Catalytic chain transfer and its derived macromonomers
Johan P. A.
Heuts
*a and
Niels M. B.
Smeets
b
aLaboratory for Polymer Chemistry and Technology, Eindhoven University of Technology, PO Box 513, 5600, MB, Eindhoven, The Netherlands. E-mail: j.p.a.heuts@tue.nl
bDepartment of Chemical Engineering, Queen's University, Kingston Ontario, Canada K7L 3N6
First published on 26th July 2011
Abstract
An overview is given of cobalt-catalyzed chain transfer in free-radical polymerization and the chemistry and applications of its derived macromonomers. Catalytic chain transfer polymerization is a very efficient and versatile technique for the synthesis of functional macromonomers. Firstly the mechanism and kinetic aspects of the process are briefly discussed in solution/bulk and in emulsion polymerization, followed by a description of its application to produce functional macromonomers. The second part of this review briefly describes the behavior of the macromonomers as chain transfer agents and/or comonomers in second-stage radical polymerizations yielding polymers of more complex architectures. The review ends with a brief overview of post-polymerization modifications of the vinyl endfunctionality of the macromonomers yielding functional polymers with applications ranging from initiators in anionic polymerization to end-functional lectin-binding glycopolymers.
 Hans Heuts | Hans Heuts has been an academic in the field of (controlled) radical polymerization in Eindhoven since November 2005. Before taking up this position he held a Ramón y Cajal research fellowship at the University of Murcia (2003–2005), a senior lectureship at the University of New South Wales (2000–2003) and worked as a process development engineer at GE Plastics (1999–2000). He obtained his PhD from the University of Sydney after which he worked on CCT as a post-doc with Tom Davis at UNSW. His current research interests include: controlled polymer synthesis, block copolymer surfactants, functional polymer colloids and radical polymerization kinetics. |
 Niels Smeets | Niels Smeets obtained his MSc in applied organic chemistry in 2005 and a PhD in polymer chemistry and polymer reaction engineering in 2009, both from Eindhoven University of Technology. His thesis focused on the implementation of CCT in emulsion polymerization under the supervision of Jan Meulijk, Hans Heuts and Alex van Herk. One year of his PhD was spent at Queen's University with Michael Cunningham. Currently he holds a “Queen's University—Province of Ontario” fellowship with Timothy McKenna. His current research interests include: synthesis of functional and responsive polymers, polymer colloids and polymer micro/nanogels from a range of different chemistries. |
Introduction
About three decades ago, catalytic chain transfer (CCT) emerged as a very efficient method for producing low molecular weight functional polymers in free-radical polymerization.1 Smirnov and Marchenko discovered that certain low-spin CoII complexes, in particular CoII porphyrins (1), efficiently catalyze the chain transfer reaction to monomer reaction.2–8 Further studies and developments by the Russian group (most notably by Gridnev),9–12 the Glidden Paint company,13–15 DuPont,16,17 ICI/Zeneca18,19 and Ken O'Driscoll20,21 have led to considerable insight into the catalytic process and to the very active cobal-oxime catalysts (2, 3) (Scheme 1).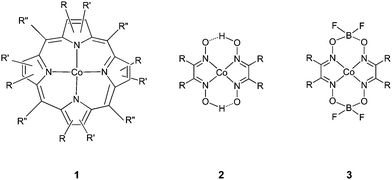 | ||
| Scheme 1 A selection of cobalt catalysts. | ||
Cobaloximes (2) are very sensitive to hydrolysis and oxidation. This sensitivity has been significantly reduced by the introduction of BF2 bridges, after which the catalysts (3) can be readily handled as a solid (even in aerobic conditions). In solution these BF2-bridged catalysts are still sensitive to acid hydrolysis or oxidation by peroxides and other oxygen-centred radicals, but much less than catalyst 2. To further reduce the sensitivity to oxidation it is possible to use the alkylated CoIII derivative of 3 (CoIII-alkyl) which will dissociate into the active CoII catalyst and an alkyl radical.22 The most commonly used catalysts at present are those with structure 3, where the substituent R can be used to tailor solubility and activity. Structure 3 with R = CH3 and R = phenyl is the most widely reported and is commonly denoted by COBF (or CoBF) and COPhBF (or CoPhBF), respectively.
A wide range of commonly used monomers can be efficiently polymerized in the presence of the catalytic chain transfer agents with (functional) methacrylates being the most widely used.23–29α-Methyl styrene and methacrylonitrile, having an α-methyl substituent in common with the methacrylates, are also very active monomers in CCT. Styrenes, acrylates, vinyl acetate and acrylonitrile, i.e., monomers without the α-methyl group show a much lower activity; styrene, with a relatively stable propagating radical, still shows a high activity as compared to the other examples given.
The overall reaction is given by eqn (1), where Rn• is a polymeric radical of chain length n, M is the monomer, CoII is the low-spin CoII catalyst, Pn is a dead polymer chain containing a vinyl ω-endgroup and R1• a monomeric radical.
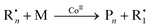 | (1) |
The reaction proceeds via the abstraction of a β-hydrogen atom from the growing radical as shown in Scheme 2, for methyl methacrylate (MMA) and styrene (STY) polymerizations.
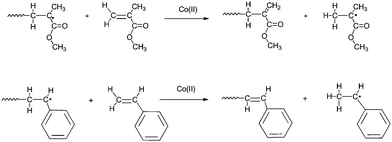 | ||
| Scheme 2 Catalytic chain transfer in the free-radical polymerization of MMA and STY. | ||
It can be seen that for monomers containing an α-methyl group (such as MMA), hydrogen abstraction takes place from the α-methyl group, whereas for monomers without an α-methyl group (such as STY), abstraction takes place from the backbone. Considering that most of the chains are formed by the CCT process, the great majority of chains are macromonomers with a hydrogen atom as the α and a vinyl group as the ω-endgroup. These macromonomers can be used in further reactions as described in one of the following sections (see below).
As with any “ordinary” chain transfer process, the (instantaneous) number average degree of polymerization (DPn) is well described by the Mayo equation (eqn (2)).30
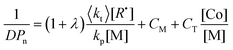 | (2) |
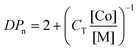 | (2a) |
| Monomera | Reaction medium | Catalyst b | Temp./°C | C T/103 | References | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a MMA = methyl methacrylate; EMA = ethyl methacrylate, n-BMA = n-butyl methacrylate; t-BMA = t-butyl methacrylate; BzMA = benzyl methacrylate; 2-EHMA = 2-ethyl hexyl methacrylate; LMA = lauryl methacrylate; EαHMA = ethyl α-hydroxymethacrylate; POEMA = 2-phenoxyethyl methacrylate; GlyMA = glycerol methacrylate; HEMA = 2-hydroxyethyl methacrylate; DMAEMA = 2-dimethylaminoethyl methacrylate; BHPMA = 2-[3-(2H-benzotriazol-2-yl)-4-hydroxyphenyl]ethyl methacrylate; PIEMA = 2-phtalimidoethyl methacrylate; DEGMA = di(ethylene glycol)methyl ether methacrylate; PEGxMA = poly(ethylene glycol)xmethyl ether methacrylate; MAA = methacrylic acid; TRIS = 3-[tris-(trimethylsilyloxy)silyl]propyl methacrylate; DMI = dimethyl itaconate; STY = styrene; AMS = α-methyl styrene; PAA = phenyl allyl alcohol; MA = methyl acrylate; BA = butyl acrylate; AAm = acrylamide. b COBF = bis[(difluoroboryl)dimethylglyoximato]cobalt(II); COPhBF = bis[(difluoroboryl)diphenyl glyoximato]cobalt(II); Co(dmg)2 = bis[dimethyl glyoximato]cobalt(II); cycCOBF = bis[(difluoroboryl) cyclobutylglyoximato]cobalt(II); CoP = cobalt(meso-Ph4-porphyrin); CoTMHP = tetramethyl ether of cobalt hematoporphyrin IX; CoTFPP = cobalt tetrafluorophenyl porphyrin; Co(ipp)BF = bis[(difluoroboryl)dimethyl glyoximate]isopropyl pyridine cobalt(II); CoPc = cobalt(II) 2,16-bis(4-butanamidoyl)phthalocyanine. c Most likely long-chain value. d Pressure dependent. e Values against PMMA calibration. f M n determined viaNMR for polymers at high conversion. g Most likely value in the absence of Co–C bond formation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Bulk | COBF | 60 | 24–40 (40)c | 21,31 and 53–56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Bulk | COPhBF | 60 | 18–24 | 55–58 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Bulk | Co(dmg)2 | 60 | 2.2–20 (2.2)c | 20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Bulk | cycCOBF | 60 | 13.7 | 56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Bulk | CoP | 60 | 3.6 | 59 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Bulk | CoTMHP | 60 | 2.4 | 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Toluene | COBF | 60 | 41–60 | 60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Butanone | COBF | 60 | 26.5 | 56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Methanol | COBF | 60 | 10.1 | 56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | Ethanol | COBF | 60 | 16–25 | 61 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | scCO2 | COPhBF | 50 | 110 | 62 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMA | scCO2 | CoTFPP | 60 | 1.3 | 63 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EMA | Bulk | COBF | 60 | 27 | 55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EMA | Bulk | COPhBF | 60 | 18 | 55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| n-BMA | Bulk | COBF | 60 | 16–28 | 55 and 60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| n-BMA | Bulk | COPhBF | 60 | 10 | 55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| n-BMA | scCO2 | COPhBF | 50–60 | 11.9–46.7d | 64 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| n-BMA | Bulk | CoTMHP | 60 | 0.7 | 65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| t-BMA | Bulk | COBF | 60 | 14.8–16.8 | 66 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BzMA | Bulk | COBF | 60 | 5.7–6.9 | 67 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2-EHMA | Bulk | COBF | 60 | 11.9 | 60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LMA | 2-Butanone | COBF | 60 | 20 | 68 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EαHMA | Bulk | COBF | 60 | 0.7e | 69 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEMA | Bulk | COPhBF | 60 | 2.0e | 70 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GlyMA |
H2O/methanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
COBF | 80 | 1.0f | 71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HEMA | Bulk | COBF | 40, 60 | 0.6 | 61 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HEMA |
H2O/methanol (2:1) |
COBF | 60 | 1.1f | 71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HEMA | H2O | Co(ipp)BF | 60 | 0.9 | 72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DMAEMA | Bulk | COBF | 70 | 4.4 | 73 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BHPEMA | Toluene | COBF | 60 | 4 | 68 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PIEMA | 30% toluene | COBF | 80 | 0.5 | 74 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DEGMA | Acetonitrile | COBF | 70 | 7.6 | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PEG475MA | Acetonitrile | COBF | 70 | 1.8 | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PEG1100MA | Acetonitrile | COBF | 70 | 0.2 | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PEG2000MA | H2O | Co(ipp)BF | 60 | 0.02 | 72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MAA | H2O | COBF | 55 | 1.1f | 71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TRIS | Toluene | COBF | 60 | 0.8–1.7 | 76 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TRIS | 2-Butanone | COBF | 60 | 7.5 | 68 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DMI | Bulk | COBF | 60 | 7.3–9.5 | 66 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| STY | Bulk | COBF | 60 | 0.4–8.3 (∼8)g | 31,54,56,60,77 and 78 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| STY | scCO2 | COPhBF | 50 | 0.1–1.2d | 64 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AMS | Bulk | COBF | 50 | 89.3 | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PAA | 10% in MMA | COBF | 60 | 138–157 | 79 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MA | Bulk | COBF | 60 | 0.008–0.022 | 53 and 80 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BA | Bulk | COBF | 60 | 0.7 | 60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AAm | Acetic acid | CoPc | 60 | 0.1 | 81 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
An alternative procedure to the Mayo method is the chain length distribution (CLD) method, first introduced by Gilbert and co-workers.34,35 In this method one uses the slope, Λ, of the chain length distribution, P(M), plotted as ln (P(M)) vs. M, which in the limit of high molecular weight (ΛH) is given by eqn (3):
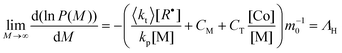 | (3) |
In what follows we will present a brief overview of mechanistic and synthetic aspects of cobaloxime-mediated catalytic chain transfer and the chemistry related to the resulting macromonomers, with a focus on more recent studies. We refer to earlier reviews for more detailed overviews, in particular to the excellent review by Gridnev and Ittel,26 which also extensively covers related Co chemistry. An important aspect covered in Gridnev's and Ittel's review is the effect of ligands on the catalytic activity. In addition to the equatorial ligand system, the CoII complexes in Scheme 1 also contain two axial ligands, which can have dramatic effects on the catalytic activity.10 These ligands are initially introduced in the synthesis of the complexes and may be exchanged for solvent or monomer molecules once in solution. Very strongly coordinating molecules such as phospines and pyridines can indeed affect the catalytic activity tremendously and for cobaloximes in the presence of dimethyl formamide catalytic inhibition has even been reported.37 Considering that catalysts with different ligands can have different catalytic activity, it is important to comment on the values reported in Table 1. Most of the references related to the BF2-bridged cobaloximes use catalysts that were synthesized according to a procedure similar to that described by Bakac and Espenson38 after which a recrystallization in methanol took place. Hence, many of the listed catalysts would contain two axial methanol ligands in the solid state.
In addition to the described CoII catalysts, other transition metal complexes have also been reported to catalyze chain transfer to monomer.39 These include Cr complexes,40–47Mo complexes,47,48Fe complexes49–51 and V complexes.52 All the systems display lower catalytic activities than the cobaloximes, and therefore we will limit ourselves to the cobaloximes in the remainder of this review.
To conclude the introduction, we refer the reader also to a review by Haddleton and co-workers set to appear in the new second edition of Comprehensive Polymer Science,29 which contains complementary information (especially on cobalt catalysts and more detailed descriptions of the applications) to that provided in the current review.
Mechanistic aspects of catalytic chain transfer
All experimental evidence to date indicates that cobaloxime-mediated CCT proceeds via a two-step radical process in which the CoII complex first abstracts a hydrogen atom from the growing radical leading to a CoIIIH complex and a macromonomer. The CoIIIH complex then reacts with a monomer molecule yielding back the original CoII catalyst and a monomeric radical, which can start growing. This mechanism has been studied in detail1–12,20,21,23–28,57,60,65,67,82 and can be summarized in the catalytic cycle of Scheme 3.1,27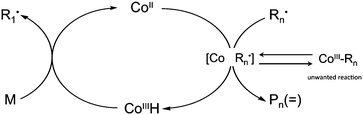 | ||
| Scheme 3 Main catalytic cycle for cobaloxime mediated catalytic chain transfer. | ||
In polymerization kinetic terms, the CCT process is just a chain transfer process with fast re-initiation and to a first approximation does not affect the rate of polymerization in any other way than do ordinary chain transfer agents (with the obvious exception of catalytic inhibition).26,37,83 As such, the standard kinetic equations of free-radical polymerization84 (taking into account the significant amount of monomer consumption by the CCT process) would have to be applicable. With increasing amount of catalyst (and hence an increase in the shorter radical population), a decrease in polymerization rate is observed,6–8,10,20,31,37,54,58 which has commonly be ascribed to a higher termination rate coefficient.20,31,54,58 This view is an over-simplification. First of all the smaller radicals will also propagate faster,85–89 and this effect will be more significant the more catalyst is used. Secondly, the kinetics of the chain transfer process may show a chain-length dependence, but the extent to which the transfer reaction itself shows a chain length dependence is not yet clear, as both an increase5 and a decrease20,21 in CT have been reported with increasing chain length. Thirdly with an increase in catalyst concentration more monomer is being regenerated from the CCT reaction of monomeric radicals (thus slowing down the net monomer consumption).90 And finally, both catalytic inhibition37,83 and reversible Co–C bond formation10,11,58,91 (see below) may take propagating radicals out of the system, which will also slow down the polymerization. A recent kinetic modelling study by Nikitin et al. showed that the incorporation of all these effects was necessary to provide an adequate description of the polymerization kinetics.90
Returning to the catalytic scheme of Scheme 3 it should be noted that Co–C bonding does not form part of the catalytic cycle of CCT, but that it is an unwanted side reaction which takes active catalyst out of the catalytic cycle.1,11,26,27 The main pathway for hydrogen abstraction is unlikely to involve a β-hydrogen elimination from a coordinated radical, but involves a direct reaction with the propagating radical in a close-contact radical pair through a Co⋯H⋯C transition state.92–95 The effect of Co–C bond formation is not very large in the polymerization of methacrylates (where EPR studies show the catalyst to be present in its CoII state),58 but very much in that of the polymerization of monomers forming secondary radicals,11,53,58,59,91–101 such as styrene (EPR signal of CoII is virtually absent)58 and acrylates (polymer chains with attached catalyst have even been observed in MALDI-TOF).80 When expressing the equilibrium constant, K (which can be of the order of 107 to 108 L mol−1 for secondary radicals),91,92,99 of this reversible reaction in the reactant concentrations (eqn (4)), it is clear that the concentration of active catalyst, [CoII], depends on the overall radical concentration, [R•].78
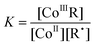 | (4) |
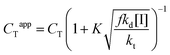 | (5) |
As is also shown in Scheme 3, the Co–C bond formation is reversible and the bond can be broken by heat or by light.78,100 This latter effect is illustrated by the Mayo plots in Fig. 1, where the polymerizations carried out under UV light conditions appear to have a more active catalyst (instead, [CoII] is higher under light than under dark conditions).78
![Effect of light on the chain transfer behaviour in the catalytic chain transfer polymerization of styrene (bulk) in the presence of COBF at 60 °C, [AIBN] = 2.93 × 10−3 mol L−1; (□) reactions carried out under light conditions, CT ≈ 5 × 103. (■) reactions carried out under dark conditions, CT ≈ 1 × 103. The microscopic (true) CT ≈ 8 × 103 for COBF in styrene at 60 °C. Data taken from ref. 78.](/image/article/2011/PY/c1py00224d/c1py00224d-f1.gif) | ||
| Fig. 1 Effect of light on the chain transfer behaviour in the catalytic chain transfer polymerization of styrene (bulk) in the presence of COBF at 60 °C, [AIBN] = 2.93 × 10−3 mol L−1; (□) reactions carried out under light conditions, CT ≈ 5 × 103. (■) reactions carried out under dark conditions, CT ≈ 1 × 103. The microscopic (true) CT ≈ 8 × 103 for COBF in styrene at 60 °C. Data taken from ref. 78. | ||
The extent to which Co–C bond formation plays a role in the polymerization of several different monomers was investigated by NMR studies on a CoIIporphyrin (1 with R = R′ = H and R′′ = p-methoxyphenyl, (TAP)Co) in CDCl3 at 60 °C in the presence of the initiator 2,2′-azobis(isobutyronitrile) (AIBN) and various different vinylic monomers. The extent of Co–C bond formation was found to increase in the order: MMA ≪ EαHMA < methacrylonitrile (MAN) ≪ STY < cyclohexene ≪ methyl crotonate ≪ MA, vinyl benzoate, vinyl acetate (VAc), cis-2-pentenenitrile.101
Whereas this Co–C bond formation negatively affects the catalytic chain transfer ability of the CoII complexes in these systems (especially in the acrylates and VAc), the reversibility of this bond formation has been used successfully to induce living radical polymerization. First successes in this field were achieved for the living radical polymerization of acrylates. Harwood and co-workers successfully used cobaloximes in combination with light102 and Wayland and co-workers used hindered porphyrins at elevated temperatures.103–105 In more recent years, Jérôme and co-workers have been successful in using CoII complexes for the living radical polymerization of VAc.106 Albeit very interesting, living radical polymerization mediated by Co-complexes lies beyond the scope of the current review and we refer the reader to an excellent recent review on the topic.107
To conclude this discussion on Co–C bond formation, it is probably also interesting to note here that when comparing CT values for methacrylates and styrene in the literature, there is significantly more scatter in the styrene than in the methacrylate data. Large part of this scatter is caused by the use of varying initiator concentrations, light conditions and, what has not been mentioned so far, different conversions. The Co–C bond formation equilibrium takes time to establish itself and for a typical styrene polymerization it was found that a monomer conversion of about 5% was required for the equilibrium to be established. Hence, whereas one would normally wish to determine chain transfer constants at monomer conversions as low as possible, in the case of styrene it is actually better to do this at 5%.58,82
Returning now to CCT in methacrylates, where there is no significant Co–C bonding, it is probably safe to state that the true rate coefficient for chain transfer is given by ktr = CT × kp. When substituting CT ≈ 104 and kp ≈ 103 dm3 mol−1 s−1 one obtains a typical value of 107 dm3 mol−1s−1 for ktr which is of the same order as the rate coefficient for termination kt, which is known to be diffusion controlled. This fact, together with the consistently lower CT values for larger cobaloximes,55,56activation energies for transfer close to those of termination,21,54,55,60 led to the conclusion that CCT may also be diffusion-controlled.55,56 Studies carried out to investigate whether CCT is indeed diffusion-controlled clearly showed that there was no dependence on the bulk viscosity60,67 of the medium, but a dependence on the monomer viscosity (ηmon) was found (eqn (6)), suggesting that there is an effect of the microviscosity.55
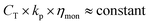 | (6) |
Except in monomer systems where other specific interactions may play a role (such as coordination of the hydroxyl group in hydroxyethyl methacrylate, HEMA,61 or very hindered monomers such as dimethyl itaconate,66 DMI), the relationship of eqn (6) seems to hold, as clearly illustrated by the entries collected in Table 2.
| Monomer | R | C T k p η mon | Reference system | C T k p η mon | Ref. |
|---|---|---|---|---|---|
| EMA | Me | 9.6 × 106 | MMA | 1.0 × 107 | 55 |
| BMA | Me | 9.5 × 106 | MMA | 1.0 × 107 | 55 |
| EMA | Ph | 6.3 × 106 | MMA | 5.9 × 106 | 55 |
| BMA | Ph | 6.0 × 106 | MMA | 5.9 × 106 | 55 |
| POEMA | Ph | 4.8 × 106 | MMA | 5.9 × 106 | 70 |
| BzMA | Me | 1.2 × 107 | MMA | 1.0 × 107 | 67 |
| 2EHMA | Me | 1.5 × 107 | n-BMA | 1.4 × 107 | 60 |
| t-BMA | Me | 6.7 × 106 | n-BMA | 9.5 × 106 | 66 |
| HEMA | Me | 4.3 × 106 | MMA | 1.0 × 107 | 61 |
| DMI | Me | 0.4 × 106 | MMA | 1.0 × 107 | 66 |
The fact that the chain transfer activity of cobaloximes in methacrylate polymerizations seems to only depend on monomer viscosity (i.e., the microviscosity108–111 as also used in the theory of viscoelasticity) and not on bulk viscosity implies that changing solvents should not change the catalytic activity. This is in general indeed the case for not strongly coordinating solvents (see above) and any possible solvent effects in these systems are most likely due to impurities in the solvent that may poison the catalyst.59 There seems to be only one exception to this rule and that is when supercritical CO2 (scCO2) is used as the solvent.62,64,112 In this very low-viscosity medium the measured chain transfer constants are much higher than in bulk or common solvents and with only small effects on kp this means that ktr is much higher.
To conclude this section on the mechanism of CCT we wish to spend a few words on a still unsolved issue. It is well-established that the process is catalytic and that the catalyst does not end up as forming part of the product polymer chain (as do, for example, initiator fragments and common chain transfer agents). This means that according to eqn (2), the DPn should decrease with increasing conversion (as [Co]/[M] increases), but in practice an almost unchanging MWD or only very small changes are observed during the polymerization. To the best of our knowledge there still only exists a single study which indeed shows the behaviour as predicted in eqn (2) and this study used a CoIII–R catalyst.113 One has speculated much as to why the predicted behaviour in general is not observed, e.g., higher viscosities decreasing CT or catalyst poisoning decreasing [Co].82 We could spend many words on this issue here, but the simple fact is that it is not clearly known why the evolution of the MWD does not follow eqn (2). It should be noted, however, that the large kinetic modelling study of Nikitin et al.,90 as already mentioned above, was able to adequately describe the experimental molecular weight results as well (but also this study suggests that the process is far more complex than what would be expected from eqn (2)).
Catalytic chain transfer copolymerization
Thus far we have described kinetic and mechanistic aspects of CCT in free-radical homopolymerization, but in practice many polymers are made by copolymerization of two or more monomers, which complicates the system. In addition to the “traditional” copolymer composition and rate control in a free-radical copolymerization, catalytic chain transfer copolymerization (CCTcP) also aims at molecular weight, and potentially end-group control. The most important thing to realize in CCTcP is that the observed chain transfer constant (used for molecular weight control) is an average and depends on monomer feed composition,32,77,114,115 as illustrated in Fig. 2 for the COPhBF-mediated CCTcP of MMA and STY, where the average chain transfer constant 〈CT〉 decreases from its value in the homopolymerization of MMA to that in the homopolymerization of STY.32 This dependence of 〈CT〉 on monomer feed composition implies that a significant composition drift in the copolymerization may automatically lead to a significant broadening of the MWD.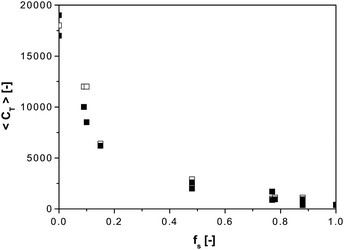 | ||
| Fig. 2 Average chain transfer constants in the COPhBF-mediated chain transfer copolymerization of styrene and methyl methacrylate at 40 °C. Closed symbols: determined via the CLD method using ΛP, open symbols: determined via the Mayo method using Mw/2. Data taken from ref. 32. | ||
The average chain transfer constant as shown in Fig. 2 is defined as the ratio of the average chain transfer rate coefficient and the average propagation rate coefficient (eqn (7)), which depend on the relative contributions of the reactions of the two different radicals (in the terminal model formulation) to the overall chain transfer and propagation rates, respectively.32,77,114,115
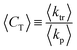 | (7) |
The average chain transfer rate coefficient in a CCTcP of monomers A and B can then be expressed in terms of the mole fractions of radicals ∼A• and ∼B• (resp. ϕA and ϕB) and the transfer rate coefficients of these radicals to CoII (resp. ktr,A and ktr,B):32,77,114,115
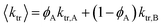 | (8) |
Hence, in order to predict the average chain transfer constant, it is necessary to predict the average propagation rate coefficient and via the relative radical concentrations, the average chain transfer rate coefficient. This is not a trivial exercise (not even in a conventional radical copolymerization) and is still subject to debate.114 The earliest attempts to model CCTcP32,77,114,115 used a very simple, qualitatively insightful approach to predict average chain transfer constants and the relative fractions of endgroups. This latter parameter is very important in the copolymerization of methacrylates (or α-methyl styrene) with styrenes or acrylates, as the endgroups resulting from the methacrylates are more accessible for post-polymerization reactions than those of the styrenes or acrylates (see Scheme 2). The simple models did a reasonable job in qualitatively describing the available experimental data on endgroups,116,117 but failed in quantitative predictions. One of the reasons lies in the fact that these models ignored Co–C bond formation (its importance was not realized until a few years later), which was later included in the models of Pierik et al.53,118 Two further oversimplifications in the models were the assumptions of a simple steady-state of propagating radical concentrations and the validity of the long-chain approximation, either of which were shown not to be applicable.119 A more elaborate kinetic model, able to quantitatively describe the experimental results, has been published by Moad and co-workers.119 Finally a very extensive modelling study by Nikitin et al.90 was able to describe the polymerization rates and MWDs of a copolymerization well, but no results on endgroups were reported.
Catalytic chain transfer in emulsion polymerization
Most of the earlier studies on CCT have been reported for solution or bulk polymerization, and indeed CCT has been used industrially in solution applications. Many industrial free-radical polymerization processes, however, are carried out in dispersed systems, most notably in suspension and emulsion. These dispersed systems offer the advantage of high rates of polymerization and a low overall viscosity in an environmentally benign solvent (water). It is therefore not surprising that there is a larger industrial interest in applying CCT in dispersed systems, especially in emulsion polymerization. When applying CCT in emulsion polymerization, one commonly observes an apparent lower catalytic activity of the catalyst or, better formulated, one obtains higher molecular weights than what would be expected on the basis of the Mayo equation (eqn (2)) using the overall Co-concentration. Furthermore, the efficiency of the chain transfer process and the observed polymerization kinetics were found to depend greatly on the process conditions and the used catalyst. These observations and the industrial interest have sparked academic interest and most of the more recent publications on CCT deal with its application in emulsion polymerization.In emulsion polymerization a sparingly water soluble monomer is dispersed in a continuous aqueous phase containing initiator and surfactant. The surfactant stabilizes the relatively large monomer droplets (∼1 μm) which act as monomer reservoirs and forms monomer swollen micelles (∼10 nm), the loci of polymerization. Radicals generated from the decomposition of the initiator in the aqueous phase nucleate the monomer swollen micelles, forming polymer particles (∼50 to 400 nm). During the polymerization, the polymer particles continuously capture radicals from the aqueous phase by radical entry and lose radicals to termination and desorption to the aqueous phase. The radical concentration is expressed as the average number of radicals per particle (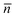 ). This compartmentalization behavior of radicals in emulsion polymerization ultimately results in high rates of polymerization and high molecular weights. The mechanism of miniemulsion polymerization is very similar with the main exception that small monomer droplets (∼100 to 300 nm) are generated and subsequently converted into polymer particles.
). This compartmentalization behavior of radicals in emulsion polymerization ultimately results in high rates of polymerization and high molecular weights. The mechanism of miniemulsion polymerization is very similar with the main exception that small monomer droplets (∼100 to 300 nm) are generated and subsequently converted into polymer particles.
Catalytic chain transfer was first introduced in a dispersed system roughly 10 years after its initial discovery.17 Initial studies on the application of catalytic chain transfer in emulsion polymerization showed that low molecular weight polymer could readily be obtained in seeded emulsion,120 semi-batch emulsion120–123 and miniemulsion polymerization124 using COBF or COPhBF. As stated above the observed chain transfer activity in emulsion polymerization is lower than that in bulk and solution polymerization,120–123 and the reduction in molecular weight comes at the expense of a decreased rate of polymerization.120–125 Both these observations can be explained by the fact that the catalysts are present in both the dispersed and the aqueous phases.
In a dispersed system, the presence of the catalyst at the locus of polymerization is required to achieve proper control over the molecular weight distribution. Mass transport of the catalyst between monomer droplets, aqueous phase and polymer particles is therefore a prerequisite. The most commonly used catalysts possess some solubility in both the aqueous and dispersed phase and will consequently display partitioning.120,122,123 The extent to which catalyst partitioning occurs depends both on the catalyst structure and the hydrophobicity of the monomer, see Table 3.
| Complex | R | Monomer | m Co a | Ref. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a m Co is the partition coefficient. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| COBF | Me | MMA | 0.31–0.72 | 60,120,123 and 126 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMA | 0.035 | 127 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| STY | 0.052 | 128 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| COEtBF | Et | MMA | 19–60 | 60 and 129 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMA | 8.4 | 127 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| COPhBF | Ph | MMA | ∞ | 60 and 120 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The partition coefficient (mCo) is given by eqn (9),
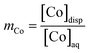 | (9) |
As is clear from the data in Table 3, a significant part of the added amount of catalyst resides in the aqueous phase, especially when considering COBF. This means that the actual amount of catalyst at the locus of polymerization (i.e., the concentration that should really be used in eqn (2)) is lower than the overall concentration. Hence any data analysis (or prediction of DPn) based on eqn (2) and the overall catalyst concentration should use an apparent chain transfer constant (CTapp), similar to what was done above when discussing the chain transfer constants observed in styrene.123 Considering that the overall chain transfer frequency is determined by the product of the chain transfer constant and the catalyst concentration at the site of polymerization (i.e., CT[Co]actual = CTapp[Co]0), there is a significant effect of catalyst partitioning—captured in the partition coefficient (mCo) and the phase ratio (β; the volumetric ratio of dispersed and aqueous phase)—on the observed CTapp. A relatively simple modification of the Mayo equation (eqn (2)) taking these effects into account was derived for relating the number-average degree of polymerization (DPn) to the overall catalyst concentration ([Co]) and the true (microscopic) chain transfer constant (CT; this value may actually be different to the bulk value because of axial ligand exchange in water):126
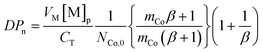 | (10) |
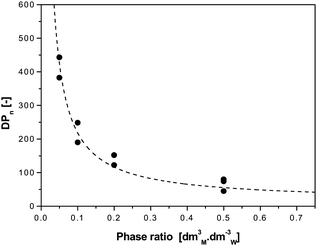 | ||
Fig. 3 Effect of CCTA partitioning on the number-average degree of polymerization of COBF-mediated miniemulsion polymerizations of MMA with varying monomer to water ratios. (●) Experimentally observed DPn and (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) theoretical fit using eqn (8) and mCo = 0.72 and CT = 15 × 103. Data taken from ref. 126. ) theoretical fit using eqn (8) and mCo = 0.72 and CT = 15 × 103. Data taken from ref. 126. | ||
Although partitioning appears to be a dominant factor for the lower observed chain transfer activities in dispersed media, aqueous phase deactivation and mass transport limitations can also contribute to lower CTapp values. While the introduction of BF2-bridges significantly increases the stability of compound 3 as compared to compound 2, the CoII complexes remain susceptible towards oxidation by oxygen26,27 or (peroxide) radicals12,26,101 and hydrolysis.26 Although catalyst deactivation in the aqueous phase cannot be excluded, the effects can be minimized by working at basic pH,128oxygen-free conditions and circumventing the use of peroxide initiators.124 Furthermore, a continuous feed of the CCTA could allow for maintaining a constant chain transfer activity throughout the course of the polymerization,123 preventing an increase in DPn and a consequent broadening of the MWD.
The rate of mass transfer in CCT mediated polymerizations in a dispersed phase is important as these polymerizations typically proceed in a regime where there are less catalyst molecules than polymer particles (10−2 ≤ NCo/Np ≤ 10−1). This implies that mass transport has to be sufficiently fast to allow a single catalyst molecule to mediate the MWD in multiple polymer particles. Controlling the viscosity of the polymer particles has proven to be crucial to allow for effcient CCT in emulsion polymerization. In semi batchemulsion polymerization monomer-flooded conditions have to be maintained throughout the course of the polymerization. Once the polymer particles become starved for monomer their glass transition temperature (Tg) increases and the particles become glassy. It is assumed that this restricts the mobility of the CCTA which causes a decrease in the observed chain transfer activity and broadens the MWD.120,121,123 Efficient CCT conditions can be achieved by lowering the Tg of the polymer particles by copolymerizing low Tg monomers such as BMA120 or by adding an initial shot of monomer prior to the monomer feed.121 However, as the presence of a CCTA causes a reduction in the rate of polymerization it auto-enhances its activity by plasticizing the polymer particles. In ab initio (mini)emulsion polymerization where the viscosity in the polymer particles is relatively low, monomodal MWDs are observed with a DPn that can be predicted by the modified Mayo equation (eqn (10)).126,130 The dispersed phase can be regarded as a continuous phase with respect to CCT, where the MWD is mediated by an average number of catalyst molecules per particle, i.e. DPn ∝ [Co]p ∝ NCo/Np.131 Incidentally, in the early stages of the polymerization the MWDs from CCT mediated emulsion polymerizations can display the presence of a polymer population with a DPn significantly higher than expected based on eqn (10).120,130 This suggests a mass transport limitation as the ratio of catalyst molecules to monomer swollen micelles is far below unity (NCo/Np < 10−3). Under these conditions, two consecutive radical entry events may occur prior to the entry event of a catalyst, resulting in a conventional free radical polymerization within that polymer particle.
Mass transfer limitations also arise when COPhBF is used to mediate a dispersed phase polymerization.132 This catalyst is extremely hydrophobic and has no detectable water solubility.120,126 Mass transport of this catalyst cannot proceed via the aqueous phase and is thought to occur through collisions between e.g. two polymer particles.127 As exchange of COPhBF molecules can only occur when in the proximity of the surface of a monomer droplet or polymer particle, a fraction of the CCTA located in the bulk of the monomer droplets will be unavailable for mass transport. Consequently the dispersed phase cannot be regarded as one continuous phase with respect to CCT. Mass transport of COPhBF is hampered in the initial stages of the polymerization up to the disappearance of the monomer droplets, resulting in an continuous decrease in the instantaneous DPn to the expected DPn value based on eqn (10).132 Despite the existence of mass transport limitations in the early stages of the polymerization, proper molecular weight control using COPhBF is possible and possibly even preferred due to the reduced impact on the polymerization kinetics.
The increasing viscosity of the polymer particles also affects the activity of the catalyst. The CLD method was used to determine the chain transfer activity of COBF as a function of the polymer weight fraction in seeded emulsion polymerization.133 The results showed that the value of ΛP (see eqn (3)), which reflects the value of CT[Co]p/[M]p, decreases as the conversion increases.133 The diffusion coefficient of low molecular weight species, such as COBF, decreases strongly with increasing polymer weight fraction. It is therefore not unlikely that this could ultimately affect the value of CT. Although there is no hard evidence for diffusion-controlled rate-determining chain transfer reaction, these results seem to suggest that this might be the case.
High viscosity of the polymer particles can also severely limit the exchange of catalyst molecules, resulting in compartmentalization of the CCTA.131 Using COBF in seeded emulsion polymerization resulted in multimodal MWDs which are attributed to a statistical distribution of CCTA molecules over the polymer particles. Polymer particles growing in the presence of 0, 1, 2, …, n CCTA molecules (nCo) will exhibit MWDs with different average molecular weights, depending on the number of catalyst molecules, i.e. DPn ∝ [Co]p ∝ nCo = 0, 1, 2, 3, … Population balance calculations on the effects of CCTA compartmentalization on the MWD illustrated that diffusional resistance (i.e. reduced entry and desorption of the CCTA from the polymer particles) resulted in multimodal MWDs.134 Both the experimental and theorical results suggest that there are two limiting cases in CCT-mediated emulsion polymerization: one at low particle viscosity where the MWD is governed by a global CCTA concentration and one at high particle viscosity where the MWD is governed by a discrete distribution of the CCTA.
The control of the MWD in CCT-mediated polymerizations in dispersed media affects the rate of polymerization and the final latex properties in terms of the particle size distribution (PSD) and the final particle number (Np). Fig. 4 presents an example of the effect of increasing levels of a CCTA on the conversion-time history in an MMA emulsion polymerization. The rate of polymerization decreases as the COBF concentration is increased from 0.5 to 10.0 ppm, illustrating the effect of a CCTA on the emulsion polymerization kinetics. Although the addition of a CCTA affects the emulsion polymerization process, it has been established that for CCT mediated emulsion polymerization the classical theoretical framework can be used to describe the polymerization kinetics.130
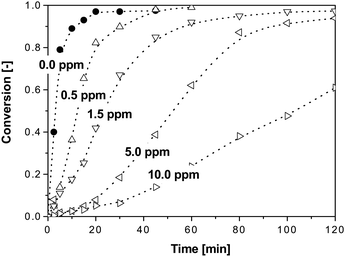 | ||
| Fig. 4 Conversion-time histories for COBF-mediated emulsion polymerizations of MMA. Data taken from ref. 130. | ||
An overview of the effect of a CCTA on the emulsion polymerization kinetics is presented in Scheme 4. It was immediately recognized that monomeric radicals, originating from the CCT process, can readily desorb from the polymer particles,120 lowering and the volumetric growth rate of the polymer particles (see Scheme 4).135 The extent to which radical desorption (exit) occurs depends on the water-solubility of the monomer and the chain transfer activity in the polymer particles, which can be expressed as the chain transfer frequency (ftr ∝ CT[Co]p).135 Depending on the partitioning behavior of the used catalyst the aqueous phase kinetics can also be affected. Water-soluble oligomeric radical species propagating in the aqueous phase can undergo chain transfer prior to entry into a polymer particle, lowering ![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) and the rate of particle nucleation.135 However, experimental results suggest that the rate of radical desorption is the main kinetic event in CCT mediated emulsion polymerizations.135 The presence of a catalyst affects both and Np thereby lowering the rate of polymerization, the magnitude of which is governed by the partitioning behavior of the catalyst. However, as the production of monomeric radicals is inherent to the CCT process lower rates of polymerization are characteristic to the CCT process.
and the rate of particle nucleation.135 However, experimental results suggest that the rate of radical desorption is the main kinetic event in CCT mediated emulsion polymerizations.135 The presence of a catalyst affects both and Np thereby lowering the rate of polymerization, the magnitude of which is governed by the partitioning behavior of the catalyst. However, as the production of monomeric radicals is inherent to the CCT process lower rates of polymerization are characteristic to the CCT process.
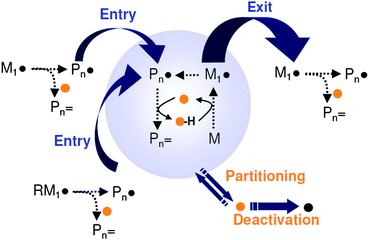 | ||
| Scheme 4 Effect of a catalytic chain transfer agent on the emulsion polymerization kinetics. | ||
Applications of catalytic chain transfer
Considering the fact that CCTP follows conventional radical polymerization kinetics, small amounts of chain transfer agents are required and existing industrial equipment remains adequate when changing over to CCTP, it is only logical that CCT has found its use in applications requiring low molecular weight and/or vinyl endfunctional polymers. Polymers synthesized from CCTP have found direct use in thermoforming sheets for mould manufacturing136 and as additives for road pavement manufacturing.137Most notable is its application in the production of low-VOC high solids coatings, especially in the automotive industry.138 In these applications a functional monomer, such as hydroxyethyl methacrylate (HEMA), is copolymerized to provide functional groups that can be used for crosslinking reactions after application of the coating.138,139 Furthermore, the CCT-derived dimer of HEMA can be used to introduce hydroxyl groups in the α and ω endgroups of an acrylic polymer by copolymerizing it with other methacrylates (see below), so ensuring that at least two reactive groups are present in each chain.138,140
For applications in which specific ω-endfunctional polymers are required, one can use a suitable copolymerization in which one monomer is highly reactive towards propagation and not towards chain transfer and a second monomer which readily undergoes chain transfer, but not propagation.32,53,77,115,119,141 Comonomer pairs that fulfill these criteria are for example MA/MMA53 and STY/AMS.77 A very specific end-group that can be introduced is the aldehyde end-group (i.e., a versatile synthon in synthetic chemistry), which can be introduced directly via a tautomeric rearrangement of the endgroup that is produced in the polymerization of α-hydroxymethyl-substituted monomers (Scheme 5), such as ethyl-α-hydroxymethacrylate (EαHMA, 4)69 and α-hydroxymethyl styrene (PAA, 5).79 We will briefly discuss an alternative way of introducing an aldehyde endgroup in the final section of this review.
When polymerizing α-hydroxymethyl-substituted monomers, abstraction of a hydrogen atom from the terminal of the α-hydroxymethyl group in the propagating radical leads to an enol endgroup which rearranges to an aldehyde (see Scheme 6). This process has been termed CCT isomerism by Gridnev.1
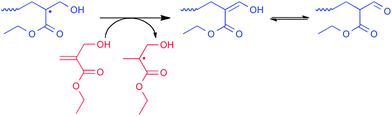 | ||
| Scheme 6 Catalytic chain transfer isomerism of EαHMA.69 | ||
The introduction of specific endgroups in both the α and ω positions can be achieved by using an unreactive functional olefin as described by Gridnev and co-workers.142 When a functional olefin which is normally considered as unreactive in free-radical polymerization is added to a CCTP of a “normal” monomer, then predominantly a polymer is produced with the initiating (i.e., α) endgroup being the unreactive olefin and the remainder of the chain, including the unsaturated ω-endgroup, being monomer units. Used non-polymerizable olefins are 2-pentenenitrile, 2-cyano-2-butene, crotonaldehyde, dimethyl maleate, ethyl crotonate and cyclopentene-1-one. α,ω-Difunctional polymers can also be produced via the addition–fragmentation mechanism as described in the following section.140,143
Another direct application of CCTP to produce functional polymers is found in the area of hyperbranched polymers as dendrimer analogues. These polymers can be produced by addition of a CCTA to the free-radical polymerization of dimethacrylates. The first report of branched polymers was reported by Guan144 on the homopolymerization of ethylene glycol dimethacrylate (EGDMA) in the presence of a CCTA. Appropriate levels of CCTA yield trimerization144,145 of dimethacrylates which, if followed by repetative trimerization, ultimately results in the formation of hyperbranched polymers. The branched polymers formed by this “cascade polymerization” process not only possess properties that resemble those of dendrimers but also offer the benefit of being synthesized in an industrially viable way.
This method has been further explored in the groups of Sherrington, Haddleton and Estrina by extending the cascade polymerization process to copolymerizations of different vinyl monomers to enhance or decrease the level of branching. Sherrington and co-workers copolymerized vinyl monomers and cross-linkers in the presence of a chain transfer agent, a process which is often referred to as the “Strathclyde methodology”.146 A balanced amount of cross-linker and chain transfer agent prevents macrogelation and in turn introduces control over the degree of branching to the polymer architecture. Costello et al. reported the first use of CCT for polymer architecture control in a methyl methacrylate (MMA)—tripropylene glycol diacrylate (TPGDA) solution copolymerization.147 When compared to conventional chain transfer agents, synthesizing hyperbranched polymers using CCT as the chain transfer chemistry has the advantage that only minor amounts of CCTA are required and that no adverse organic functionalities are incorporated into the polymer backbone.147 The potential of CCT for the synthesis of hyperbranched polymers was further explored by Camerlynck et al.148 (COPhBF), Haddleton145,149 (COBF) and Kurmaz et al.150–155 (cobalt(II) tetramethyl hematoporphoryn-IX). Although the majority of the reports on architecture control using CCT to date have focussed on methacrylates, the viability of this method has also been estabilished for styrene150 and arcylates.154 A further advantage of using CCT for the synthesis of branched polymers is that significant amounts of unsaturated vinyl end-groups remain in the polymer structure.147,148,150,151 Haddleton et al.145,149 utilized the pendant vinyl unsaturations of a hyperbranched poly(EGDMA) to obtain multi-arm star block copolymers. Hereto, hyperbranched poly(EGDMA) polymer was end-functionalized via a thiol-Michael addition.145,149 CCT has proven to be a very attractive chemistry for the synthesis of hyperbranched polymers, especially as interest in the synthesis of responsive polymer hyperbranched materials has recently emerged.156,157
Interesting applications of CCT in atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer (RAFT) have also been reported. In these studies, a CCT agent is added at the end of the ATRP158 or RAFT polymerization159 in order to remove the halogen (in ATRP) or RAFT endgroups and to introduce the typical methacrylic vinyl endgroup (see Scheme 7). This has the benefits of replacing the fairly unstable RAFT endgroup by a more stable one and, if necessary, allows for the synthesis of macromonomers with a narrow molecular weight distribution (reported PDIs lie in the range of 1.2–1.5); macromonomers prepared via “normal” CCT typically have a PDI = 2.
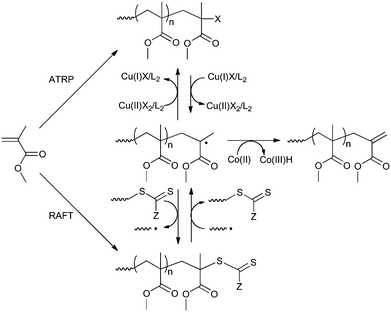 | ||
| Scheme 7 CCT conversion of ATRP and RAFT polymers to methacrylic macromonomers (example shown for MMA). | ||
CCTP has also been used directly in heterogeneous applications. Macromonomers containing both hydrophobic and hydrophilic groups have been used as pigment dispersants160 and reactive surfactants in emulsion polymerization.160–162CCTP has also been used successfully for the production of inorganic–organic hybrid nano-composites (nanosilica,163 semi-conductors164 and photonic crystals165) and sediment-free toner for electrographic imaging and printing processes.166
The use of CCT agents has also been very useful in the production of concentrated translucent nanolatexes.167 Typically latexes with particle sizes in the range of 20 to 50 nm are prepared from microemulsion polymerization, which suffers from the drawbacks that large amounts of surfactant are required and that the polymer to surfactant ratio is relatively low. The potential of CCT to enhance the rate of radical desorption (exit) can be utilized to synthesize nanolatexes from emulsion polymerization. COPhBF-mediated emulsion polymerization of MMA, BMA and STY resulted in 20 nm nanolatexes which are translucent in appearance.167 The synthesis of these nanolatexes requires 8 wt% surfactant (with respect to the amount of monomer), which is substantially less than the >100% which is used in microemulsion polymerization. Furthermore, the polymer concentration could readily be increased to 40 w/w% without compromising the properties of the nanolatexes. The optical transmittance of the nanolatexes was correlated to the particle size distribution using Mie theory.
Finally, the most important application of CCTP is probably the production of functional macromonomers, which are used in a second polymerization step. Their polymerization chemistry is quite complex and will be treated in more detail in the following section.
Radical polymerization of macromonomers
Initial studies on CCT-derived methacrylic macromonomers indicated that in a copolymerization with (other) methacrylic comonomers they acted as chain transfer agents22,140,143,168,169 and could even induce a living-like polymerization.22,170 With acrylates or styrenic monomers the macromonomers were found to copolymerize leading to graft copolymers.15,171 Both reactions have since been exploited extensively, but, as we will show below, the copolymerization behaviour with acrylates and styrene is much more complicated than suggested by the early studies and this is still not widely appreciated.172,173The chain transfer mechanism that is operative in a copolymerization of macromonomers with methacrylates involves a (reversible) addition and fragmentation sequence as shown in Scheme 8 for the polymerization of BMA in the presence of a PMMA macromonomer. Upon the addition of a propagating methacrylate radical to the macromonomer double bond, a relatively unreactive intermediate radical is formed, which subsequently undergoes a β-scission reaction resulting in a new propagating radical and a new macromonomer. The net result of this addition–fragmentation chain transfer (AFCT) process is the transfer of the unsaturated ω-endgroup from one chain to the other.
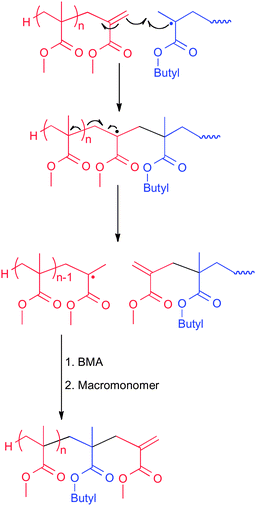 | ||
| Scheme 8 Addition–fragmentation chain transfer leading to telechelic (n = 1) and block (n > 1) copolymers in the free-radical copolymerization of butyl methacrylate and an MMA-derived macromonomer. | ||
The resulting radical after the AFCT step will add to the “second stage” monomer and will keep growing until it undergoes an AFCT reaction sequence (or terminates with other radicals). When the starting macromonomer is a dimer (n = 1 in Scheme 8), then the resulting polymer is a telechelic polymer, as clearly demonstrated by Haddleton and co-workers.140,143 In the case of larger macromonomers, block copolymers will result as reported by Moad and co-workers, who demonstrated that by using low monomer concentrations it is possible to achieve low PDI and a linear increase in Mn with conversion.21,170 In fact, this was one of the early demonstrations of what later was termed Reversible Addition–Fragmentation chain Transfer (RAFT), and which is more effectively carried out using di- or trithioesters.174 Whereas these latter compounds induce living-like behaviour by increasing the chain transfer rate over propagation via a high chain transfer constant, the use of macromonomers (with CT ≈ 0.04)169 requires low monomer concentrations to reduce propagation relative to chain transfer.21
A relatively straightforward application of this AFCT chemistry is the use of α-methyl styrene dimer as a chain transfer agent in free-radical polymerization.175,176 Examples of this include its use in the manufacturing of polystyrene,177 dental applications,178 paper production,179food packaging180 and the production of poly(bromo-styrene) as a flame retardant.181
As stated above, copolymerization with styrenes and acrylates leads to graft copolymers and applications have been found in several different areas. As one example can be mentioned the copolymerization of hydrophobic PMMA macromonomers with hydrophilic N-vinyl pyrrolidone182 and N,N-dimethyl acrylamide183 to form self-reinforcing hydrogels for contact lens applications. The PMMA-segments impart the mechanical strength to these hydrogels with very high equilibrium water content. Another example is the copolymerization of macromonomers with diacrylates, which leads to star polymers184,185 with potential applications as rheology modifiers, tougheners in plastics and adhesives.184
The main application of this chemistry can be found in the area of waterborne coatings. Here, the applications have been two-fold, firstly as polymeric stabilizers160,161 and secondly as polymeric binders with improved properties.186–189 In the first case, water-soluble or amphiphilic block copolymers are used as so-called surfmers190 (i.e., surfactants with a monomer functionality) in emulsion polymerization, leading to latex particles stabilized by covalently attached polymeric (or oligomeric) surfactants. In the second case, a macromonomer seed is prepared by emulsion polymerization in the presence of a catalytic chain transfer agent, as discussed in detail in a previous section. This seed is used in a second stage semi-batch emulsion polymerization where an acrylate is added. This procedure then leads to a polymer latex consisting of graft copolymers which may form microdomains containing different glass transition temperatures than the surrounding matrix. Final (often improved) coating properties were found to depend on the macromonomer chain lengths.
Although this graft copolymerization has already found industrial use and commercial products exist on this basis, it is highly unlikely that these products are as well-defined as the reported studies suggest. From detailed studies on the reactivity of the macromonomer double bond by Yamada and co-workers, it can be concluded that the graft copolymers mentioned above are not likely to consist of an acrylate backbone with methacrylate grafts, but are much more complex.172,173
Yamada and co-workers showed that the polymerization behaviour of the terminal double bond highly depends on the penultimate monomer unit in the macromonomer (X in the structures in Scheme 9). An all-methacrylic macromonomer, i.e., with a methacrylic penultimate unit, almost exclusively undergoes an AFCT process irrespective of the attacking radical, whereas only those macromonomers with an acrylate penultimate unit undergo a true copolymerization reaction with an attacking acrylate radical. These results have been summarized in Table 4.173
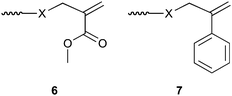 | ||
| Scheme 9 Schematic representation of MMA and AMS derived macromonomers. The nature of the penultimate group X determines their copolymerization behaviour. | ||
Considering the results shown in Table 4, a more likely polymerization behavior of an acrylate in the presence of a methacrylate macromonomer is shown in Scheme 10, where MMA and BA were taken as examples.
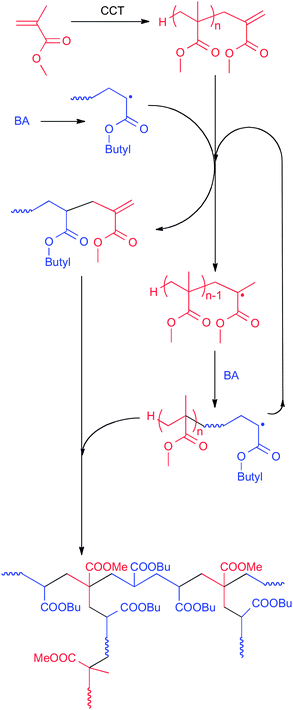 | ||
| Scheme 10 Probable mechanism of the graft copolymerization of butyl acrylate and MMA-derived macromonomer, based on the data in Table 4. | ||
First all of the all-methacrylate macromonomers will undergo an AFCT reaction with an attacking BA radical (which may or may not contain a PMMA block) converting this radical into a macromonomer with a BA penultimate unit, which will subsequently undergo a copolymerization reaction with another BA radical. When considering the mechanism of Scheme 10, one would expect the final polymer structure to also depend strongly on the second-stage polymerization conditions (e.g., whether the reaction is carried out in batch or a feeding strategy is used—and with what feed rates).
For better defined grafts one should therefore have macromonomer 6 or 7 with X = acrylate. Since the terminal group is derived from a methacrylate (or AMS), this means that such macromonomers need to be synthesized via a two-step procedure, for example by one of the methods as schematically shown in Scheme 7, or via an appropriate copolymerization in which the dominant terminal unit in the radical is the methacrylate or AMS (see above).115 We previously studied this second route via a COBF-mediated copolymerization (Scheme 11) at 125 °C of butyl acrylate (BA) with either benzyl methacrylate (BzMA) or AMS as the CCT-active comonomer.141 In both cases, NMR and MALDI-TOF analyses showed that macromonomers with the desired endgroups were produced, but the copolymerization with AMS yielded macromonomers containing 1–3 AMS units exclusively with AMS endgroups (8), the copolymerization with BzMA yielded a larger variety of macromonomers (9a and 9b), including those with the undesired (because unreactive) acrylate endgroup (9b). These results can be explained using simple kinetic arguments based on reactivity ratios and chain transfer constants.
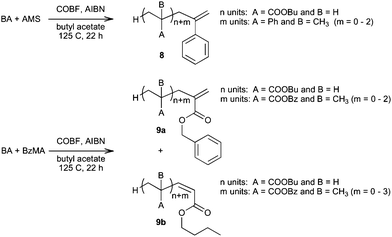 | ||
| Scheme 11 COBF-mediated copolymerization of butyl acrylate with α-methyl styrene or benzyl methacrylate in butyl acetate and resulting macromonomers.141 | ||
An alternative route to the desired macromonomers has been used by Yamada and co-workers who also successfully exploited the route of using dimeric macromonomers as AFCT agent in the polymerization of acrylates.191–194
The final procedure for the preparation of methacrylic macromonomers with acrylic penultimate unit (i.e., 6 with X = acrylate, Scheme 9) which we will describe has been explored by the CSIRO team194–196 and Barner-Kowollik and co-workers.197 These groups have exploited the fact that the mid-chain radicals that are formed in the free-radical polymerization of acrylates (and which are responsible for branching in polyacrylates) can undergo a β scission resulting in the (for this purpose) desired macromonomer as schematically shown in Scheme 12.
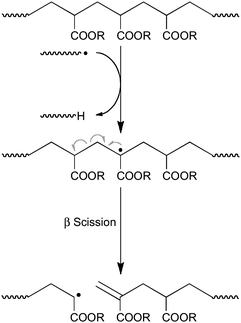 | ||
| Scheme 12 Formation of methacrylic macromonomers with acrylic penultimate units in high temperature acrylate polymerization. | ||
Typically, this process involves the free-radical polymerization of virtually any acrylate194–197 at temperatures between 80 and 240 °C, preferably using a low radical flux,196 yielding macromonomer purities typically greater than 90%.194–197 In this process, the DPn of the macromonomers can be controlled by temperature and monomer concentration.195 Under otherwise unchanged conditions, an increase in temperature reduces DPn as an increase in temperature increases the formation of mid-chain radicals that can undergo β-scission (Scheme 12) and an increase in monomer concentration (increasing the propagation rate relative to chain stopping rates) causes an increase in DPn.
Post-polymerization modification of macromonomers
Recently, an increasing number of studies has been published in which the unsaturated endgroup of the macromonomers has been used as a reactive endgroup in other reactions than second-stage radical polymerizations. One of the first examples to be published was the use of the macromonomer endgroup in thiol–ene “click” reactions. Thiol–ene addition reactions can be performed via two routes, e.g. anti-Markovnikov radical addition or base/nucleophile catalyzed Michael addition. The Michael addition reaction typically is the method of choice as it requires mild reaction conditions and the end-functionalized polymer is obtained in high yield with minimal byproduct formation.198 Different catalysts, primary and tertiary amines and phosphines, can be used as well as a range of different thiols to synthesize well-defined block copolymers.198 A schematic representation of the thiol-Michael functionalization of macromonomers is shown in Scheme 13.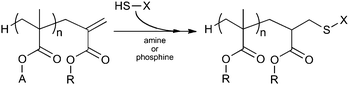 | ||
| Scheme 13 Thiol-Michael addition to methacrylic macromonomer. | ||
Haddleton and co-workers have used the thiol-Michael addition to end-functionalize CCT-derived branched PEGDMA polymers with an alkylthiol to form multi-arm star block copolymers.149 The thiol–ene addition reaction was catalyzed by dimethylphenylphosphine (DMPP) and complete conversion of the pendant double bounds was achieved in 1 h.149 This post-modification has been futher utilized to end-functionalize both branched PEGDMA149,199,200 and linear methacrylate macromonomers such as PMMA,198PHEMA,198M(EO)2MA,75,201 and PEGMA475,75,198 with a range of different functional groups. Nurmi et al. synthesized glycopolyers from a combination of thiol–ene Michael addition and Huisgens cycloaddition.202
Staying with the theme of “click” chemistry, a very versatile synthon and “clickable” group is the aldehyde group,203 which can be introduced as an endgroup viacatalytic chain transfer isomerism as already discussed above (see Scheme 6).69 Since this procedure requires the presence of an α-hydroxymethyl group in the monomer and not many commercial monomers exist with this group we used an alternative route to introduce this endgroup.204 The unsaturated endgroup of macromonomers can be converted into a pendant aldehyde group by performing rhodium catalyzed hydroformylation in supercritical carbon dioxide (Scheme 14). This synthetic pathway was used to synthesize PMMA and PSTY aldehyde end-functionalized polymers in high yields and high chemoselectivity, and the position of the double bond (internal or external as dictated by the CCT chemistry of STY and MMA) was found to have little effect on the chemoselectivity of the hydroformylation.204
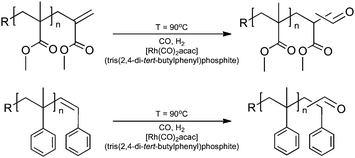 | ||
| Scheme 14 Synthesis of aldehyde end-functional polymersviahydroformylation. | ||
We finish this brief overview of macromonomer chemistry with a very recent example of converting methacrylic macromonomers into macro-initiators for the anionic polymerization of methacrylates. It was shown by Sanders et al. that after addition of α-lithioisopropylisobutyrate to a methacrylic macromonomer, a macro-initiator was formed capable of initiating MMA polymerization leading to a stereo-block copolymer of PMMA (Scheme 15).205
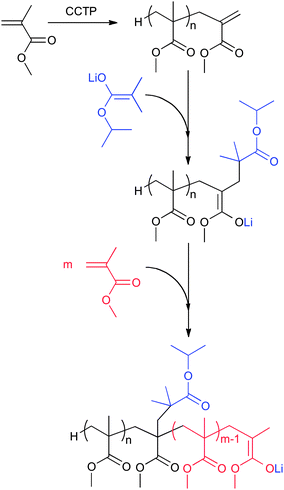 | ||
| Scheme 15 Schematic representation of the use of macromonomers as macro-initiators for the anionic polymerization of methacrylates. Example shows PMMA macromonomer in the anionic polymerization of MMA. | ||
Conclusions
Catalytic chain transfer polymerization is a very efficient and versatile free-radical polymerization technique for the synthesis of functional low molecular weight macromonomers, which have found applications in a wide range of fields. The mechanism of CCTP in both homogeneous and heterogeneous systems is largely known and so are the effects of the addition of a CCT agent on the overall polymerization kinetics of these systems. The derived macromonomers (which would normally have a polydispersity index of about 2) can be used in second stage radical polymerization leading to block or graft copolymers, depending on the nature of the macromonomer and the comonomers. For applications in which lower polydispersity indices of the macromonomers are required, performing ATRP or RAFT followed by CCT is a handy solution. Finally, an increasing number of studies is appearing in the literature in which the functional endgroups of the macromonomers are used for “click” reactions or modified for further reactions, including as macroinitiators for anionic polymerization.Acknowledgements
The authors gratefully acknowledge the pleasant collaborations and useful discussions with past and present co-workers, whose contributions can be found in the list of references. We wish to especially thank Tom Davis, Dave Haddleton and Alexei Gridnev whose numerous contributions to the field have been, and still are, invaluable.Notes and references
- A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753 CrossRef CAS.
- B. R. Smirnov, I. S. Morozova, A. P. Marchenko, M. A. Markevich, L. M. Pushchaeva and N. S. Enikolopyan, Dokl. Akad. Nauk SSSR, 1980, 253, 891 CAS.
- B. R. Smirnov, I. M. Belgovskii, G. V. Ponomarev, A. P. Marchenko and N. S. Enikolopyan, Dokl. Akad. Nauk SSSR, 1980, 254, 127 CAS.
- B. R. Smirnov, I. S. Morozova, L. M. Pushchaeva, A. P. Marchenko and N. S. Enikolopyan, Dokl. Akad. Nauk SSSR, 1980, 255, 609 CAS.
- B. R. Smirnov, A. P. Marchenko, V. D. Plotnikov, A. I. Kuzayev and N. S. Yenikolopyan, Polym. Sci. U.S.S.R., 1981, 23, 1168 Search PubMed.
- B. R. Smirnov, V. D. Plotnikov, B. V. Ozerkovskii, V. P. Roshchupkin and N. S. Yenikolopyan, Polym. Sci. U.S.S.R., 1981, 23, 2807 CrossRef.
- B. R. Smirnov, A. P. Marchenko, G. V. Korolev, I. M. Belgovskii and N. S. Yenikolopyan, Vysokomol. Soedin., Ser. A, 1981, 23, 1042 CrossRef CAS.
- N. S. Enikolopyan, B. R. Smirnov, G. V. Ponomarev and I. M. Belgovskii, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 879 CrossRef CAS.
- A. A. Gridnev, P. M. Bel'govskii and N. S. Enikolopyan, Vysokomol. Soedin., Ser. B, 1986, 28, 85 CAS.
- A. A. Gridnev, Polym. Sci. U.S.S.R., 1989, 31, 2369 CrossRef.
- A. Y. Nokel, A. A. Gridnev and A. F. Mironov, Izvestia Vissh. Uchebn.Zaved., Khim. Khim. Teknol., 1990, 33, 57 CAS.
- A. A. Gridnev, Polym. J., 1992, 24, 613 CrossRef CAS.
- G. M. Carlson and K. J. Abbey, US Pat., 4526945, 1985.
- J. C. Lin and K. J. Abbey, US Pat., 4680354, 1987.
- K. J. Abbey, D. L. Trumbo, G. M. Carlson, M. J. Masola and R. A. Zander, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3417 CrossRef CAS.
- A. H. Janowicz and L. R. Melby, US Pat., 4680352, 1987.
- A. H. Janowicz, US Pat., 4694054, 1987.
- D. M. Haddleton, A. V. G. Muir and S. W. Leeming, WO Pat., 95/17435, 1995.
- A. V. G. Muir, J. R. Lawson and D. M. Haddleton, WO Pat., 95/27737, 1995.
- A. F. Burczyk, K. F. O'Driscoll and G. L. Rempel, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 3255 CrossRef CAS.
- R. Amin Sanayei and K. F. O'Driscoll, J. Macromol. Sci., Part A: Pure Appl. Chem., 1989, 26, 1137 CrossRef.
- J. Krstina, C. L. Moad, G. Moad, E. Rizzardo, C. T. Berge and M. Fryd, Macromol. Symp., 1996, 111, 13 CrossRef CAS.
- L. V. Karmilova, G. V. Ponomarev, B. R. Smirnov and I. M. Bel'govskii, Russ. Chem. Rev., 1984, 53, 132 CrossRef.
- T. P. Davis, D. M. Haddleton and S. N. Richards, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1994, C34, 243 CAS.
- T. P. Davis, D. Kukulj, D. M. Haddleton and D. R. Maloney, Trends Polym. Sci., 1995, 3, 365 CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611 CrossRef CAS.
- J. P. A. Heuts, G. E. Roberts and J. D. Biasutti, Aust. J. Chem., 2002, 55, 381 CrossRef CAS.
- J. Chiefari and E. Rizzardo, in Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. P. Davis, Wiley-Interscience, Hoboken, 2002, p. 629 Search PubMed.
- S. Slavin, K. McEwan and D. M. Haddleton, in Comprehensive Polymer Science, 2nd edn, in press Search PubMed.
- F. R. Mayo, J. Am. Chem. Soc., 1943, 65, 2324 CrossRef CAS.
- K. G. Suddaby, D. R. Maloney and D. M. Haddleton, Macromolecules, 1997, 30, 702 CrossRef CAS.
- J. P. A. Heuts, D. Kukulj, D. J. Forster and T. P. Davis, Macromolecules, 1998, 31, 2894 CrossRef CAS.
- J. P. A. Heuts, T. P. Davis and G. T. Russell, Macromolecules, 1999, 32, 6019 CrossRef CAS.
- B. Y. C. Whang, M. J. Ballard, D. H. Napper and R. G. Gilbert, Aust. J. Chem., 1991, 44, 1133 CrossRef CAS.
- P. A. Clay and R. G. Gilbert, Macromolecules, 1995, 28, 552 CrossRef CAS.
- G. Moad and C. L. Moad, Macromolecules, 1996, 29, 7727 CrossRef CAS.
- K. G. Suddaby, K. F. O'Driscoll and A. Rudin, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 643 CrossRef CAS.
- A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1984, 106, 5197 CrossRef CAS.
- F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959 CrossRef CAS.
- A. H. Janowicz, US Pat., 4746713, 1988.
- G. P. Abramo and J. R. Norton, Macromolecules, 2000, 33, 2790 CrossRef CAS.
- L. Tang, E. T. Papish, G. P. Abramo, J. R. Norton, M.-H. Baik, R. A. Friesner and A. Rappé, J. Am. Chem. Soc., 2003, 125, 10093 CrossRef CAS.
- L. Tang, J. R. Norton and J. C. Edwards, Macromolecules, 2003, 36, 9716 CrossRef CAS.
- L. Tang and J. R. Norton, Macromolecules, 2006, 39, 8229 CrossRef CAS.
- L. Tang and J. R. Norton, Macromolecules, 2006, 39, 8236 CrossRef CAS.
- J. Choi, L. Tang and J. R. Norton, J. Am. Chem. Soc., 2007, 129, 234 CrossRef CAS.
- L. Tang and J. R. Norton, Macromolecules, 2004, 37, 241 CrossRef CAS.
- E. Le Grognec, J. Claverie and R. Poli, J. Am. Chem. Soc., 2001, 123, 9513 CrossRef CAS.
- V. C. Gibson, R. K. O'Reilly, D. F. Wass, A. J. P. White and D. J. Williams, Macromolecules, 2003, 36, 2591 CrossRef CAS.
- L. E. N. Allan, M. P. Shaver, A. J. P. White and V. C. Gibson, Inorg. Chem., 2007, 46, 8963 CrossRef CAS.
- M. P. Shaver, L. E. N. Allan and V. C. Gibson, Organometallics, 2007, 26, 4725 CrossRef CAS.
- J. Choi and J. R. Norton, Inorg. Chim. Acta, 2008, 361, 3089 CrossRef CAS.
- B. Pierik, D. Masclee and A. van Herk, Macromol. Symp., 2001, 165, 19 CrossRef CAS.
- D. Kukulj and T. P. Davis, Macromol. Chem. Phys., 1998, 199, 1697 CrossRef CAS.
- J. P. A. Heuts, D. J. Forster and T. P. Davis, Macromolecules, 1999, 32, 3907 CrossRef CAS.
- D. M. Haddleton, D. R. Maloney, K. G. Suddaby, A. V. G. Muir and S. N. Richards, Macromol. Symp., 1996, 111, 37 CrossRef CAS.
- J. P. A. Heuts, D. J. Forster and T. P. Davis, Macromol. Rapid Commun., 1999, 20, 299 CrossRef CAS.
- J. P. A. Heuts, D. J. Forster, T. P. Davis, B. Yamada, H. Yamazoe and M. Azukizawa, Macromolecules, 1999, 32, 2511 CrossRef CAS.
- R. Vollmerhaus, S. Pierik and A. van Herk, Macromol. Symp., 2001, 165, 123 CrossRef CAS.
- S. C. J. Pierik, Shining a Light on Catalytic Chain Transfer, PhD thesis, TU, Eindhoven, 2002 Search PubMed.
- J. D. Biasutti, G. E. Roberts, F. P. Lucien and J. P. A. Heuts, Eur. Polym. J., 2003, 39, 429 CrossRef CAS.
- D. J. Forster, J. P. A. Heuts, F. P. Lucien and T. P. Davis, Macromolecules, 1999, 32, 5514 CrossRef CAS.
- S. A. Mang, P. Dokolas and A. B. Holmes, Org. Lett., 1999, 1, 125 CrossRef CAS.
- G. Zwolak and F. P. Lucien, Macromolecules, 2006, 39, 8669 CrossRef CAS.
- V. Y. Mironychev, M. M. Mogilevich, B. R. Smirnov, Y. Y. Shapiro and I. V. Golikov, Polym. Sci. U.S.S.R., 1986, 28, 2103 CrossRef.
- G. E. Roberts, T. P. Davis, J. P. A. Heuts and G. E. Ball, Macromolecules, 2002, 35, 9954 CrossRef CAS.
- G. E. Roberts, T. P. Davis, J. P. A. Heuts and G. T. Russell, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 782 CrossRef CAS.
- A. G. Steward, D. M. Haddleton, A. V. G. Muir and S. L. Willis, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1998, 39(2), 459 CAS.
- T. P. Davis, M. D. Zammit, J. P. A. Heuts and K. Moody, Chem. Commun., 1998, 2383 RSC.
- D. J. Forster, J. P. A. Heuts and T. P. Davis, Polymer, 1999, 41, 1385 CrossRef.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378 CrossRef CAS.
- K. S. Kazanskii, G. V. Rakova, S. I. Kozlov, E. V. Stegno and G. Lapiensis, J. Polym. Sci., Part A: Polym. Chem., 2004, 46, 214 Search PubMed.
- M. D. Eason, D. M. Haddleton and E. Khoshdel, Polym. Prepr., 1998, 39(2), 455 CAS.
- E. J. Kelly, D. M. Haddleton and E. Khoshdel, Polym. Prepr., 1998, 39(2), 453 CAS.
- A. H. Soeriyadi, G.-Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Polym. Chem., 2011, 2, 815 RSC.
- L. M. Muratore, J. P. A. Heuts and T. P. Davis, Macromol. Chem. Phys., 2000, 201, 985 CrossRef CAS.
- D. Kukulj, J. P. A. Heuts and T. P. Davis, Macromolecules, 1998, 31, 6034 CrossRef CAS.
- G. E. Roberts, J. P. A. Heuts and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 752 CrossRef CAS.
- D. A. Morrison, L. Eadie and T. P. Davis, Macromolecules, 2001, 34, 7967 CrossRef CAS.
- G. E. Roberts, J. P. A. Heuts and T. P. Davis, Macromolecules, 2000, 33, 7765 CrossRef CAS.
- A. Martchenko, T. Bremner and K. F. O'Driscoll, Eur. Polym. J., 1997, 33, 713 CrossRef CAS.
- J. P. A. Heuts, D. J. Forster and T. P. Davis, ACS Symp. Ser., 2000, 760, 254 CrossRef CAS.
- B. R. Smirnov, L. M. Pushchayeva and V. D. Plotnikov, Polym. Sci. U.S.S.R., 1989, 31, 2607 CrossRef.
- G. T. Russell, Aust. J. Chem., 2002, 55, 399 CrossRef CAS.
- G. B. Smith, G. T. Russell, M. Yin and J. P. A. Heuts, Eur. Polym. J., 2005, 41, 225 CrossRef CAS.
- G. B. Smith, J. P. A. Heuts and G. T. Russell, Macromol. Symp., 2005, 226, 133 CrossRef CAS.
- J. P. A. Heuts and G. T. Russell, Eur. Polym. J., 2006, 42, 3 CAS.
- J. P. A. Heuts, G. T. Russell, G. B. Smith and A. M. van Herk, Macromol. Symp., 2007, 248, 12 CrossRef CAS.
- J. P. A. Heuts, G. T. Russell and G. B. Smith, Aust. J. Chem., 2007, 60, 754 CrossRef CAS.
- A. N. Nikitin, M. C. Grady, G. A. Kalfas and R. A. Hutchinson, Macromolecular Reaction Engineering, 2008, 2, 422 CrossRef CAS.
- G. E. Roberts, C. Barner-Kowollik, T. P. Davis and J. P. A. Heuts, Macromolecules, 2003, 36, 1054 CrossRef CAS.
- F. T. T. Ng, G. L. Rempel, C. Mancuso and J. Halpern, Organometallics, 1990, 9, 2762 CrossRef CAS.
- A. A. Gridnev, S. D. Ittel, M. Fryd and B. B. Wayland, Organometallics, 1996, 15, 5116 CrossRef CAS.
- A. A. Gridnev, S. D. Ittel, M. Fryd and B. B. Wayland, Organometallics, 1996, 15, 5116 CrossRef CAS , ESI.
- B. de Bruin, W. I. Dzik, S. Li and B. B. Wayland, Chem.–Eur. J., 2009, 15, 4312 CrossRef CAS.
- A. A. Gridnev, S. D. Ittel, M. Fryd and B. B. Wayland, Organometallics, 1993, 12, 4871 CrossRef CAS.
- A. A. Gridnev, S. D. Ittel, M. Fryd and B. B. Wayland, Organometallics, 1996, 15, 222 CrossRef CAS.
- F. T. T. Ng, G. L. Rempel, C. Mancuso and J. Halpern, J. Am. Chem. Soc., 1982, 104, 621 CrossRef CAS.
- B. B. Wayland, A. A. Gridnev, S. D. Ittel and M. Fryd, Inorg. Chem., 1994, 116, 7943 CAS.
- S. C. J. Pierik, R. Vollmerhaus, A. M. van Herk and A. L. German, Macromol. Symp., 2002, 182, 43 CrossRef CAS.
- D. A. Morrison, T. P. Davis, J. P. A. Heuts, B. Messerle and A. A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6171 CrossRef CAS.
- L. D. Arvanitopoulos, M. P. Greuel, B. M. King, A. K. Shim and H. J. Harwood, ACS Symp. Ser., 1998, 685, 316 CrossRef CAS.
- B. B. Wayland, G. Poszmik, S. L. Mukerjee and M. Fryd, J. Am. Chem. Soc., 1994, 116, 7943 CrossRef CAS.
- B. B. Wayland, L. Basickes, S. Mukerjee and M. Wei, Macromolecules, 1997, 30, 8109 CrossRef CAS.
- B. B. Wayland, S. Mukerjee, G. Poszmik, D. C. Woska, L. Basickes, A. A. Gridnev, M. Fryd and S. D. Ittel, ACS Symp. Ser., 1998, 685, 305 CrossRef CAS.
- A. Debuigne, J.-R. Caille and R. Jérôme, Angew. Chem., Int. Ed., 2005, 44, 1101 CrossRef CAS.
- A. Debuigne, R. Poli, C. Jérôme, R. Jérôme and C. Detrembleur, Prog. Polym. Sci., 2009, 34, 211 CrossRef CAS.
- T. P. Lodge, J. A. Lee and T. S. Frick, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 2607 CAS.
- D. J. Grisser, B. S. Johnson, M. D. Ediger and E. D. von Meerwall, Macromolecules, 1993, 26, 512 CrossRef.
- M. B. Wisnudel and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2999 CrossRef CAS.
- M. B. Wisnudel and J. M. Torkelson, Macromolecules, 1996, 29, 6193 CrossRef CAS.
- G. Zwolak and F. P. Lucien, Macromolecules, 2008, 41, 5141 CrossRef CAS.
- C. Kowollik and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3303 CrossRef CAS.
- J. P. A. Heuts, M. L. Coote, T. P. Davis and L. P. M. Johnston, ACS Symp. Ser., 1998, 685, 120 CrossRef CAS.
- J. P. A. Heuts, D. A. Morrison and T. P. Davis, ACS Symp. Ser., 2000, 768, 313 CrossRef CAS.
- J. Chiefari, J. Jeffery, G. Moad, E. Rizzardo and S. H. Thang, Polym. Prepr., 1990, 40(2), 344 Search PubMed.
- S. D. Ittel, A. A. Gridnev, C. L. Moad, G. Moad, E. Rizzardo and L. Wilczek, WO Pat., 9731030, 1997.
- S. C. J. Pierik and A. M. van Herk, Macromol. Chem. Phys., 2003, 204, 1406 CrossRef CAS.
- J. Chiefari, J. Jeffery, J. Krstina, C. L. Moad, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2005, 38, 9037 CrossRef CAS.
- D. Kukulj, T. P. Davis, K. G. Suddaby, D. M. Haddleton and R. G. Gilbert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 859 CrossRef CAS.
- D. M. Haddleton, D. R. Morsley, J. P. O'Donnell and S. N. Richards, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3549 CrossRef CAS.
- S. A. F. Bon, D. R. Morsley, J. L. Waterson and D. M. Haddleton, Macromol. Symp., 2001, 165, 29 CrossRef CAS.
- K. G. Suddaby, D. M. Haddleton, J. J. Hastings, S. N. Richards and J. P. O'Donnell, Macromolecules, 1996, 29, 8083 CrossRef CAS.
- D. Kukulj, T. P. Davis and R. G. Gilbert, Macromolecules, 1997, 30, 7661 CrossRef CAS.
- S. C. J. Pierik, B. Smeets and A. M. van Herk, Macromolecules, 2003, 36, 9271 CrossRef CAS.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk and A. M. van Herk, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5839 CrossRef CAS.
- N. M. B. Smeets, T. G. T. Jansen, A. M. van Herk, J. Meuldijk and J. P. A. Heuts, Polym. Chem., 10.1039/c1py00127b.
- N. M. B. Smeets, U. S. Meda, J. P. A. Heuts, J. T. F. Keurentjes, A. M. van Herk and J. Meuldijk, Macromol. Symp., 2007, 259, 406 CrossRef CAS.
- J. L. Waterson, D. M. Haddleton, R. J. Harrison and S. N. Richards, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1998, 39, 457 CAS.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. van Herk, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5078 CrossRef CAS.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. van Herk, Macromolecules, 2009, 42, 7332 CrossRef CAS.
- N. M. B. Smeets, T. G. T. Jansen, J. Meuldijk, A. M. van Herk and J. P. A. Heuts, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1038 CrossRef CAS.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. van Herk, Macromolecules, 2009, 42, 6422 CrossRef CAS.
- M. E. Thomson, N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk and M. F. Cunningham, Macromolecules, 2010, 43, 5647 CrossRef CAS.
- N. M. B. Smeets, T. G. T. Jansen, T. J. J. Sciarone, J. P. A. Heuts, J. Meuldijk and A. M. van Herk, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1038 CrossRef CAS.
- J. P. Lynch, D. J. Irvine and G. M. Beverly, WO Pat., 9804603, 1998.
- G. D. Airey, J. Wilmot, J. R. A. Grenfell, D. J. Irvine, I. A. Barker and J. E. Harfi, Eur. Polym. J., 2011, 47, 1300 CrossRef CAS.
- K. Adamsons, G. Blackman, B. Gregorovich, L. Lin and R. Mathesons, Prog. Org. Coat., 1998, 34, 64 CrossRef CAS.
- J. P. A. Heuts, L. Muratore and T. P. Davis, Macromol. Chem. Phys., 2000, 201, 2780 CrossRef CAS.
- D. M. Haddleton, C. Topping, J. J. Hastings and K. G. Suddaby, Macromol. Chem. Phys., 1996, 197, 3027 CrossRef CAS.
- T. Y. J. Chiu, J. P. A. Heuts, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Chem. Phys., 2004, 205, 752 CrossRef CAS.
- A. A. Gridnev, W. J. Simonsick, Jr and S. D. Ittel, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1911 CrossRef CAS.
- D. M. Haddleton, C. Topping, D. Kukulj and D. Irvine, Polymer, 1998, 39, 3119 CrossRef CAS.
- Z. Guan, J. Am. Chem. Soc., 2002, 124, 5616 CrossRef CAS.
- K. A. McEwan and D. M. Haddleton, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2010, 51(1), 721 CAS.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027 CrossRef CAS.
- P. A. Costello, I. K. Martin, A. T. Slark, D. C. Sherrington and A. Titterton, Polymer, 2002, 43, 245 CrossRef CAS.
- S. Camerlynck, P. A. G. Cormack, D. C. Sherrington and G. Saunders, J. Macromol. Sci., Part B: Phys., 2005, 45, 881 Search PubMed.
- D. M. Haddleton, K. McEwan and J. P. Menzel, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2010, 51(1), 310 CAS.
- S. V. Kurmaz, E. O. Perepelitsina, M. L. Bubnova, G. A. Estrina and V. P. Roshchupkin, Mendeleev Commun., 2002, 12, 21 CrossRef.
- S. V. Kurmaz, E. O. Perepelitsina, M. L. Bubnova and G. A. Estrina, Mendeleev Commun., 2004, 14, 125 CrossRef.
- S. V. Kurmaz, E. O. Perepelitsina, M. L. Bubnova and G. A. Estrina, e-Polym., 2004, no. 47 Search PubMed.
- S. V. Kurmaz, E. O. Perepelitsina, M. L. Bubnova and G. A. Estrina, Vysokomol. Soedin., Ser. A, 2006, 48, 1081 CAS.
- S. V. Kurmaz and E. O. Perepelitsina, Russ. Chem. Bull. Int. Ed., 2006, 55, 835 CrossRef CAS.
- S. V. Kurmaz, V. P. Grachev, I. S. Kochneva, E. O. Perepelitsina and G. A. Estrina, Vysokomol. Soedin., Ser. A, 2007, 49, 1480 CAS.
- M. Luzon, C. Boyer, C. Peinado, T. Corrales, M. Whittaker, L. Tao and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2783 CrossRef CAS.
- J. V. M. Weaver, R. T. Williams, B. J. L. Royles, P. H. Findlay, A. I. Cooper and S. P. Rannard, Soft Matter, 2008, 4, 985 RSC.
- J. Norman, S. C. Moratti, A. T. Slark, D. J. Irvine and A. T. Jackson, Macromolecules, 2002, 35, 8954 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, J. Burns, C. R. Becer, M. R. Whittaker, D. M. Haddleton and T. P. Davis, Chem. Commun., 2010, 46, 6338 RSC.
- J. Huybrechts, P. Bruylants, K. Kirshenbaum, J. Vrana and J. Snuparek, Prog. Org. Coat., 2002, 45, 173 CrossRef CAS.
- J. Huybrechts, P. Bruylants, A. Vaes and A. De Marre, Prog. Org. Coat., 2000, 38, 67 CrossRef CAS.
- L. Chen, L. Yan, Q. Li, C. Wang and S. Chen, Langmuir, 2010, 26, 1724 CrossRef CAS.
- Z. Lu, J. Wang, Q. Li and S. Chen, Eur. Polym. J., 2009, 45, 1072 CrossRef CAS.
- C.-F. Wang, Y.-P. Cheng, J.-Y. Wang, D. Zhang, L.-R. Hou, L. Chen and S. Chen, Colloid Polym. Sci., 2009, 287, 829 CAS.
- L. Yan, Z. Yu, L. Chen, C. Wang and S. Chen, Langmuir, 2010, 26, 10657 CrossRef CAS.
- C.-M. Cheng and D. J. Tshudy, US Pat., 5928829, 1999.
- N. M. B. Smeets, R. P. Moraes, J. A. Wood and T. F. L. McKenna, Langmuir, 2011, 27, 575 CrossRef CAS.
- D. M. Haddleton, D. R. Maloney and K. G. Suddaby, Macromolecules, 1996, 29, 481 CrossRef CAS.
- C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1996, 29, 3119 Search PubMed.
- G. Moad, A. G. Anderson, F. Ercole, C. H. J. Johnson, J. Krstina, C. L. Moad, E. Rizzardo, T. H. Spurling and S. H. Thang, ACS Symp. Ser., 1998, 685, 332 CrossRef CAS.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A: Pure Appl.Chem., 1986, 23, 839 CrossRef.
- B. Yamada, F. Oku and T. Harada, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 645 CrossRef CAS.
- B. Yamada, P. B. Zetterlund and E. Sato, Prog. Polym. Sci., 2006, 31, 835 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- A. A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1366 CrossRef CAS.
- A. A. Gridnev, WO Pat., 9941218, 1999.
- H. Ishigaki, H. Okkada and S. Suyama, JP Pat., 03212402, 1991.
- M. Nagashima and H. Kazama, JP Pat., 11071220, 1997.
- H. Shinike, K. Takeda and N. Sakurabam, JP Pat., 11124791, 1997.
- K. Yoshimi, O. Kazeto and M. Katayama, EP Pat., 1101773, 1999.
- W. Meyer, J. L. Bohan, L. D. Timberlake and J. D. Siebecker, WO Pat., 2006/039602, 2006.
- L. M. Muratore, K. Steinhoff and T. P. Davis, J. Mater. Chem., 1999, 9, 1687 RSC.
- L. M. Muratore and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 810 CrossRef CAS.
- J. A. Antonelli and C. Scopazzi, US Pat., 5310807, 1994.
- E. M. Coen, J. F. Quinn, F. Dehghani, N. R. Foster and T. P. Davis, Polymer, 2003, 44, 3477 CrossRef CAS.
- W. Lau, P. R. Van Rheenen, K. A. Bromm, D. Fasano and T. R. Tepe, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2005, 46(2), 197 CAS.
- W. Lau, K. A. Bromm and P. R. Van Rheenen, US Pat., 2004/0167269, 2004.
- T. Nabuurs, S. Van der Slot and A. Overbeek, Prog. Org. Coat., 2007, 58, 80 CrossRef CAS.
- S. Van der Slot and T. Nabuurs, WO Pat., 2006/007999, 2006.
- J. M. Asua and H. A. S. Schoonbrood, Acta Polym., 1998, 49, 671 CrossRef CAS.
- T. Harada, P. B. Zetterlund and B. Yamada, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 597 CrossRef CAS.
- E. Sato, P. B. Zetterlund and B. Yamada, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6021 CrossRef CAS.
- E. Sato, P. B. Zetterlund and B. Yamada, Macromolecules, 2004, 37, 2363 CrossRef CAS.
- J. Chiefari, G. Moad, E. Rizzardo and A. A. Gridnev, WO Pat., 9847927, 1998.
- J. Chiefari, J. Jeffery, R. T. A. Mayadunne, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 7700 CrossRef CAS.
- J. Chiefari, J. Jeffery, R. T. A. Mayadunne, G. Moad, E. Rizzardo and S. H. Thang, ACS Symp. Ser., 2000, 768, 297 CrossRef CAS.
- A.-A. Zorn, T. Junkers and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 2028 CrossRef CAS.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
- J. P. Menzel, D. M. Haddleton and E. Khoshdel, PMSE Prepr., 2010, 102, 18 Search PubMed.
- K. A. McEwan and D. M. Haddleton, Polym. Chem., in press 10.1039/C1PY00221J; K. A. McEwan and D. M. Haddleton, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2010, 51(1), 721 CAS.
- G.-Z. Li and D. M. Haddleton, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2010, 51(2), 555 CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 19, 2727 RSC.
- R. C. Li, R. M. Broyer and H. D. Maynard, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5004 CrossRef CAS.
- N. M. B. Smeets, J. Meuldijk, J. P. A. Heuts and A. C. J. Koeken, Polym. Chem., 2010, 1, 1102 RSC.
- G. C. Sanders, T. J. Sciarone, H.M.L. Lambermont-Thijs, J. P. A. Heuts and R. Duchateau, 2011, submitted; G. C. Sanders, T. J. Sciarone, J. P. A. Heuts, R. Duchateau, A. M. van Herk and C. E. Koning, Book of Abstracts Macro, 2010, C11_P27 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |