Novel soluble polyazomethines with pendant carbazole and triphenylamine derivatives: preparation, characterization, and optical, electrochemical and electrochromic properties†
Haijun
Niu
*ab,
Hongqiang
Kang
ac,
Jiwei
Cai
a,
Cheng
Wang
a,
Xuduo
Bai
*a and
Wen
Wang
d
aKey Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University), Ministry of Education, Key laboratory macromolecular chemistry, Harbin, 150086, P R China. E-mail: haijunniu@hotmail.com; xuduobai@hotmail.com; Fax: +086-0451-86608131; Tel: +086-0451-86608131
bKey Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, Shanghai, 200237, P R China
cSchool of Chemical Engineering, Harbin Institute of Technology, Harbin, 150080, P R China
dSchool of Material Science and Engineering, Harbin Institute of Technology, Harbin, 150080, P R China. E-mail: Wangwen@hit.edu.cn; Fax: +086-0451-86414291; Tel: +086-0451-86402040
First published on 5th October 2011
Abstract
Two series of novel polyazomethines (PAMs) P1–P5, Pa–Pe with an Mn of 1522–7879 g mol−1 were prepared via the direct polycondensation from N,N-Bis(4-aminophenyl)-4-N′-carbazolyl-1,4- phenylenediamine, and N,N-Bis (4-aminophenyl)-N′,N′-di-phenyl-1,4-phenylenediamine with various dicarboxaldehyde such as phthaldialdehyde, 1,3-isophthalaldehyde, terephthalaldehyde, 4,4′-diformyltriphenylamine, 3,5-thiophenedialdehyde. All the polymers were amorphous with good solubility in many organic solvents, such as CHCl3, tetrahydrofuran (THF), N-methyl-2-pyrrolidinone (NMP) and N,N-dimethylacetamide (DMAc), and could be solution-cast into polymer films. All polymers displayed outstanding thermal stabilities, i.e. 20% wt loss in excess of 470 °C under nitrogen. A hybrid density functional theory (DFT) method was used to calculate the optimized geometry and electronic structure of PAMs. The variation of the backbone ring significantly affected the dihedral angles and resulted in the variation of electronic properties. The HOMO and LUMO energy levels of these polymers calculated by electrochemistry and absorption spectra were in the range 4.687–5.189 and 2.366–2.989 eV below the vacuum level, respectively. Our study demonstrated the backbone ring can tune the electronic properties of conjugated PAMs effectively. All obtained PAMs revealed stability of electrochromic characteristics, changing color from original yellowish to red or violet or blue. And multiple reversible colours states were observed upon chemical doping. The properties prove that the polyazomethines are multipurpose materials which will be used in hole-transporting, electrochromic and chemical sensor applications in the near future.
Introduction
Conjugated polymers are of great interest because they are useful electronic, optoelectronic, electrochemical, and nonlinear optical conductors,1-5 due to low cost synthesis, and ease of processing via solution-based casting or printing technologies. There have been several reports on poly(1,4-phenylenevinylene) (PPV),6 polythiophene (PTh),7 and polyfluorene (PF)8 derivatives and so on which are available mostly via Suzuki-type aryl–aryl coupling methods, Wittig-type coupling techniques, Gilch and Horner–Emmons protocols. These methods require rigorous reaction conditions such as noble catalyst, anhydrous solvents and inert atmospheres, and the resulting polymers are sporadically contaminated with trace metals or other byproducts with difficult purifications, which will influence the optoelectronic properties in the devices.Polyazomethine (PAM) containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds connection which is isoelectronic with the CHCH group, having the similar planar molecular structure and the absorption maximum peak to CHCH, capable of protonation and complexation,9 would be suitable alternatives to conjugated polymers mentioned above. This is because that the synthesis requires the mild reaction conditions required (not needing noble catalyst and monomers, and stringent conditions) leading to high yields and purity via the condensation between an amine and an aldehyde. Additionally, the robust azomethine bond exhibits a high stability toward hydrolysis and reduction and the resulting polymers are easily purified through being extracted with Soxhlet apparatus and dried under vacuum. Polyazomethine derivatives are further advantageous because they are easy to be processed with functional group linked to the end of oligomer such as aldehyde or amine according the design.10
N bonds connection which is isoelectronic with the CHCH group, having the similar planar molecular structure and the absorption maximum peak to CHCH, capable of protonation and complexation,9 would be suitable alternatives to conjugated polymers mentioned above. This is because that the synthesis requires the mild reaction conditions required (not needing noble catalyst and monomers, and stringent conditions) leading to high yields and purity via the condensation between an amine and an aldehyde. Additionally, the robust azomethine bond exhibits a high stability toward hydrolysis and reduction and the resulting polymers are easily purified through being extracted with Soxhlet apparatus and dried under vacuum. Polyazomethine derivatives are further advantageous because they are easy to be processed with functional group linked to the end of oligomer such as aldehyde or amine according the design.10
However, like PPV and other conjugated polymers, insolubility of polyazomethines in common organic solvents limits their processability, characterization and application. There are three main methods to improve the processability of polyazomethine: one is introducing long large alkyl, or aryloxy groups into the main-chain aromatic benzene rings,11 but which will reduce the thermal stability, influence the conjugation, lower the crystallinity and chain-to-chain conduction and cause photooxidation of polymers; another is selection of polymer structure;12 the third approach is based on the reversible Lewis acid–base complexation which has been successfully applied to the processing of poly(azomethines).13 One method which does not sacrifice high thermal stability is the introduction of bulky, packing disruptive groups into the polymer backbone.14 The triarylamine unit is easy to oxidize in the nitrogen center and has the excellent ability to transport charge carriers via the radical cation species with high stability and low ionization potential.15 The compounds containing triarylamine are widely investigated and applied in various electro-optical materials such as organic light emitting diodes (OLEDs),16 organic field-effect transistors,17 non-linear optical materials,18 and xerography,19dye- sensitized solar cell (DSCs).20 The triphenylamine derivatives with the propeller-shaped molecular structure are bulky, easy dissolve in organic solvent which would increase solubility and decrease intermolecular aggregation dramastically.21
The polyazomethine contains triphenylamine and the nitrogen atom of the imine group (CN) with a lone pair electrons in the backbone can form intra- or inter-molecular interactions such as hydrogen bonding with other groups. Furthermore, it also has the capabilities of protonation and complexation with metal ions, acid and iodine.22
So the polyazomethine will give novel interesting optical properties such as electrochromism and absorption controlled by oxidation or environmental stimuli.
From about 2005, Liou has investigated deeply on a series of TPA-based high-performance polyimides, polyamides, polyazomethines with attractive electrochromic properties bearing triphenylamine derivatives such as N,N-Bis(4-aminophenyl)-4-N′-carbazolyl-1,4-phenylenediamine,23N,N-bis(4-aminophenyl)-N′,N′-diphenyl-1,4-phenylenediamine24 and other substituted triphenylamines,25etc. For example, they synthesised a series of poly(amine-imide)s by the reaction of N,N-Bis(4-aminophenyl)-4-N′-carbazolyl-1,4-phenylenediamine or N,N-bis(4-aminophenyl)-N′,N′-diphenyl-1,4-phenylenediamine with various dianhydrides. All poly(amino-imide)s had an excellent stability of electrochromic characteristics, changing colour from the pale-yellowish neutral form to the green and to blue oxidised forms when scanning potentials changed positively from 0 V to 1.15 V. They found that the polymer based on triphenylamine as core exhibited stable, excellent, multi-color, electrochromic characteristic, furthermore the incorporation of electron-donating substituents at the para positions of triarylamines afforded stable radical cations.
Recently, we have reported the synthesis of soluble aromatic polyazomethines bearing triphenylamine units in the main chain based on diaminephenylamine and dialdehyde triphenylamine.26 During preparation of the manuscript, a series of similar oligomers and polymers based on 4,4′-diformyltriphenylamine were reported by Iwan and co-workers.27 They observed hypsochromic shift of UV-vis spectra in comparison to the pristine ones which is reverse to the result obtained by Jeneke and co-workers28 and us.26
Owing to the distinguished stability and optical properties, the thiophene derivatives have attracted intensive attention. Destri's group have designed and studied a great deal of electrochromic materials made from thiophene derivatives.29
Skene has also synthesized a series of polythiopheno azomethines which exhibit low optical band gaps (1.3–2.0 eV), and all polymers showed excellent thermal properties as well.30 In addition, the polymers were claimed to be chemically and photochemically stable. The authors also showed that this class of polymers could be used in both n- and p-type devices. The thiophene improves the overall conjugation and associated opto-electronic properties. There is a strong absorption at 510 nm shows the high degree of conjugation which is a result of the high degree of coplanarity of the thiophenes and the imine bond. This is in contrast to their homoaryl azomethine counterparts that absorb only at ca. 300 nm, owing to their limited degree of conjugation and twisting between the aryl and imine planes. Reversible oxidation and electrochromic properties dependent on structure and heterocycles demonstrate the suitability of azomethines as functional materials.
Like triphenylamine, carbazole is also a conjugated unit with planar structure that has interesting optical and electronic properties such as photoconductivity and photorefractivity.31 It is well known that the 1,3-isophthalaldehyde, terephthalaldehyde, phthaldialdehyde with the different substitution position on the benzene ring have various degrees of conjugation. Herein, we propose a new, facile, low-cast, noble metal catalyst-free way for mass production to design triphenylamine-based and carbazole-based alternating copolyazomethine family with fine color control, that is, for obtaining degree color tuning by varying the substitution patterns on the incorporating dialdehyde units. And, in order to investigate the role of the monomer unit and azomethine linkage on the optical electrochemical properties, we also tuned different degrees of conjugation on the backbone of polymers by introducing 2,5-thiophenedialdehyde and 4,4′-diformyltriphenylamine, and studied the relationships of structure and electro-properties of the polyazomethine which has potential applications in chemical sensors and electrochromic devices.
Experimental
Materials
Terephthalaldehyde, 1,3-isophthalaldehyde, phthaldialdehyde, 2,5-thiophenedialdehyde were purchased from Acros, Aldrich. 4,4′-Diformyltriphenylamine (4,4′-(phenylimino)bisbenzaldehyde) was prepared by the Vilsmeier reaction according to the method described in the literature we had reported,264-nitrotriphenylamine and N-(4-nitrophenyl) carbazole were prepared by the Ullman reaction. 4-Aminotriphenylamine and N-(4-aminophenyl)-carbazole were prepared according to the procedure given in the literature,23,24 as shown in Scheme 1, by the arylation of diphenylamine and carbazole with 4-fluoronitrobenzene, followed by H2Pd/C-catalyzed reduction in autoclave of the intermediate compound, 4-nitrotriphenylamine and N-(4-nitrophenyl) carbazole.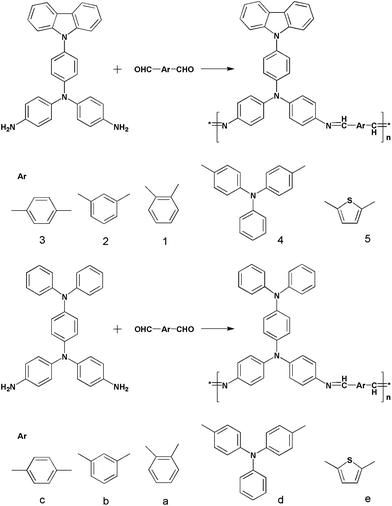 | ||
| Scheme 1 The synthetic routes for polyazomethines with different monomers. | ||
Measurements and characterization
Fourier transform infrared (FT-IR) spectra were recorded on a PerkinElmer Spectrum 100 Model FT-IR spectrometer. 1H NMR spectra were measured on a Bruker AC-400 MHz spectrometer in CDCl3, using tetramethylsilane as an internal reference. Gel permeation chromatography (GPC) analysis was performed on a Waters instrument connected with one refractive index detector. All GPC analyses were performed using a polymer/THF solution at a flow rate of 1 mL min−1 at 70 °C and calibrated with polystyrene standards. Thermogravimetric analysis (TGA) was conducted with a PerkinElmer Pyris 6 TGA. Experiments were carried out on approximately 6∼8 mg powder samples heated in flowing nitrogen or air (flow rate = 20 cm3 min−1) at a heating rate of 20 °C min−1. DSC analyses were performed on a PerkinElmer Pyris diamond DSC at a scan rate of 10 °C min−1 in flowing nitrogen (20 cm3 min−1). Cyclic voltammetry (CV) measurements were conducted on a CH Instruments 660A electrochemical analyzer at a scan rate of 50 mV s−1 with a 0.1 M solution of LiClO4 as an electrolyte under nitrogen atmosphere in dry acetonitrile (CH3CN). The oxidation and reduction potentials of polymer film coated on a ITO disk were measured using a Pt wire and a Ag/AgCl electrode as a counter electrode and a quasi reference electrode, respectively. Under these conditions, the onset of oxidation and reduction potentials of the polymer thin films against the Ag/AgCl quasi reference electrode were measured and calibrated against the ferrocene/ferrocenium (Fc/Fc+) redox couple. The HOMO and LUMO levels of the polymers were calculated by assuming the absolute energy level of Fc/Fc+ as −4.80 eV to vacuum. X-ray diffraction patterns were recorded using powder samples on a wide-angle D/max-γB diffractometer working in typical Bragg geometry with Cu-Kα radiation (λ = 0.154nm). Density-functional theory (DFT) calculations were performed on a computer. Geometry optimizations were carried out using the B3LYP functional as implemented in Gaussian 98 program.32Monomer synthesis
N,N-Bis(4-aminophenyl)-4-N′-carbazolyl-1,4-phenylenediamine was prepared according to the method described as N-(4-aminephenyl)- carbazole.24 The product was collected by filtration and washed with ethanol, and dried in vacuo at 60 °C to give white needles with a mp of 192–195 °C, 68% in yield.IR (KBr, cm−1):3434, 3361(N–H stretch), 3222,1622, 1508,1422 (benzene stretch), 1328, 1263, 1222, 822, 749, 716.1H NMR (CDCl3, ppm): 8.10, 8.13(d,2H), 7.39(s,H), 7.26(s,4H), 7.07,7.05, 7.02(t,6H), 6.69,6.67(d,4H),3.71(4H, NH2).
The crude N,N-bis(4-aminophenyl)-N′,N′-diphenyl-1,4-phenylenediamine was prepared according to the method described as N-(4-aminephenyl)-diphenylamine, and similar to literature except the reduction method, and was washed with methanol and recrystallized from toluene in nitrogen and dried in vacuo at 60 °C to give 7.02 g (yield: 89%) of light-beige product; mp: 245–247 °C. IR (KBr, cm−1): 3450, 3361 (N–H stretch), 1622, 1590, 1500 (benzene stretch), 1263, 822, 749, 692. 1H NMR (CDCl3, ppm):7.26 (s,2H), 7.20 (s,4H), 7.08 (s,4H), 6.92 (s,8H), 6.61 (s,4H), 3.53 (s, 4H, NH2).
Polymer synthesis
The synthesis of P1 was used as an example to illustrate the general synthetic procedure. A three necked 50 ml glass reactor fitted with a magnetic stirrer, Dean–Stark trap and reflux condenser was charged with N,N-bis(4-aminophenyl)-4-N′-carbazolyl-1,4-phenylenediamine (0.8804 g, 2 mmol), 10 ml DMAc, and 2 ml of toluene containing 5% dry lithium chloride. 0.2682 g (2 mmol) of o-phthalaldehyde was added to the stirred solution three times every 20 min, and the solution was heated at 160 °C for 15 h to remove water under nitrogen reflux. After the reaction, the obtained polymer solution was poured slowly into 400 ml of ice frozen methanol. The precipitate was collected by filtration, washed thoroughly with hot methanol in a Soxhlet apparatus for 48 h, then was dried under vacuum at 70 °C overnight. Reprecipitations of the polymer by NMP/methanol were carried out twice for further purification. The inherent viscosity of the obtained polymer (denoted as P1) was 0.14 dl/g (measured at a concentration of 0.5 g/dl in NMP at 25 °C). Yield: 0.9642g, 83.5%, red dish brown. FTIR spectrum (KBr pellet): 1715 (ν CO), 1623 (ν CN), 1595, 1503, 1479, 1448 (ν Ph), 1H NMR (400 MHz, CDCl3, ppm): δ 8.14 (H–CN); 7.98–7.81 (HC–ph–CH, ph); 7.63–6.71 (H in carbazole).
The other polymers were prepared by an analogous procedure and shown as Scheme 1.
O), 1623 (ν CN), 1598, 1509, 1499, 1451(ν Ph), 1H NMR (400 MHz, CDCl3, ppm): δ 10.13(HCO); 8.64 (H–CN); 8.46–8.00 (HC–ph–CH, ph); 7.69–7.24 (H in carbazole).
O), 1623 (ν CN), 1585, 1559, 1509, 1451 (ν Ph), 1H NMR (400 MHz, CDCl3, ppm): δ 10.11 (HCO); 8.63 (H–CN); 8.18–8.00 (HC–ph–CH, ph); 7.51–7.21 (H in carbazole).
O), 1623 (ν CN), 1588, 1506, 1451 and 1417 (ν Ph), 1H NMR (400 MHz, CDCl3, ppm): δ 9.89 (HCO); 8.48 (H–CN); 8.16–7.76 (HC–ph–CH, ph); 7.49–7.13 (H in carbazole).
O), 1605 (ν CN), 1513, 1492, 1451 (ν Ph), 1H NMR (400 MHz, CDCl3, ppm): δ 8.89 (H–CN); 8.24–7.94 (HC–thiophene–CH, H in thiophene); 7.81–6.62(H in carbazole).
O), 1649 (ν CN), 1584, 1494, 1313(ν Ph), 1256, 747, 829, and 689. 1H NMR (400 MHz, CDCl3, ppm): δ: 8.03(H–CN),8.00–7.89(HC–ph–CH, ph), 7.87 (H in triphenylamine).
O), 1625 (ν CN), 1584, 1491, 1317 (ν Ph), 1267, 836, 754, and 682.1H NMR (400 MHz, CDCl3, ppm): δ 10.21 (HCO), 8.60 (H–CN), 7.57–8.41 (HC-ph-CH, ph), 6.90–7.34 (H in triphenylamine).
O), 1613 (ν CN), 1584, 1498, 1317 (ν Ph), 1267, 833, 747, and 689. 1H NMR (400 MHz, CDCl3, ppm): 10.09 (HCO), 8.57 (H–CN), 7.97–8.10 (HC–ph–CH, ph), 6.90–7.94 (H in triphenylamine).
O), 1621 (ν CN), 1592, 1498, 1310 (ν Ph), 1260, 829, 747, and 696. 1H NMR (400 MHz, CDCl3, ppm):δ 9.87 (HCO), 8.45 (H–CN), 7.71–7.85 (HC–ph–CH, ph), 6.92–7.42 (H in triphenylamine).
O), 1609 (ν CN), 1491, 1317 (ν Ph), 1260, 827, 754, and 696. 1H NMR (400 MHz, CDCl3, ppm): δ 9.96 (HCO), 8.63 (H–CN), 7.44–8.40 (HC–ph–CH, ph), 6.81–7.46 (H in triphenylamine).
Results and discussion
Synthesis of monomers and polyazomethines
IR and 1H NMR spectroscopic techniques were used to identify the structures of the intermediate compounds and the diamine monomers. The transformation of nitro to amino group could be revealed by the change of IR spectra. The nitro groups of intermediate compounds gave two bands at about 1580 and 1313 cm−1 (–NO2 characteristic symmetric and symmetric stretching vibration). After reduction, the typical absorptions of the nitro group disappeared and the amino group displayed the typical two N–H stretching absorption peaks in the region of 3300–3500 cm−1. The 1H NMR spectra confirm that the nitro groups have been completely transformed into amino groups by the resonance signals at around 3.5 ppm corresponding to the amino protons.
 | ||
| Fig. 1 1H NMR spectra of P1 to P5. | ||
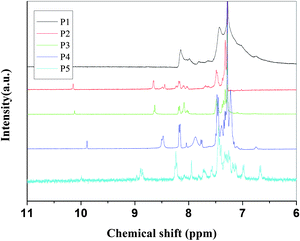
 | ||
| Fig. 2 FT-IR spectra of P1 to P5. | ||
Fig. 1 shows the 1H NMR spectra of the polymers in CDCl3; the main peaks at about 8.58 ppm (except P1 at 8.14, P5 at 8.85) corresponding to the proton resonance of the imine group (CHN) confirm the formation of the imine linkage. The small signal at 9.85 ppm of P4 and 10.08 ppm of the others are attributed to the terminal aldehyde groups which are not reacted. The chemical shifts of the protons on phenylene rings are located at the range 7.0–8.3 ppm. It was found that the absorptions in 1650∼1400 cm−1 (CN stretching and benzene ring absorptions) were apparently changed during doping in Fig. 3. When CN bond was influenced by conjugation which usually appeared between 1630 and 1680 cm−1, its stretching would move to lower wave number. So, the CN stretching in the original polymer appeared at 1620 cm−1, indicating delocalization of CN bond along the polymer chain. When the polymer was doped with HCl and I2, the strength of CN stretching decreased, and new peak at 1658 cm−1 appeared which may be attributed to CN stretching in the complex of PSB/dopants according to the literature.33
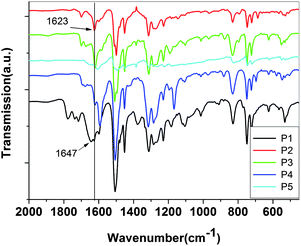
 | ||
| Fig. 3 FT-IR spectra of original, I2 and HCl doped P3. | ||
Since nitrogen is a very electronegative element, the iodine doping occurs not only on the nitrogen atoms but also on the triphenylamine or carbazole rings (Scheme 2).
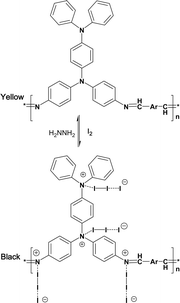 | ||
| Scheme 2 Possible doping reaction of the polyazomethines. | ||
But since the low basicity of nitrogen atom of carbazole and triphenylamine is not very strong due to delocalization of the pair of electrons on N atom, the H+ can only coordinate with CHN. In this way, the following doping reaction is suggested: the new peaks at 1658, 1689 cm−1, related to CNIn− stretching with the influence of different anions (I−, I3−, or I5−), represent CT complex formation between the polymer and iodine. The absorption of the benzene ring in the polyazomethine at 1584 cm−1, decrease with increase of doping extent, and a new peak appears strong at 1579 cm−1 with doping as seen from Fig. 3, which may be attributed to the quinoid structure. The result is in good agreement with the results of the literature.34
The ratio number of iodine atoms to polyazomethine unit are introduced by the doping lies between 7–20 (containing triphenylamine unit) and 1.5–3 (containing carbazole unit), depending on the polymer structures.
However, as for polyazomethines doped with HCl molecule, the ratio lies in the range 1.5–6 (containing triphenylamine unit) and 2–7.5 (containing carbazole unit). The result shows that the steric hindrance existing in polyimines (containing carbazole unit) may be accounted for in these values of iodine doped atoms.
Basic characterization
Solubility, GPC and TG
The solubility behavior of the polyazomethines depended on their chain packing ability and intermolecular interactions which were affected by the rigidity, symmetry, and regularity of the molecular backbone. The introduction of TPA group increases solubility of the polymers. So, the resulting PAMs showed good solubility in common organic solvents, such as CHCl3, DMSO, NMP, DMAC, DMF, THF. It is facility to fabricate film for device by spinning.The molecular weight of PAMs was measured by GPC, using polystyrenes as standard and THF as eluent. The weight-average molecular weight (Mw) and polydispersity (PDI) were determined as 2117–25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 905 and 1.39–2.82, respectively (Table 1). In general, the Mws of polymers containing triphenylamine are larger than the ones containing carbazole units which may be due to steric hindrance that makes the latter possessing high weight more insoluble than the former. From the Mn calculated, the polymer has about average 5 units.
905 and 1.39–2.82, respectively (Table 1). In general, the Mws of polymers containing triphenylamine are larger than the ones containing carbazole units which may be due to steric hindrance that makes the latter possessing high weight more insoluble than the former. From the Mn calculated, the polymer has about average 5 units.
| 5% | 20% | 50% | M n | M W | M z | PDI polydispersity | |
|---|---|---|---|---|---|---|---|
| P1 | 400 | 515 | >700 | 2238 | 3860 | 5757 | 1.72 |
| P2 | 459 | 503 | >700 | 4942 | 13908 | 25905 | 2.81 |
| P3 | 352 | 506 | >700 | 1522 | 2117 | 2828 | 1.39 |
| P4 | 448 | 513 | >700 | 3276 | 5328 | 7982 | 1.62 |
| P5 | 433 | 523 | >700 | 4007 | 6985 | 10311 | 1.74 |
| Pa | 252 | 479 | >650 | 2441 | 4521 | 6602 | 1.85 |
| Pb | 323 | 492 | >650 | 3629 | 7930 | 13793 | 2.18 |
| Pc | 323 | 492 | 625 | 7414 | 13138 | 20536 | 1.77 |
| Pd | 425 | 479 | 625 | 4909 | 9702 | 15652 | 1.97 |
| Pe | 402 | 479 | >650 | 7879 | 14234 | 23186 | 1.81 |
As shown in Table 1, which shows the thermal properties compared with the analogous poly(amine-imide)s, the series of PAMs did not show clear Tg but showed an enhanced thermal stability (Fig. 4 and Fig. S3†). Because of the decreased conformational flexibility or free volume caused by the introduction of planar carbazole groups in the repeat unit, the P1–P5 series are more thermally stable than the Pa–Pe series. All the polymers indicated no clear melting endotherms up to the decomposition temperatures on the DSC thermograms.
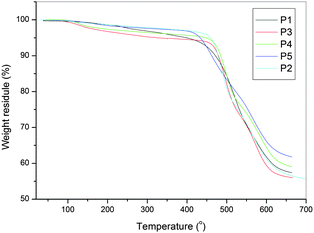 | ||
| Fig. 4 TG diagrams of P1 to P5. | ||
According to the literature,35 the weight loss below 400 °C could be attributed to out-gassing of residual monomer species (TPA) and H2O. The latter is thought to be due to a post-polycondensation process, where the polymer amine and aldehyde end groups continue to react and water is expelled.
XRD analysis
In order to gain insight into the molecular packing preferences of the polymers, all polymers were investigated by WAXD. The WAXD (seen from Fig. 5) studies of the PAMs powder indicated that there were some crystalline in carbazole-based polymers (P2, P5) due to the planar structure of carbazole unit, but all the triphenylamine-based polymers were amorphous which displayed broad wide-angle reflections with a maximum around 2θ = 19.51°(See Fig. S4†), which corresponded to a lateral intermolecular spacing of 0.46 nm. The amorphous nature can be attributed to the introduction of bulky, twisted, three dimensional triphenylamino units along the polymer backbone.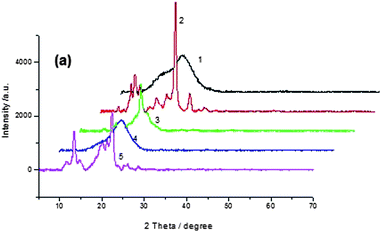 | ||
| Fig. 5 XRD spectra of P1 to P5. | ||
Geometric structure calculations and electronic structure
To gain insight into the polymer structure–properties relationships, ground-state geometry optimizations of oligomer of P1 to P5, were carried out using density functional theory at the B3LYP/6-31G level (Table 2).| R C1– N2(Å) |
R
N2C3(Å) |
R C3– C4(Å) | δ | Φ1(deg) | Φ2(deg) | |
|---|---|---|---|---|---|---|
| P1 | 1.411 | 1.291 | 1.473 | 0.182 | 152.104 | 148.607 |
| P2 | 1.410 | 1.292 | 1.470 | 0.178 | 153.885 | 175.043 |
| P3 | 1.410 | 1.292 | 1.471 | 0.179 | 153.147 | 170.239 |
| P4 | 1.409 | 1.293 | 1.467 | 0.174 | 179.760 | 179.917 |
| P5 | 1.409 | 1.293 | 1.452 | 0.159 | 153.972 | 176.236 |
| Pa | 1.410 | 1.290 | 1.472 | 0.182 | 179.814 | 179.972 |
| Pb | 1.410 | 1.292 | 1.470 | 0.178 | 154.410 | 174.352 |
| Pc | 1.408 | 1.296 | 1.465 | 0.169 | 179.908 | 179.921 |
| Pd | 1.408 | 1.296 | 1.463 | 0.167 | 179.905 | 179.579 |
| Pe | 1.409 | 1.293 | 1.451 | 0.158 | 157.693 | 176.168 |
The average C–C bond lengths (RC3–C4) between the C2N3 linkage and phenylene ring of P1–P5 are in the range 1.452∼1.473 Å, while the CN bond lengths (N2C3) are in the range 1.290∼1.296 Å. The torsional angles (Φ2) between the CN linkage and adjacent phenylene or thiophene ring (with aldehyde group) are in the range 170.239∼179.917 (with the exception of 148.607 in P1), which suggest that monomer (with aldehyde group) unit part has a more planar conformation than the part of monomer (N-phenylene ring) unit.
The replacement of the phenylene ring (with aldhyde) by thiophene and triphenylamine or alternation of the site of substitution of group, has no influence on the dihedral angle Φ2. However, it leads to a larger dihedral angle Φ1. The Φ1s of P2 and Pb are 153.885 and 154.410, respectively, which are less than that of P4 and Pd by 25.875, 25.495.
This indicates that the meso-site substitution of phenylene ring significantly enhances the nonplanarity of the polyazomethine backbone. The PAM with different conformations also has effect on the dihedral angle Φ1, for example, Φ1 of P3 is 153.147 while the one of Pc with the corresponding unit is 179.908 due to that the carbazole plays a role on the nonplanarity of the backbone.
To better understand the evolution of the oxidation and reduction potentials, the electronic structures of the polymer series (as isolated oligomers) were evaluated by using DFT at the B3LYP/6-31G level of theory; the highest-occupied molecular orbital (HOMO) and lowest-unoccupied molecular orbital (LUMO) energies and HOMO–LUMO energy gaps (Eg) are given in Table 3. Pictorial representations of the frontier molecular orbitals of P1 to P5 and Pa to Pb are shown in Fig. 6.
| λ abs,max,solution(CHCl3)/film | λ PL , max solution (CHCl3) | λ abs,max solution THF | λ PL , max THF(EX) | Φ (CHCl3) | Exited most at intensity | λ abs,onset,solution/film onset | E g solution/film | E peak onset | E HOMO onset 0.506 | E LUMO (EHOMO − Eg(film)) | E LUMO quan | E HOMO quan | E g quan | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 431/436 | 466(330) | 430 | 473(335) | — | 335 | 530/563 | 2.34/2.20 | 0.895 | −5.189 | −2.989 | −1.873 | −4.739 | 2.865 |
| P2 | 413/419 | 460(375) | 406 | 487(330) | — | 330 | 468/489 | 2.65/2.54 | 0.832 | −5.126 | −2.586 | −2.136 | −4.626 | 2.491 |
| P3 | 444/450 | 560(340) | 442 | 527(295) | 0.00454 | 295 | 518/543 | 2.39/2.28 | 0.767 | −5.061 | −2.781 | −2.319 | −4.734 | 2.414 |
| P4 | 419/456 | 488(375) | 425 | 461(285) | 0.00665 | 285 | 483/506 | 2.57/2.45 | 0.973 | −5.187 | −2.737 | −1.975 | −4.712 | 2.737 |
| P5 | 474/482 | 604(355) | 475 | 594(305) | 0.00665 | 305 | 564/580 | 2.20/2.14 | 0.729 | −5.023 | −2.883 | −2.441 | −4.785 | 2.343 |
| Pa | 446/494 | 506(335) | 441 | 510(295) | — | 295 | 525/592 | 2.36/2.09 | 0.393 | −4.687 | −2.597 | −2.368 | −4.375 | 2.007 |
| Pb | 422/414 | 596(335) | 419 | 572(285) | 0.00103 | 285 | 493/523 | 2.52/2.37 | 0.536 | −4.83 | −2.46 | −1.877 | −4.489 | 2.611 |
| Pc | 456/431 | 602(335) | 461 | 607(305) | 0.00111 | 305 | 549/560 | 2.26/2.21 | 0.508 | −4.802 | −2.592 | −2.332 | −4.381 | 2.049 |
| Pd | 429/431 | 539(345) | 424 | 459,581(370) | 0.00282 | 370 | 500/507 | 2.48/2.45 | 0.522 | −4.816 | −2.366 | −2.037 | −4.357 | 2.320 |
| Pe | 500/499 | 635(355) | 491 | 653(280) | 0.000905 | 280 | 595/603 | 2.08/2.06 | 0.516 | −4.81 | −2.75 | −2.282 | −4.587 | 2.304 |
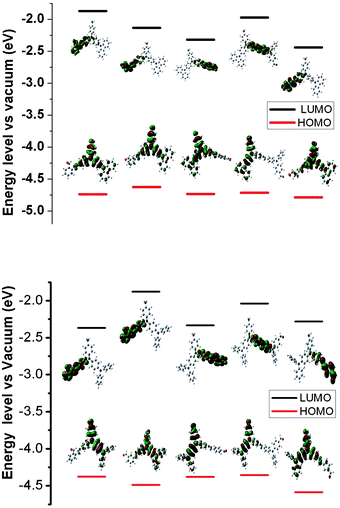 | ||
| Fig. 6 Pictorial representations of the electron density in the frontier molecular orbitals of repetition units for P1 to P5 (upper) and Pa to Pe (lower) in order. | ||
In the polymers with triphenylamine or carbazole units, the electron cloud distribution is strongly confined to the triphenylamine or carbazole ring containing end amine group in the HOMO, whereas for LUMO, the electron clouds delocalize over all chromophores including one aromatic ring of the triphenylamine or carbazole, aldehydes, and bridge bond (CHN) of the inter-rings, as shown in Fig. 6. All frontier orbitals in the oligomers spread over the whole p-conjugated backbones, with largest contributions coming from different chromophores, either bridge bond (CHN) or ring of dialdehyde moiety. This leads to an intramolecular charge transfer with significant distribution of the electronic density over the entire molecule. The large conjugated area provides an extended region across which the excited state can be efficiently dissipated. The quantum calculation suggests that the fluorescence is efficiently quenched by intramolecular photoinduced electron transfer (PET) that occurs from the amine HOMO to the LUMO of both the CH-aryl and azomethine moiety.36 The concentration-dependent fluorescence of the fluorenone azomethines was further examined. The experiment showed that the fluorescence peak did not decrease when the concentration increased. This was to ensure that the reduced azomethine fluorescence was not from self-quenching or from other intermolecular deactivation effects. The investigation of the quenching mechanism should be further carried out.
The large twist caused by triphenylamine along the polymer backbones prevents further conjugation across the π-system. The trend in Eg for the oligomer series follows P1 > P4 > P2 > P3 > P5, which basically corresponds with the experimental data (Table 3). The bias may be due to the adjacent ring which plays extra interaction on the energies. It should be noted that the calculated results are based on monomer unit and cis form, in fact, the experiments data are obtained from oligomer of polymer and more complicated.
The deeper energetic stabilization of the LUMO energies of P3 and Pc can be attributed to the more planar structure, thus allowing for stronger coupling between neighboring azomethine units. The HOMO–LUMO gap of P3 (Eg = 2.414 eV) is lower than the gap of P2 (Eg = 2.491 eV), and reflects the higher degree of conjugation of polymer P3 than P2. P2 and P4 have larger energy gaps than P5 which is attributed to the interruption of the linearity of the conjugated backbone.
For polymers, this implies that triphenylamine and carbazole units can serve as electron- donating functionalities due to the presence of the electron-rich nitrogen heteroatoms.
Electrochemical properties
Cyclic voltammograms (CVs) between −2.0 and 2.0 V of thin layer of P1–P5 and Pa–Pe coated from a polymer solution in chloroform onto ITO electrode are carried out and shown in Fig. 7. In comparison, the redox behavior of the two monomers has also been investigated by CV conducted in the solution state under the same conditions.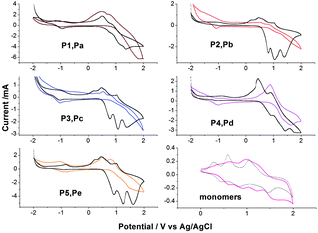 | ||
| Fig. 7 C–V curves of polyazomethines and monomers (curves in color are P1 to P5 and in black are Pa to Pb). | ||
Thus, the electrochemical behavior of triphenylamine-based polymers has, in common, three reversible oxidation peaks, near 0.9, 1.1 and 1.4 V. According to the electrochemical behavior of polyimides containing pendent triphenylamine,23 the first and second peaks can be attributed to the successive oxidations of the triphenylamine included in the oligoazomethine in the radical-cation and then the dication, and the third peak may be ascribed to oxidation of –CHN–. The first oxidation of polyazomethine is assumed to occur at the nitrogen center of the triphenylamine, which is linked to three electron-rich phenyl groups in a propeller-like geometry at 0.84 V. Polymer films degraded and dissolved in solution under higher potential during the oxidation process.
Comparing the electrochemical data with the monomer, the three oxidation steps of the PAMs unit are slightly more difficult than the electrochemical oxidation of pendent triphenylamine monomer. So, an electronic attractive effect of CN group on the triphenylamine moiety increases each characteristic oxidation potential.
The presence of moieties with different degree conjugation along the polymer backbone alters the bandgap energy when compared with the two other polyimides23,24 (Table 3).
In comparison, the Pa–Pe series were not only more readily oxidized and more stable than the P1–P5 series, but also exhibited lower HOMO values than the P1 series. This could be attributed to the triphenylamine (Ep,a = 0.98V in CH3CN)23 being easier to oxidize than N-phenylcarbazole (Ep,a = 1.35 V in CH3CN). The external ferrocene/ferrocenium (Fc/Fc+) redox standard Eonset (Fc/Fc+) is 0.506V vs.Ag/AgCl in CH3CN with respect that the HOMO energy for the Fc/Fc+ standard is − 4.80 eV to the zero vacuum level. As a result, the HOMO energy values were calculated using the equation
| EHOMO = −e (Eox + 4.296) |
| ELUMO = EHOMO + Egopt |
The energy of the HOMO and LUMO levels of the investigated PAMs can be determined from the oxidation onset potentials and the onset absorption wavelength and the results are listed in Table 3.
All of the calculated resulting HOMO of polyazomethines are higher than the corresponding poly(amine-amide-imide)s, poly(amine-amide)s and poly(amine-imides)s reported by Liou,23 which means the –CHN– could play the role of tuning the energy level of polymers.
As a result of CV measurements (Fig. 7), in the negative regime, the reversible peaks which reflect the reduction of imine (–CHN–) with peak potential of −1.05 and −1.00 V are observed for radical anion formation of P1 series and Pa series, respectively. The result shows that the carbazole-based PAMs are more difficult to reduction than the corresponding triphenyl-based ones. And the PAMs could also be used as n-type semconductivity.
Optical properties
The optical properties of the PAMs were investigated by UV-vis and photoluminescence (PL) spectroscopy (shown in Fig. 8). The results are summarized in Table 3. The organosoluble P1–P5 exhibited UV-vis absorption bands with λmax at 413–474 nm in CHCl3 solutions, assignable to the π–π* transition resulting from the conjugation between the triphenylamine moieties and aromatic rings linked by CN double bond.
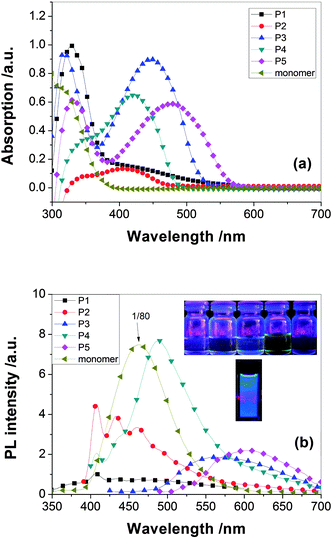 | ||
| Fig. 8 UV and PL spectra of P1–P5. | ||
Comparing with the absorption of the diaminetriphenylamine and dialdehyde monomers, the absorption of the novel polyazomethines in solution is different from that of the constitute chromophores. The color of the PAMs family of conjugated polymers is governed by the presence of two types of chromophore groups in their macromolecules, namely the aromatic rings and the imine groups. It implies that there is interaction between the chromophores in their ground states. It is also explained according to the molecular conformation.
In ref. 23, 24, because the spacer between the triphenylene or carbazole blocks out the transfer of electrons in the N atom, the absorption maximum peaks of the polyimide- or polyamide-containing triphenylamine or carbazole are in the range 295–350 nm. For comparison, the peaks of PAMs are significantly red-shifted to 420–500 nm, longer than polyimide and polyamide which suggested the conjugated structure had come into being. About 40 nm bathchromic shift of the absorption maxima is observed comparing the peaks of the P1–P5 with their TPA-containing analogues of PPV.37 In comparison with Pc (containing triphenylamine), P3 (containing carbazole units) shows a slight blue spectral shift both in absorption and emission in solution, that is, the blue-shifted spectra maxima being 2 nm for absorption and 17 nm for emission in film state. The spectra difference between Pc and P3 could also be explained by the slightly increased steric hindrance by the incorporation of carbazole group onto the rigid backbond. It increases the torsion angles between the carbazole and the CH–phenyl units and results in the reduction of effective conjugation along the polymer backbone (shown in Fig. 9). In addition, due to the steric hindrance, P3 achieved lower molecular-weight (Mw 5328) than Pc (Mw 13138), which also results in lower conjugation length for P3. Therefore, both increased steric hindrance and lower molecular-weight of P3 lead to its blue-shifted spectra in comparison with Pc. The results are in consistent with the XRD data.
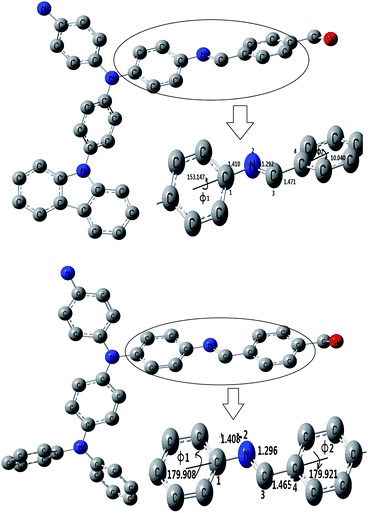 | ||
| Fig. 9 Calculated optimum configuration of P3 (upper) and Pc (below). | ||
The maximum absorption peaks of P1 series in solid film bathochromically shifted by 6–8nm with respect to that in solution due to aggregation of the planar carbazole units, which is contrary to the Pa series which are not apt to aggregation due to the propeller structure of triphenylamine.
The replacement of phenylene linkage by thiophene in the backbone produced a significant red shift of λmax by 30–60 nm and a reduction of the band gap by 0.14–0.4 eV. The incorporation of the thiophene units in the conjugated PAMs backbone could dramatically enhance the coplanarity of the flexible terthiophene rings with the rigid phenylene azomethine (CHNC6H4) units and improve the π-electron delocalization along the polymer backbone, which may involve the possible delocalization of the lone pair electrons of the sulfur atom in a thiophene ring.38
The PL emission results in solution also indicate that the optical properties of carbazole-based copolymers can be well tuned by properly rational design of polymer architectures, such as the incorporation of different phenylene units into the polymer backbone so as to adjust their effective conjugation length along polymer main chain.
The photoluminescence quantum yields of P1–P5 in chloroform solutions were measured relative to 10−5 M quinine sulfate in 0.1 M H2SO4. The results are summarized in Table 3. All polymers achieve low quantum yields in the solution state, ranging from 0.001–0.004, which are inferior to poly(arylenevinylene)s.22 The poor emission efficiency of the conjugated PAMs is similar to those of oligothienylene-based PAMs, phenylene-based conjugated PAMs, carbazole-linked PAMs and thiophene-linked PAMs.39 It may be due to the non-fluorescent n–p* transition of imine group of the polyazomethines which the heteroatomic azomethine bond efficiently quenches both the singlet excited and triplet states by intramolecular photoinduced electron transfer (PET) and energy transfer, respectively.38 The experimental results are consistent with those theoretical electronic properties mentioned above.
Spectroelectrochemical and electrochromic properties
The spectroelectrochemical properties of the polymer solution were monitored by a UV–vis spectrometer at different applied potentials. The absorption spectral changes of P3, P4 and P5 are shown in Fig. 10, which exhibit that λmax values for the π–π* transitions of P3, P4 and P5 are centered at 444, 419 and 474 nm, respectively. When the applied external potential was gradually raised from 0.6 to 1.2 V, the absorption intensity at 419 nm of P4 slightly decreased, while two new peaks at about 313 nm and 527 nm appeared. The new spectrum was assigned to the cationic radical polyazomethine+˙.23 Meanwhile, the P4 film color changed from yellow to red. When the applied potential was higher than 1.5 V, the film color was changed from red to purple which was different to the one changed from green to dark blue in the ref. 23. The purple film was not very stable and dissolved in solution.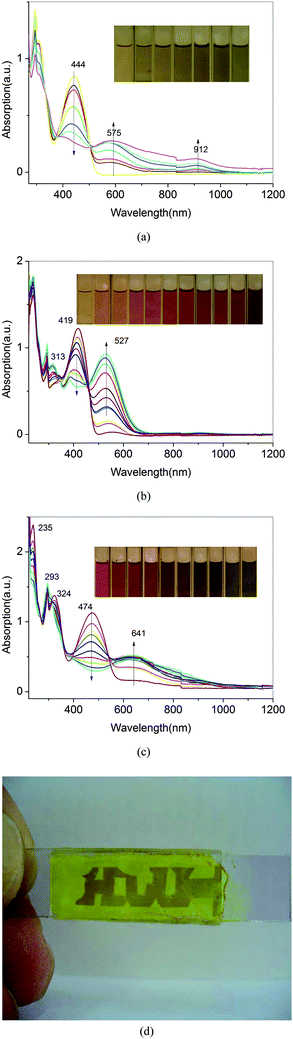 | ||
| Fig. 10 Electrochemical p-type doping electronic absorption spectra of P3(a), P4(b) and P5 (c) between 0.0 and 1.2 V with 0.05 V potential intervals in solution. (d) Photos of a sandwich-type ITO/P4/CH3CN–LiClO4/ITO device in oxidized semiconducting state. | ||
Stepwise oxidation of the polymer films by applied external potentials also resulted in a decrease in the π–π* transitions and their lower energy transitions intensified at 575,912 for P3 and 641 nm for P5, respectively. Since neutral state absorptions for P3 and P5 were at 444 and 474 nm and lower energy transition emerged at 575 and 641 nm, partially oxidized polymer solution revealed blue-green color for P3 and purple for P5. Further oxidation of P3 and P5 films resulted in blue color due to diminished π–π* transition intensity, respectively. A similar spectral change was observed for other polyazomethines of P1–P5 series and the corresponding Pa–Pe series.
Fig. 10(d) demonstrated the sandwich-type electrochromic cell in oxidized semiconducting state, in which the color can be sustained all the time without additional external potential and would not change until the opposite potential was applied, which suggests that the electrochromic cell fulfils the needs for adaptive camouflage.
The stability and response time upon electrochromic switching of the P5 film between its neutral and oxidized forms in the visible (474 and 641 nm) was monitored (Fig. 11). The color switching times were estimated by applying a potential step, and the absorbance profiles were followed. The switching time was defined as the time required to reach 90% of the full change in absorbance after switching potential. The thin P5 film would require 1.0 s at 1.10 V for switching absorbance at 641 nm and 1.0 s for bleaching. After ten continuous cyclic scans between 0.00 and 1.10 V, the polymer films still exhibited good stability of electrochromic characteristics.
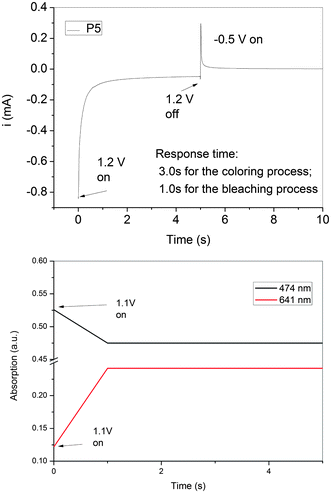 | ||
| Fig. 11 Dynamic changes of the current and transmittance upon switching the potential between −0.5 and 1.1 V (vs.Ag/AgCl) with a pulse width of 10 s applied to the cast film of P5 on the ITO-coated glass slide in MeCN containing 0.1 M LiClO4. The absorption was recorded at 474 and 641 nm, respectively. | ||
During the CV experiments, the PAM films dissolved quickly when the potential was more than 2.0V, and the solution turned to green, then to blue quickly which meant some degradation on the overoxidation.
Doping with acid
As seen from the spectra (Fig. 12), initially neutral P3, P4 and P5 in solution have absorption maxima at 444, 419 and 474 nm, respectively. Upon continual exposure to HCl vapor, new absorption bands emerged at 600, 540 and 650 nm which were accompanied by a decrease in neutral state absorptions. Neutral solutions of the three kinds of polymers were orange, yellow and red, however upon the exposure of HCl vapor, the solution turned into blue, purple and blue as is shown as inset picture in Fig. 12 for P3, P4 and P5, respectively. It is probably because the protonation on the nitrogen of the imine linkage by acid results in the backbone planarization, which leads to an increased π-electron delocalization. Absorption spectra for P3, P4 and P5 reveal isosbestic point in solution form which confirms the coexistence of discrete chromophores. It also eased the detection of different colors for P3, P4 and P5 solutions upon HCl vapor or acid addition in solution.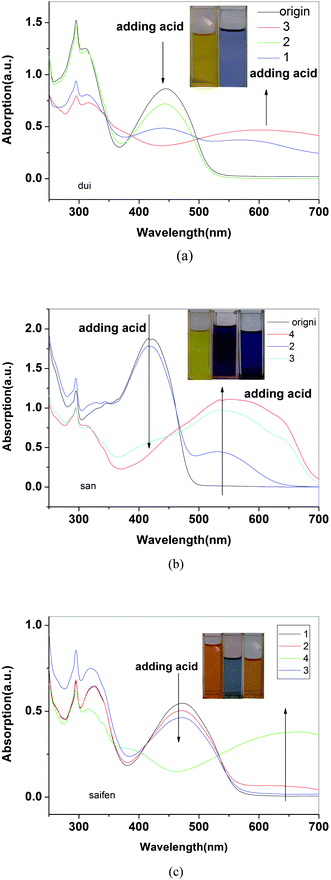 | ||
| Fig. 12 The UV-vis absorption spectra of solutions of P3(a), P4(b) and P5(c) in various concentrations of HCl vapor, respectively. | ||
Further exposure of the solution to ammonia vapor reversed the polymer to the initial colors for the polymers (shown in the inset of Fig. 12(c)). The ammonium chloride formed during the deprotonation steps remained trapped in the solution, which made some opaque in the solution. This indicates the reversibility of solution doping process by adding HCl and pH responsive characters of polymers.
Doping with oxidation by FeCl3 and I2
The compounds were additionally oxidized chemically with FeCl3 to confirm their stability. As seen in Fig. 13, the azomethines undergo significant colour changes similar to those obtained anodically. The absorbance at 444 nm, corresponding to the neutral state of P3, decreases concomitant with the formation of a new peak at 600 nm. It can be concluded that the oxidized species are stable since their color is persistent and does not dissipate with time, which otherwise is indicative of decomposition. The stability of P3 and its oxidized form are further epitomized by the reversible change in absorbance intensity with the addition of hydrazine. The same phenomena take place in P1–P5 series and Pa–Pe series. Upon doping with iodine, the solution-cast films turn dark brown (shown in Fig. 14). The π–π* absorption band of P3 at 2.75 eV disappears upon oxidation and a new absorption bands appear at higher energy (1.93eV) and a new low-energy absorption appears at 3.17 eV.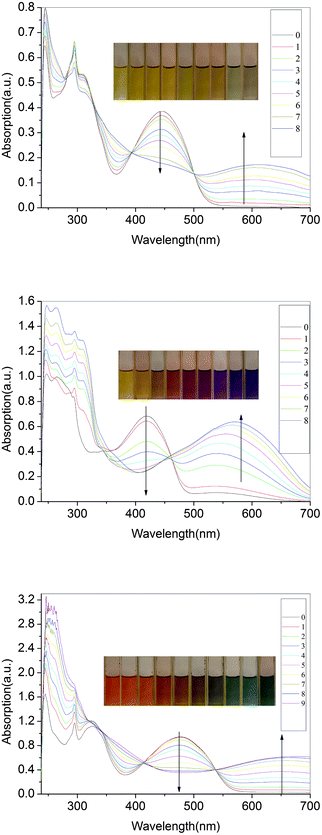 | ||
| Fig. 13 Spectra of P3(a), P4(b) and P5(c) recorded in anhydrous and deaerated dichloromethane with FeCl3 for 0, 2, 4, 6, 8, 10, 12, 14, 16, 18 min. | ||
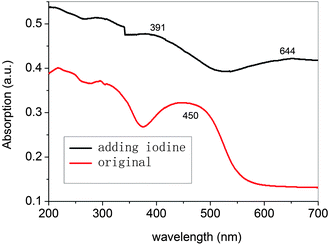 | ||
| Fig. 14 UV-visible absorption spectra of the CHCl3 solution-cast thin films of P3 in (A) neutral form and (B) iodine-doped form. | ||
In comparison with that of being doped with HCl vapor, the colors of polymers doped with I2 vapor were deeper, and the recovered-time was also longer. Even more, the reversible property of being doped with HCl was better. After long time, the polymers doped with I2 became some sticky suggesting that there was some cross linking in the complex.
Irradiation under the UV
Fig. 15 shows the UV-vis absorption spectra of the polyazomethine in CHCl3 under UV irradiation, respectively.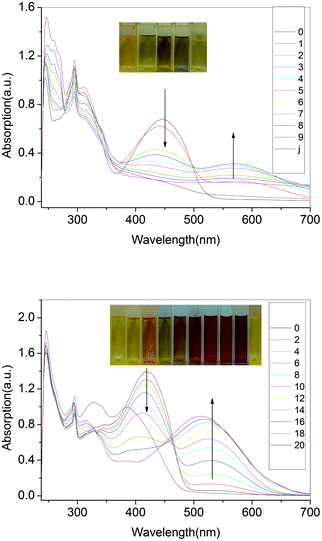 | ||
| Fig. 15 Spectra of P3(a), P4(b) recorded in anhydrous and deaerated CHCl3 under UV at 345 nm for 0, 2, 4, 6, 8, 10, 12, 14, 16, 18, 20 s. | ||
By prolonging the irradiation time, the absorption maxima were red shifted from 444 to 589 nm for P3 and 419 to 530 nm for P4, respectively. Similar phenomena were depicted in ref. 26 and 40. The authors ascribed the result to formation of highly stable trityl cations which contributed to the extensive delocation of the charge onto the phenyl rings and TPA. We also determined the polymers in NMP solution and PMMA matrix in the same concentration, however, it was strange that no such phenomenon took place. The CHCl3 seemed to decompose and emit HCl under the UV irradiation. So the result was attributed to the doping reaction of HCl with the polymers.
The variation of the absorption spectra of the carbazole-based PAMs with acidity, metal complex and HCl suggests that these polymers may have potential applications for chemical sensors.
Conclusions
Multicolored electrochromic alternating triphenylamine and N-carbazol copolymers of P1 and Pa series were synthesized and investigated in terms of their electronic and optical properties. Introduction of electron-donating triphenylamine or carbazole groups to the main chain of polymer not only affords good thermal stability but also leads to good solubility of the polyazomethines. The P1 and Pa series show multicolored states when doped with dopants such as FeCl3 and I2 or when the pH is altered by addition of various acids or removal of acid dopant by ammonia, making the polymers responsive chromophores that can be used in different applications in due course. The spectroelectrochemical results show the good electrochromic and effective hole-transporting properties of PAMs in displays. The reported properties prove that the polyazomethines are multipurpose materials which will be the subject of further research interest in the near future.Acknowledgements
The authors are grateful for the support of the National Science Foundation of China (Grant No. 21074031, 50872024, 20872030), Heilongjiang University Outstanding Youth Science Foundation, Foundation of Heilongjiang Education Bureau (12511379), Foundation of Heilongjiang University Innovator Group (No. hdtd-2010-11).Notes and references
- W. H. Chen, K. L. Wang, W. Y. Hung, J. C. Jiang, D. J. Liaw, K. R. Lee, J. Y. Lai and C. L. Chen, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4654 CrossRef CAS
; A. Kaeser and A. P. H. J. Schenning, Adv. Mater., 2010, 22, 2985 CrossRef
- J. Song, Q. Cheng, S. Kopta and R. C. Stevens, J. Am. Chem. Soc., 2001, 123(14), 3205 CrossRef CAS
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028 CrossRef CAS
- C. S. Wu and Y. Chen, Macromolecules, 2009, 42(11), 3729 CrossRef CAS
- J. Y. Kim, Y. Qin, D. M. Stevens, V. Kalihari, M. A. Hillmyer and C. D. Frisbie, J. Phys. Chem. C, 2009, 113(52), 21928 CAS
- Z. K. Chen, N. H. Sim Lee, W. Huang, Y. S. Xu and Y. Cao, Macromolecules, 2003, 36(4), 1009 CrossRef CAS
- G. Sang, Y. Zou and Y. Li, J. Phys. Chem. C, 2008, 112(31), 12058 CAS
- Y. Tian, C. Y. Chen, H. L. Yip, W. C. Wu, W. C. Chen and A. K. Y. Jen, Macromolecules, 2010, 43(1), 282 CrossRef CAS
- C. J. Yang and S. A. Jenekhe, Macromolecules, 1995, 28, 1180 CrossRef CAS
- L. Song, C. Tu, Y. Shi, F. Qiu, L. He, Y. Jiang, Q. Zhu, B. Zhu, D. Yan and X. Zhu, Macromol. Rapid Commun, 2009, 30(5), 443 Search PubMed
- H. Kim, S. B. Park, J. C. Jung and W. C. Zin, Polymer, 1996, 37, 2845 CrossRef CAS
- P. Gutch, S. Banerjee, D. C. Gupta and D. K. Jaiswal, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 383 CrossRef CAS
- G. Zotti, A. Randi, S. Destri, W. Porzio and G. Schiavon, Chem. Mater., 2002, 14, 4550–4557 CrossRef CAS
- G. S. Liou, H. Y. Lin, Y. L. Hsieh and Y. L. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4921 CrossRef CAS
- Q. X. Tong, S. L. Lai, M. Y. Chan, K. H. Lai, J. X. Tang, H. L. Kwong, C. S. Lee and S. T. Lee, Chem. Mater., 2007, 19(24), 5851 CrossRef CAS
- M. W. Thesen, B. Hofer, M. Debeaux, S. Janietz, A. Wedel, A. Kohler, H. H. Johannes and H. Krueger, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3417 CrossRef CAS
- Y. Song, C. Di, X. Yang, S. Li, W. Xu, Y. Liu, L. Yang, Z. Shuai, D. Zhang and D. Zhu, J. Am. Chem. Soc., 2006, 128(50), 15940 CrossRef CAS
- X. Wang, F. Jin, Z. Chen, S. Liu, X. Wang, X. Duan, X. Tao and M. Jiang, J. Phys. Chem. C, 2011, 115(3), 776 CAS
- Q. D. Ling, F. C. Chang, Y. Song, C. X. Zhu, D. J. Liaw, D. S. H. Chan, E. T. Kang and K. G. Neoh, J. Am. Chem. Soc., 2006, 128(27), 8732 CrossRef CAS
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC
- W. H. Chen, K. L. Wang, D. J. Liaw, K. R. Lee and J. Y. Lai, Macromolecules, 2010, 43, 2236 CrossRef CAS
- S. Koyuncu, I. Kaya, F. B. Koyuncu and E. Ozdemir, Synth. Met., 2009, 159, 1034 CrossRef CAS
- G. S. Liou, S. H. Hsiao and H. W. Chen, J. Mater. Chem., 2006, 16, 1831 RSC
- S. H. Cheng, S. H. Hsiao, T. H. Su and G. S. Liou, Macromolecules, 2005, 38, 307 CrossRef CAS
- C. W. Chang, C. H. Chung and G. S. Liou, Macromolecules, 2008, 41, 8441 CrossRef CAS
- H. Niu, P. Luo, M. Zhang, L. Zhang, L. Hao, J. Luo, X. Bai and W. Wang, Eur. Polym. J., 2009, 45, 3058 CrossRef CAS
- D. Sek, A. Iwan, B. Jarzabek, B. Kaczmarczyk, J. Kasperczyk, Z. Mazurak, M. Domanski, K. Karon and M. Lapkowski, Macromolecules, 2008, 41, 6653 CrossRef CAS
- F. C. Tsai, C. C. Chang, C. L. Liu, W. C. Chen and S. A. Jenekhe, Macromolecules, 2005, 38, 1958 CrossRef CAS
- T. E. Olinga, S. Destri, C. Botta, W. Porzio and R. Consonni, Macromolecules, 1998, 31, 1070 CrossRef CAS
- A. Bolduc, S. Dufresne and W. G. Skene, J. Mater. Chem., 2010, 20, 4820 RSC
- N. Leclerc, A. Michaud, K. Sirois, J. F. Morin and M. Leclerc, Adv. Funct. Mater., 2006, 16, 1694 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G.
E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98, Gaussian, Inc., Pittsburgh PA, 1998 Search PubMed
- X. Li, Y. Jiao and S. Li, Eur. Polym. J., 1991, 27, 1353 CrossRef CAS
- O. D. Is, F. B. Koyuncu and S. K. E. Ozdemir, Polymer, 2010, 51, 1663 CrossRef CAS
- J. C. Hindson, B. Ulgut, R. H. Friend, N. C. Greenham, B. Norder, A. Kotlewskic and T. J. Dingemans, J. Mater. Chem., 2010, 20, 937 RSC
- S. Dufresne, L. Callaghan and W. G. Skene, J. Phys. Chem. B, 2009, 113, 15541 CrossRef CAS
- M. Zheng, F. Bai, Y. Li and D. Zhu, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2587 CrossRef CAS
- A. N. Bourque, S. Dufresne and W. G. Skene, J. Phys. Chem. C, 2009, 113, 19677 Search PubMed
- C. L. Liu and W. C. Chen, Macromol. Chem. Phys., 2005, 206, 2212 CrossRef CAS
- G.-S. Liou, H.-W. Chang, K.-H. Lin and Y. O. Su, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2118 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00237f |
| This journal is © The Royal Society of Chemistry 2011 |