyl azides, in aqueous solution, without the need for protecting groups and in multigram scale. Using these azides, the ligand tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) was shown to give the fastest kinetics for the ‘click’ reaction at ambient temperature, and was used to prepare homogenous oligosaccharide-modified glycopolymers. The terminal sugars of these oligosaccharides were used to introduce α-linked glucose which is typically synthetically challenging.
Introduction
Multivalent interactions between carbohydrate ligands and receptors (i.e. lectins) help mediate signal transduction in biological systems including, for example, infection, immune defence and cancer malignancy.1–3 In multivalent systems, the observed binding constant is increased by presentation of multiple copies of the same ligand, to an extent greater than the simple sum of the total number of ligands and referred to as the ‘cluster glycoside’ effect. In nature, multiple copies of carbohydrates are presented on the surface of glycoproteins, often as oligosaccharides which display the binding epitope in a precise 3-dimensional orientation. The use of polymers which present carbohydrates pendant to their backbone (glycopolymers) and thus mimic the multivalent effect seen in nature is an attractive analogue to glycoproteins for biotechnological/healthcare applications.4–9 For example, the interaction of poly-[(β-D-galactopyranosyl) oxyethylmethacrylate] with the lectin peanut agglutinin was shown to be approximately 50 times greater than the free monomer.10 We have demonstrated that mannose-bearing glycopolymers can inhibit the interactions between GP120, a HIV surface glycoprotein, and its human biological target DC-SIGN and also glycopolymer–protein conjugates for complement activation.11The synthesis of precisely defined glycopolymers represents a challenge, due to complex monomer syntheses and difficulties in maintaining stereochemical purity.12,13 Post-polymerisation modification of precursor polymer scaffolds is a powerful tool for functional polymer (library) synthesis as all the product polymers have identical chain lengths and distributions.14,15 Whitesides and coworkers first demonstrated the use of polymeric active esters to create libraries of sialic acid bearing glycopolymers for inhibiting influenza hemagglutination.16 Active esters are useful precursors, but are often hydrolytically sensitive and do not lead to quantitative conversion when using bulky functional groups.17,18 ‘Click’ chemistries have improved the scope of this procedure to give high yielding reactions without the need for protecting group chemistries, in particular the copper catalysed alkyne–azide cycloaddition19 or the radical thiol–alkene reaction.20 While convenient, it is still necessary to synthesise the glycosyl azides/thiols in multistep reactions. There are also difficulties with the addition of oligosaccharides to polymers as their coupling reactions are less efficient than monosaccharides and also there is potential for degradation/anomerisation of oligosaccharides during chemical modification. Godula and Bertozzi reported using (oligo)saccharides with reducing termini (many saccharides have a single reducing end group) to graft onto a poly(acylhydrazide) backbone.21 This method is simple, versatile and requires no activation/functionalisation of the carbohydrate, but the grafting efficiency is reduced for oligo-, or N-acetyl carbohydrates. Direct polymerisation of oligosaccharide-functional monomers has been employed, but can often require multistep synthesis/purification.22,23
While the synthesis of glycosyl azides and click post-polymerisation modification have been studied previously, our aim was the optimisation of this to facilitate its application, particularly to allow addition of larger oligosaccharides. Firstly, a facile procedure to (gram scale or larger) glycosyl azides in a single step under aqueous conditions and without any protecting groups is elaborated. Secondly, the ‘click’ reaction is optimised for a large range of carbohydrates and copper(I) ligands to allow the synthesis to be conducted at room temperature with only a small excess of azide.
Results and discussion
The synthesis of glycosyl azides was conducted using the method of Tanaka et al., Scheme 1.24 The desired carbohydrate is activated at the anomeric position of the reducing terminus by 2-chloro-1,3-dimethylimidazolinium chloride (DMC), which then undergoes nucleophilic substitution by sodium azide, possibly via a 1,2-dehydro intermediate. In the original work, this was only conducted on a small (<50 mg) scale and required HPLC purification. As our aim was to make this method generally applicable, the synthesis was scaled up to use multigram quantities of the starting carbohydrate. A modified purification protocol was also used which removed the requirement for chromatography. This was achieved by precipitating sodium azide by addition of ethanol and passage through an acidic ion-exchange column to remove the HCl salts of the base (triethylamine or diisopropylethylamine). See Experimental section for full details. The desired products were isolated as white solids with yields of up to 90%. FTIR revealed the presence of a single azide stretch at 2120 cm−1, Fig. 1, confirming no residual sodium azide was present (residual NaN3 has a resolvable peak at 2015 cm−1) and ESI-MS confirmed the structure of the glycosyl azides. 1H NMR and 13C NMR confirmed the structure, as shown in Fig. 2 for glucose-azide.![Synthetic strategies employed: (i) sugar/DMC/NEt3 [1 : 3 : 10]/H2O/0 °C, (ii) azide/Cu(i)Br/ligand [1 : 0.03 : 0.03] (see text and ESI)/DMSO/25 °C.](/image/article/2011/PY/c0py00260g/c0py00260g-s1.gif) | ||
Scheme 1 Synthetic strategies employed: (i) sugar/DMC/NEt3 [1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:10]/H2O/0 °C, (ii) azide/Cu(I)Br/ligand [1:0.03:0.03] (see text and ESI†)/DMSO/25 °C. :3:10]/H2O/0 °C, (ii) azide/Cu(I)Br/ligand [1:0.03:0.03] (see text and ESI†)/DMSO/25 °C. | ||
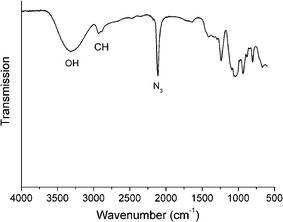 | ||
| Fig. 1 ATR-FTIR spectrum of mannose azide. | ||
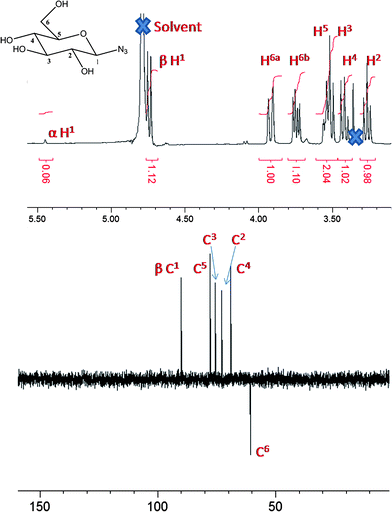 | ||
| Fig. 2 NMR spectra of β-glucose azide: top, 1H NMR; and bottom, 13C (DEPT) NMR. | ||
1H NMR was used to assign the anomeric azide as either α (axial) or β (equatorial), Table 1. In 1H NMR, higher chemical shifts (i.e. downfield, >5 ppm) and smaller coupling constants for the anomeric carbon are typical for α-linked glycosides relative to their β-isomers. All of the investigated carbohydrates gave rise to the β anomers apart from mannose which had α stereochemistry. These observations agree with the possible mechanism via a 1,2-dehydro intermediate from the 2-hydroxyl group directing the product stereochemistry. The ratio of the α:β peaks showed over 80% anomeric purity in all cases, as confirmed by the presence of a single peak between 90 and 100 ppm in 13C NMR (or 2/3 peaks for the case of di- and trisaccharides, respectively. This method represents a significant improvement over the traditional Koenigs–Knorr type glycosylation which is atom inefficient due to the need for protecting groups, additional purification and rigorously dry organic solvents and catalysts.14
| Carbohydrate | Major anomer NMR data | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1H (ppm) | J 1–2/Hz | 13C (ppm) | Assignment | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Partly obscured peak. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| D-Glucose | 4.74 | 8.78 | 90.1 | β-(95:5) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| D-Mannose | 5.44 | 1.88 | 89.7 | α-Only | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L-Fucose | 4.61 | 8.69 | 90.5 | β-(86:14) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| lactose | 4.83 | 8.80 | 90.0 | β-Only | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| D-Cellobiose | 4.94 | 8.74 | 89.9 | β-(93:7)a |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melibiose | 4.83 | 8.81 | 90.3 | β-(88:12) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Maltotriose | 4.70 | 8.78 | 89.9 | β-Onlya | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The second stage of this study was to optimise the alkyne–azide cycloaddition reaction with a poly(alkyne scaffold) (P1, Table 1). To this end, trimethylsilylpropargyl methacrylate was polymerised by copper mediated living radical polymerisation using previously published conditions,14 which following deprotection gave well-defined poly(propargyl methacrylate) with Mn = 7400 g mol−1 (DP = 60) and Mw/Mn = 1.30. DMSO was chosen as the solvent for post-polymerisation modification as it is a good solvent for both the polymer and a wide range of carbohydrates making it suitable for the glycosyl azides used in this study, and also larger branched carbohydrates. The most desirable conditions for this reaction are at ambient temperature without the need for external heating and with only a small (if any) excess of the desired carbohydrate, to facilitate the use of rare, difficult to access, carbohydrates (such as gangliosides) and to limit the opportunities for degradation of the sensitive anomeric linkages. Four common ligands, Fig. 3, for Cu(I)Br, were evaluated for their ability to promote coupling of fucose and glucose azide to poly(propargyl methacrylate) at 25 °C for 8 hours. SEC analysis of the resulting polymers revealed that approximately identical molecular weights were obtained when the ligand was varied, but with a significant difference in molecular weight between fucose and glucose functional polymers, as shown in Table 2. Fucose azide resulted in polymers with Mn ≈ 12000 g mol−1 but glucose azide gave Mn ≈ 16000 g mol−1 for each ligand. This difference reflects the lower mass of fucose due to its 6-deoxy group, and also the difference in solvation behaviour of the carbohydrates in the SEC solvent (DMF).
![Ligands used in this study: (A) tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, (B) bathophenanthrolinedisulfonic acid disodium salt, (C) bipyridine, and (D) N-ethyl-2-pyridylmethanimine.](/image/article/2011/PY/c0py00260g/c0py00260g-f3.gif) | ||
| Fig. 3 Ligands used in this study: (A) tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, (B) bathophenanthrolinedisulfonic acid disodium salt, (C) bipyridine, and (D) N-ethyl-2-pyridylmethanimine. | ||
Although the coupling reactions went to very high degrees of conversion (>90% after 92 hours), it was important to study the kinetics of this reaction as a function of the ligand used. The purpose being to identify the best ligand for addition of larger, sterically hindered oligosaccharides (vide infra). Online 1H NMR was used to monitor the kinetics of this reaction by measuring the decrease in the H-2 signal on the pyranose ring of fucose at 4.38 ppm as a function of time, Fig. 4. Ligand A gave the fastest kinetics, suggesting it is the most catalytically active in agreement with previous studies.25 It should be noted that this catalyst system is less sensitive to oxygen than others, which may contribute to the observed reaction rates.
![Kinetic measurements of consumption of fucose azide: partial time-dependent 1H NMR with (A) ligand A and (B) all ligands, 25 °C/[N3] : [alkyne] = 1.2 : 1/DMSO.](/image/article/2011/PY/c0py00260g/c0py00260g-f4.gif) | ||
| Fig. 4 Kinetic measurements of consumption of fucose azide: partial time-dependent 1H NMR with (A) ligand A and (B) all ligands, 25 °C/[N3]:[alkyne] = 1.2:1/DMSO. | ||
The ligands C and D provided good rates of reaction, but were also observed to be more sensitive to oxygen contamination, which may lead to slower rates of reaction and lower degrees of functionalisation. Ligand B was found to be particularly slow. We have previously shown ligand B to be useful for the coupling of various glycosyl azides at 50 °C, indicating that reaction temperature is important, and demonstrating the need to screen different ligands.26 The kinetics were also studied using glucosyl azide, and a less strong dependence on the nature of the ligand was found, but ligand A was still the most active (see ESI†) suggesting that both the ligand and the nature of the carbohydrate are important in this reaction. Considering the above, ligand A was the best choice as it is both highly reactive and less sensitive to oxygen impurities. Although the differences in ligand reactivity were small for monosaccharides, the choice of the most active ligand is critical when using larger branched oligosaccharides, due to the additional steric constraints of adding large moieties onto adjacent repeat units along a polymer backbone. By choosing the most active ligand it will be possible to obtain homogenous glycopolymers with different oligosaccharides allowing for structure–activity relationships without needing to consider differing degrees of functionalisation. The influence of the ligand on residual Cu(II) was not investigated and all were purified by passage through neutral alumina. This would be an important consideration for any future biological applications. Using ligand A, a library of glycopolymers with increasingly large saccharide side chains was synthesised, Table 3.
Maltotriose azide (a trisaccharide) gave a polymer with near-identical molecular weight as lactose and cellobiose (disaccharides) according to SEC. NMR analysis suggested that in all cases >90% conversion was obtained and FTIR spectroscopy of the glycopolymers did not show any residual alkyne (2130 cm−1) or azide (2115 cm−1). This collection of evidence suggests that the glycopolymers have near quantitative degrees of functionalisation (above 90% by NMR) and that the lower than expected molecular weights obtained by SEC analysis are due to the elution behaviour of the glycopolymers, which have a comb-like structure. It is also possible that the solvent (DMF) may not completely solvate the carbohydrate side chains. Nonetheless, the obtained glycopolymers retained the low PDI of the precursor polymer demonstrating that this optimised protocol can be used for a large range of carbohydrates.
There are only a few examples of the incorporation of α-gluco-/galactosides or β-mannosides into glycopolymers due to their challenging synthetic requirements.8,27,28 Interestingly, the oligosaccharides employed in this study contain some of these difficult sequences. Cellobiose contains a terminal β-D-glucopyranoside (P4), whereas maltotriose contains a terminal α-D-glucopyranoside (P5). To demonstrate that this strategy can be used to modulate and improve the binding affinities of glycopolymers we chose the lectin concanavalin A (ConA), which is specific for carbohydrates with cis equatorial hydroxyls at the 3 and 4 positions on the pyranose ring. ConA also has higher specificity for α-anomers over β.29 This was tested using P3 and P4 at equal molar concentrations (i.e. same number of saccharide moieties) using a simple turbidity assay. Following addition of P4 to the ConA solution it rapidly became turbid (Fig. 5) indicating molecular recognition was occurring. Conversely, following addition of P3 there was no increase in turbidity due to the relatively low affinity of β-D-glucosides. It should be noted that β-linked glucosides do interact with ConA,30 but with reduced avidity thus requiring either increased concentrations, or longer periods of time to allow for recognition. While simple, this test demonstrates the ability to modulate the molecular recognition properties of glycopolymers through incorporation of natural oligosaccharides. In order to exploit the oligosaccharides in this fashion, it is essential to have highly efficient coupling chemistry. Less than quantitative conversion leads to reduced sugar density, which in turn would alter the binding affinity (due to the multivalent effect), preventing quantitative structure–property relationships from being extracted.
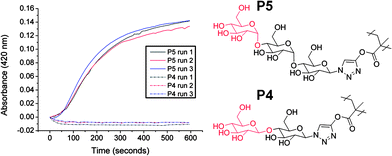 | ||
| Fig. 5 Interaction between oligosaccharide-functionalised glycopolymers with ConA. ConA 0.1 mg ml−1, total carbohydrate 0.21 mmol l−1. | ||
Conclusions
In summary, we have optimised the synthesis of glycopolymers using the azide–alkyne ‘click’ reaction. A highly efficient, aqueous, synthesis of a range of glycosyl azides has been scaled up and the purification protocols simplified. The most efficient ligand for the Cu(I) was found by screening the kinetics of a small library of ligands with two different glycoysl azides. Application of the idealised conditions allowed the synthesis of a range of structurally well-defined glycopolymers with increasing side-chain length. The efficiency of the procedure was demonstrated by the near-quantitative reaction of di- and trisaccharides onto the polymer backbone, despite their increased steric bulk. These oligosaccharides were used to introduce terminal monosaccharide functionality onto glycopolymers which is challenging when using conventional synthetic techniques. This strategy was demonstrated by the increased binding of α-glucosides to concanavalin A as compared to β derivatives. These protocols will be applicable to a wide range of oligosaccharides which will be exploited for biotechnological and biomedical applications.Experimental
Materials
Copper(I) bromide, 2-chloro-1,3-dimethylimidazolinium chloride (DMC), N,N-diisopropylethylamine (DIPEA), sodium azide, D-cellobiose, L-fucose, α-D-glucose, lactose, maltotriose, bathophenanthrolinedisulfonic acid disodium salt trihydrate (BPDS), concanavalin A and 2,2′-bipyridyl (bipyridine) were purchased from Sigma-Aldrich. Triethylamine was purchased from Fisher Scientific. D-(+)-Galactose was purchased from Alfa-Aesar. All reagents were used as received unless otherwise specified. Copper(I) bromide was purified by the method of Keller and Wycoff.31 Poly(propargyl methacrylate) was prepared according to previously published methods14 (SEC (DMF) Mn = 7400, Mn/Mw = 1.29 and satisfactory NMR data). The ligands N-ethyl-2-pyridylmethanimine32and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA)26 were prepared as described previously. Phosphate buffered saline solution was prepared by dissolving a preformulated tablet from Sigma-Aldrich in 200 ml of ultra high quality water to give a final composition of 0.01 M phosphate, 0.0027 M potassium chloride and 0.137 M sodium chloride, pH 7.4.Analytical and physical methods
1H and 13C NMR spectra were recorded on Bruker DPX-300 and DPX-400 spectrometers using deuterated solvents obtained from Aldrich. Polymerisation kinetics were followed by 1H NMR recorded on a Bruker AV-400 spectrometer. Mass spectra were recorded on an Esquire2000 using electrospray ionisation (ESI) in positive mode. FTIR spectra were recorded on a Brüker Vector 22 FTIR spectrometer. SEC was conducted on a Varian 390-LC system in DMF (1 g l−1LiBr) at 50 °C, equipped with refractive index and viscometry detectors, 2 × PLgel 5 µm mixed-D columns (300 × 7.5 mm), 1× PLgel 5 µm guard column (50 × 7.5 mm) and autosampler. Data were analysed using Cirrus 3.2 software. Molecular weight was determined relative to narrow poly(methyl methacrylate) standards. For the turbidimetry studies absorbance was measured at 420 nm on a Perkin Elmer Lambda-35 UV/VIS spectrometer using 1 cm path length glass cuvettes.Procedures
Glycosyl azides were synthesised by a modified procedure from that of Tanaka et al.24 to allow for larger scales and simplified purification.1H NMR (D2O) 400 MHz, δppm: 4.74 (1H, d, J1–2 = 8.78 Hz, H1), 3.92 (1H, dd, J6a–5 = 1.95 Hz, J6a–6b = 12.42 Hz, H6a), 3.74 (1H, dd, J6b–5 = 5.59 Hz, J6b–6a = 12.42 Hz, H6b), 3.54 (1H, m, H5), 3.51 (1H, t, J3–2 = 9.01 Hz, J3–4 = 9.01 Hz, H3), 3.41 (1H, t, J4–3 = 9.51 Hz, J4–5 = 9.51 Hz, H4), 3.25 (1H, t, J2–1 = 8.98 Hz, J2–3 = 8.98 Hz, H2).
13C NMR (D2O) 100 MHz, δppm: 90.1 (C1), 77.9 (C5), 75.7 (C3), 72.8 (C2), 69.1 (C4), 60.5 (C6).
IR ν: 2116 cm−1 (-N3).
MS (ESI) m/z = 228.0 [M + Na]+.
1H NMR (D2O) 400 MHz, δppm: 5.44 (1H, d, J1–2 = 1.88 Hz, H1), 3.90 (1H, d, J = 10.03 Hz), 3.86 (1H, dd, J2–3 = 3.25 Hz, J2–1 = 1.92 Hz, H2), 3.78 (1H, t, J = 7.11 Hz), 3.75–3.78 (1H, m), 3.72 (1H, dd, J3–4 = 9.36 Hz, J3–2 = 3.40 Hz, H3), 3.63 (1H, t, J = 9.50 Hz).
13C NMR (D2O) 100 MHz, δppm: 89.7 (C1), 74.7, 69.85, 69.80, 66.4, 60.8 (C6).
IR ν: 2110 cm−1 (-N3).
MS (ESI) m/z = 228.0 [M + Na]+.
:1 (v/v). After elution with the same solvent mixture, appropriate fractions were collected (Rf: 0.23) and the solvents were removed under reduced pressure to give β-fucose azide (2.08 g, 11 mmol, 45%) as an off-white solid.
1H NMR (D2O) 300 MHz, δppm: 4.61 (1H, d, J1–2 = 8.69 Hz, H1), 3.86 (1H, dd, J5–6 = 6.49 Hz, J5–4 = 0.91 Hz, H5), 3.76 (1H, dd, J4–3 = 3.47 Hz, J4–5 = 0.77 Hz, H4), 3.65 (1H, dd, J3–2 = 9.83 Hz, J3–4 = 3.43 Hz, H3), 3.44 (1H, dd, J2–1 = 8.72 Hz, J2–3 = 9.79 Hz, H2), 1.25 (3H, d, J6–5 = 6.51 Hz, H6).
13C NMR (D2O) 75 MHz, δppm: 90.5 (C1), 73.1, 72.8, 71.1, 70.0, 15.4 (C6).
IR ν: 2113 cm−1 (-N3).
MS (ESI) m/z = 201.1 [2M + Na + H]2+, 212.0 [M + Na]+.
1H NMR (D2O) 400 MHz, δppm: 4.83 (1H, d, J1–2 = 8.80 Hz, H1), 4.51 (1H, d, J1′–2′ = 7.78 Hz, H1′), 4.04 (1H, d, J6a–6b = 12.31 Hz, H6a), 3.99 (1H, d, J = 3.13 Hz, H4′), 3.89 (1H, d, J6′a–6′b = 11.74 Hz, H6′a), 3.70–3.85 (7H, m, H6b, H6′b, H3, H4, H5, H3′, H5′), 3.60 (1H, dd, H2′), 3.37 (1H, t, J2–1 = 8.58 Hz, J2–3 = 8.58 Hz, H2).
13C NMR (D2O) 100 MHz, δppm: 102.9 (C1′), 90.0 (C1), 77.8 (C4), 76.8 (C5), 75.4 (C5′), 74.4 (C3), 72.6 (C2), 72.5 (C3′), 71.0 (C2′), 68.6 (C4′), 61.1 (C6′), 59.9 (C6).
IR ν: 2116 cm−1 (-N3).
MS (ESI) m/z = 390.1 [M + Na]+.
1H NMR (D2O) 400 MHz, δppm: 4.94 (1H, d, J1–2 = 8.74 Hz, H1), 4.66 (1H, d, J1′–2′ = 7.85 Hz, H1′), 3.95–4.15 (4H, m, H6a, H6′a, H6b, H66′b), 3.80–3.90 (3H, m, H3, H4, H5), 3.61–3.70 (2H, m, H3′, H5′), 3.58 (1H, d, J = 9.10 Hz, H4′), 3.47 (2H, t, J = 8.61 Hz, H2, H2′).
13C NMR (D2O) 75 MHz, δppm: 102.5 (C1′), 89.9 (C1), 78.0 (C4), 76.6 (C5), 76.0 (C5′), 75.4 (C3′), 74.2 (C3), 73.1 (C2), 72.6 (C2′), 69.4 (C4′), 60.5 (C6′), 59.7 (C6).
IR ν: 2116 cm−1 (-N3).
MS (ESI) m/z = 390.1 [M + Na]+.
1H NMR (D2O) 300 MHz, δppm: 5.34 (2H, dd, J = 1.82 Hz, J = 3.85 Hz, H1′ and H1″), 4.70 (1H, d, J1–2 = 8.78 Hz, H1), 3.50–3.94 (16H, m), 3.36 (1H, t, J = 9.36 Hz), 3.24 (1H, t, J2–1 = 9.05 Hz, J2–3 = 9.05 Hz H2).
13C NMR (D2O) 75 MHz, δppm: 99.7 and 99.4 (C1′ and C1″), 89.9 (C1), 76.6, 76.4, 76.2, 76.1, 73.2, 72.8, 72.6, 71.7, 71.4, 71.1 and 69.2 (sugar-C), 60.4 (C6, C6′ and C6″).
IR ν: 2117 cm−1 (-N3).
MS (ESI) m/z = 552.2 [M + Na]+.
The precise details for each ligand are given below. The codes used for the ligands relate to the structures shown in Fig. 3.
Ligand C (2 mg, 0.013 mmol) and copper(I) bromide (2 mg, 0.014 mmol) were added to a Young's tap NMR tube, followed by stock solution (0.6 ml). The reaction was quickly placed under vacuum and then nitrogen.
SEC
Mn
= 11800, Mw = 14800, Mn/Mw = 1.25.
Ligand B (9 mg, 0.015 mmol) and copper(I) bromide (2 mg, 0.014 mmol) were added to a Young's tap NMR tube, followed by stock solution (0.6 ml). The reaction was quickly placed under vacuum and then nitrogen.
SEC
Mn
= 13000, Mw = 15000, Mn/Mw = 1.15.
Ligand D (2 µl, 2 mg, 0.015 mmol) and copper(I) bromide (2 mg, 0.014 mmol) were added to a Young's tap NMR tube, followed by stock solution (0.6 ml). The reaction was quickly placed under vacuum and then nitrogen.
SEC
Mn
= 12100, Mw = 14800, Mn/Mw = 1.22.
Ligand A (8 mg, 0.015 mmol) and copper(I)bromide (2 mg, 0.014 mmol) were added to a Young's tap NMR tube, followed by stock solution (0.6 ml). The reaction was quickly placed under vacuum and then nitrogen.
SEC
Mn
= 11600, Mw = 14900, Mn/Mw = 1.28.
Procedure 1. Stock solution (6 ml) was cannulated under nitrogen into a Schlenk tube, previously evacuated and filled with nitrogen, containing β-lactose azide (240 mg, 0.65 mmol), copper(I) bromide (22 mg, 0.15 mmol) and a magnetic follower. The reaction was left to stir for 92 hours.
Procedure 2. Stock solution (6 ml) was cannulated under nitrogen into a Schlenk tube, previously evacuated and filled with nitrogen, containing β-cellobiose azide (230 mg, 0.63 mmol), copper(I) bromide (23 mg, 0.15 mmol) and a magnetic follower. The reaction was left to stir for 92 hours.
Procedure 3. Stock solution (6 ml) was cannulated under nitrogen into a Schlenk tube, previously evacuated and filled with nitrogen, containing β-maltotriose azide (331 mg, 0.63 mmol), copper(I) bromide (22 mg, 0.15 mmol) and a magnetic follower. The reaction was left to stir for 92 hours.
Upon completion all reaction mixtures were filtered through a short column of neutral aluminium oxide, washed with DMF, and then centrifuged at 4500 rpm for 10 minutes. The resulting pellet was dissolved in H2O and dialysis carried out using dialysis tubing (MWCO = 1500 g mol−1) for 3 days; the water was changed ∼4 times every 24 hours. Following completion the contents of each dialysis bag were freeze-dried. The resulting solid was then dissolved in H2O (25 ml), centrifuged at 7000 rpm for 30 minutes and the supernatant decanted and freeze-dried to give the glycopolymer.
Each polymer solution (0.286 ml) was then added to Con A solution (0.714 ml, 0.1 mg ml−1) to give a total carbohydrate concentration of 0.21 mmol l−1 and the absorbance measured at 420 nm over a period of 10 minutes in a UV-visible spectrometer. Each polymer was tested a minimum of three times.
Acknowledgements
Equipment used was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). MIG is a Birmingham Science City Interdisciplinary Research Fellow, supported by AWM and ERDF. We thank EPSRC (Dorothy Hodgkin CASE award) and the China Scholarship Council for funding (YG) and the EU for a Marie-Curie (235999) award (CRB).Notes and references
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS.
- N. Sharon, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 527–537 Search PubMed.
- M. I. Gibson, C. A. Barker, S. G. Spain, L. Albertin and N. R. Cameron, Biomacromolecules, 2009, 10, 328–333 CrossRef CAS.
- A. Joshi, D. Vance, P. Rai, A. Thiyagajan and R. S. Kane, Chem.–Eur. J., 2008, 14, 7738–7747 CrossRef CAS.
- A. Imberty, Y. M. Chabre and R. Roy, Chem.–Eur. J., 2008, 14, 7490–7499 CrossRef CAS.
- S. G. Spain and N. R. Cameron, Polym. Chem., 2011 10.1039/c0py00149j.
- S. R. S. Ting, G. Chen and M. Stenzel, Polym. Chem., 2010 10.1039/c1030py00141d.
- S.-K. Choi, M. Mammen and G. M. Whitesides, J. Am. Chem. Soc., 1997, 119, 4103–4111 CrossRef CAS.
- M. Ambrosi, N. R. Cameron, B. G. Davis and S. Stolnik, Org. Biomol. Chem., 2005, 3, 1476–1480 RSC.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 12, 15156–15163 CrossRef.
- S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS.
- M. I. Gibson, G. J. Hunt and N. R. Cameron, Org. Biomol. Chem., 2007, 5, 2756–2761 RSC.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- M. Mammen, G. Dahmann and G. M. Whitesides, J. Med. Chem., 1995, 38, 4179–4190 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- M. I. Gibson, E. Frohlich and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4332–4345 CrossRef CAS.
- H. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- A. Gress, A. Volkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 9963 CrossRef CAS.
- A. Miyachi, H. Dohi, P. Neri, H. Mori, H. Uzawa, Y. Seto and Y. Nishida, Biomacromolecules, 2009, 10, 1846–1853 CrossRef CAS.
- T. Yoshida, T. Akasaka, Y. Choi, K. Hattori, B. Yu, T. Mimura, Y. Kaneko, H. Nakashima, E. Aragaki, M. Premanathan, N. Yamamoto and T. Uryu, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 789–800 CrossRef CAS.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S. Shoda, Chem. Commun., 2009, 3378–3379 RSC.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani, G. Chen, C. T. Sayers, G. J. Clarkson and D. M. Haddleton, QSAR Comb. Sci., 2007, 26, 1220–1228 Search PubMed.
- S. Akai, Y. Kajihara, Y. Nagashima, M. Kamei, J. Arai, M. Bito and K.-i. Sato, J. Carbohydr. Chem., 2001, 20, 121–143 CrossRef CAS.
- K. H. Mortell, R. V. Weatherman and L. L. Kiessling, J. Am. Chem. Soc., 1996, 118, 2297–2298 CrossRef CAS.
- R. D. Poretz and I. J. Goldstein, Biochemistry, 1970, 9, 2890–2896 CrossRef CAS.
- G. Pasparakis, A. Cockayne and C. Alexander, J. Am. Chem. Soc., 2007, 129, 11014–11015 CrossRef CAS.
- R. N. Keller and H. D. Wycoff, in Inorganic Syntheses,, ed. W. C. Fernelius, McGraw-Hill, New York , 1946, pp. 1–4 Search PubMed.
- D. M. Haddleton, M. C. Crossman, B. H. Dana, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, Macromolecules, 1999, 32, 2110–2119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation are included for all compounds. See DOI: 10.1039/c0py00260g |
| This journal is © The Royal Society of Chemistry 2011 |