Facile routes to star polymersvia an organocatalytic approach†
Daniel J.
Coady
a,
Amanda C.
Engler
a,
Yi Yan
Yang
b and
James L.
Hedrick
*a
a650 Harry Rd, San Jose, CA 95120, USA. E-mail: hedrick@almaden.ibm.com; Fax: +1 408 927 3310; Tel: +1 408 927 1632
b31 Biopolis Way, The Nanos, Singapore 138669. E-mail: yyyang@ibn.a-star.edu.sg; Fax: + 65 6824 7106; Tel: +65 6478 9080
First published on 16th September 2011
Abstract
Herein the synthesis of star polymers from common dendrimers and commercially available compounds via organocatalyzed ring opening polymerization is described. Through the use of selective hydrogen bonding catalysis both homo and block star polymers were generated with exceptional fidelity having specified Mn and narrow polydispersity. This report demonstrates the broad potential of these methods for the creation of uniform materials exhibiting ideal properties for designer delivery systems.
Introduction
The fact that current healthcare costs have risen dramatically and are expected to continually increase has placed paramount importance on the development of more efficacious administration of therapeutics.1,2 Through collaborative efforts, chemists, engineers, and biologists have begun assembling the tools and techniques necessary for creating synthetic biological delivery vehicles. These new vectors are expected to significantly increase therapeutic potency and bioavailability while reducing unwanted toxic side effects.3 More specifically, through alteration of composition, functionality, and architecture, polymeric delivery systems can be designed to regulate drug solubility, biodistribution, and release kinetics.4A commonly employed delivery strategy utilizes micelles loaded with therapeutic compounds. This method is characterized by amphiphilic macromolecules which undergo self-assembly upon changes in media polarity to form 3-dimensional structures.5–7 During the micellization process cargo can be sequestered into the interior thus providing a facile means for the encapsulation of therapeutics (Fig. 1). This technique has proven invaluable for administration of compounds that otherwise would be extremely difficult or impossible to deliver because of poor solubility within the body.8 Unfortunately, disparity in aggregation number during the micellization process, albeit relatively small, can cause a distribution in particle size. Considering that particle size dictates vascular circulation half-life as well as cellular uptake, any variation significantly impacts the pharmakinetic behavior.3,7 For example, systemic administration of drug-loaded micelles larger than 200 nm will be sequestered in the spleen and taken up by phagocytes.9 Conversely, if the particle size is too small (<10 nm) it is rapidly removed through extravasation and renal clearance.10 Even within the aforementioned size range, particles larger than 100 nm normally will not enter cellsviaendocytosis further exemplifying the biodistribution dependence on particle size.11 Furthermore, a critical micelle concentration (CMC) is typically found when assembling amphiphilic polymers. Upon dilution past their respective CMC, these assemblies will disintegrate prematurely randomly releasing their contents. This complication becomes extremely problematic when attempting systemic delivery within the blood stream. A myriad of clever methods have been applied to resolve this issue, many with great success.12–14 However, these strategies all require additional functionality, and often additional steps, to provide the needed stability under high dilution conditions.
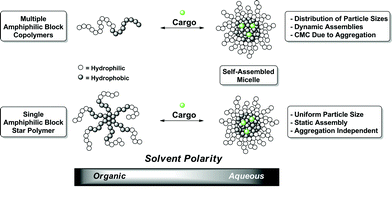 | ||
| Fig. 1 Generic illustration of the assemblies and loading of linear and star block copolymers. | ||
The use of functionalized dendrimers provides an elegant solution to the prior mentioned limitations.6,15 These globular macromolecules can undergo a similar self-assembly, however, their densely branched spherical structures afford unimolecular micelles, rather than aggregates, thereby eliminating inconsistent particle size.16 They also provide a platform for installing exceptionally regular functionality allowing for virtually limitless customization potential. Until recently, lengthy and laborious syntheses have kept dendrimer research primarily localized within a relative few research groups. This fact is now rapidly changing due to increased commercial availability combined with new advances in synthetic techniques that are ushering in a new era of dendrimer based delivery systems.17–22
Despite the tremendous utility and potential of dendrimers, they are generally at a disadvantage to commercial polymers in terms of intrinsic cost. Through the development of star polymer systems researchers can combine the advantages of polymers (inexpensive, rapidly generated, multitude of functionality, etc.) with the extremely regular and controlled nature of spherical dendrimers allowing not only a similar globular shape but also a potentially layered structure.23–26 Star polymers result from two distinct approaches - arm first27 or core first. For the purpose of this report we will only focus on the core first approach, where polyvalent initiators are used to grow outward radiating polymer arms. Production of uniform star polymers (materials having a narrow size distribution, controllable Mn, and an ability to create regular, reproducible block structures) from such initiators mandates the use of controlled or living polymerization techniques.28 Controlled radical polymerization techniques have allowed the exploration of complex architectures using the abundance of commercially available olefin monomers.29–39 Even so, petroleum based materials are not always the most desirable. Their polymerization creates exceptionally stable C–C bonds that do not undergo natural biological remediation. Perhaps even more importantly, olefin monomers and their corresponding catalysts often show unwanted toxicities reducing utility within the healthcare field. An alternative to polyolefins was found using controlled ring opening polymerization (ROP) of cyclic esters and carbonates. This method can produce highly controlled, non-toxic materials which can be entirely derived from renewable resources.40–42 The resulting polyesters and polycarbonates have been found to be completely stable under ambient conditions, yet still permit enzymatic degradation through normal metabolic pathways.43 These features provide an ideal synthetic platform for the creation of star polymer transport vectors.
Until recently organometallic catalysts have been the gold standard for polymerizing cyclic esters and carbonates. However, use of organocatalysis has been receiving increased attention because of its incredible control and simple operation.44 Unlike metallic catalysts (Sn, Zn, Ti, etc.), organocatalyts are non-oxophilic making them more readily removed from the resulting oxygen rich polymers. The removal of unwanted catalyst imparts two important benefits. First, total removal of the polymerization catalyst will significantly decrease the frequency of problematic transesterification reactions, and secondly, harmful toxicity imparted by the catalyst can be greatly reduced or eliminated. Additionally, ROP organocatalysts are generally very stable, inexpensive, and commercially available making them accessible to researchers of all experience and skill level. Finally, they provide a level of polymerization control that equals or even exceeds that of organometallic catalysts using only hydrogen bonding (Scheme 1).45–47 Herein, our efforts toward the construction of highly ordered star polymers using organocatalyzed ROP from multifunctionalized initiators are described.
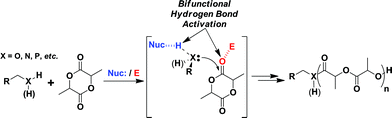 | ||
| Scheme 1 Proposed catalytic mechanism of ROP of lactide. | ||
Results and discussion
This report demonstrates possible initiators and corresponding organocatalytic polymerization conditions that yield a universal star polymer platform. Initiators evaluated in this work consist of commercially available compounds bearing multiple nucleophilic initiation sites such as dendrimers, sugars, and condensation products. This approach required matching the appropriate initiator with a corresponding organocatalyst and solvent system such that efficient initiation was followed by uniform polymer growth. Initiating moieties can be almost any nucleophile, however, we limited this study to most commonly used hydroxyl and amine functionalities (Scheme 1). Considering the vast array of possible cyclic monomers available we restricted our investigation to (L)-lactide and trimethylene carbonate (TMC) because of their routine use in polymeric delivery systems, varied reaction kinetics, commercial availability and functionalization potential.44,48 Furthermore, all initiators were tested for their ability to form both homopolymers and block copolymers demonstrating their feasibility in more advanced systems.Bis-MPA based initiators
The first initiators tested were monodisperse bis-MPA dendrimers first reported by Frechet et al.49 Using a modified literature procedure dendrimer generations 1–4 were synthesized from pentaerythritol (Scheme 2). Initial attempts using the dendrimers to polymerize (L)-lactide with (-)-sparteine/N′-(3,5-bis(trifluoromethyl)phenyl)-N-cyclohexyl-thiourea (TU) proved extremely problematic because of poor initiator solubility in DCM or other standard ROP solvents (THF, toluene, etc.). In order to circumvent this problem the initiator and catalysts ((-)-sparteine/TU) were first dissolved in a minimum amount of dimethylsulfoxide (DMSO) followed by slow addition of a separate 2M DCM solution of (L)-lactide while maintaining homogeneity. Polymerizations of (L)-lactide from the various dendrimer generations using the (-)-sparteine/TU system achieved complete conversion in less than 30 min and produced monomodal star polymers with polydispersity (PDI) values under 1.08 (targeted DP per arm was 20) (Table 1) as determined by gel permeation chromatography (GPC).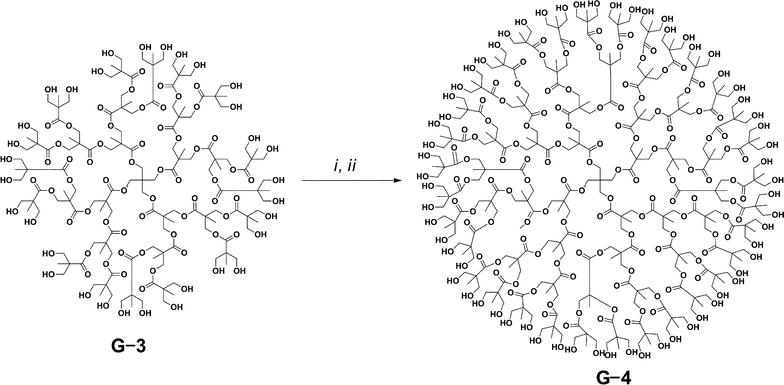 | ||
Scheme 2 Synthesis of bis-MPA generation-4 from G-3 using (i) benzylidene-2,2-bis(oxymethyl)propionic anhydride, N,N-dimethylaminopyridine, pyridine: tetrahydrofuran (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) at room temperature; (ii) Pd/C (10%), H2 (50 psi) and ethyl actetate: methanol (1:2). :4) at room temperature; (ii) Pd/C (10%), H2 (50 psi) and ethyl actetate: methanol (1:2). | ||
| Initiator | DP | Mn | Mw | Mp | PDI |
|---|---|---|---|---|---|
| a See ESI, Fig. 9 for GPC trace which shows peak front loading. | |||||
| β-Cyclodextrin | 20 | 54300 | 57300 | 61600 | 1.06 |
| Boltorn | 20 | 45400 | 52400 | 53300 | 1.15 |
| Bis MPA Dendrimers | |||||
| G-1 | 20 | 29000 | 30400 | 31700 | 1.05 |
| G-2 | 20 | 45500 | 48100 | 50800 | 1.06 |
| G-3 | 20 | 70700 | 74600 | 78200 | 1.05 |
| G-4 a | 20 | 74700 | 80700 | 69600 | 1.08 |
| PPI Dendrimers | |||||
| G-1 | 20 | 29400 | 31000 | 31000 | 1.06 |
| G-2 | 20 | 46600 | 48100 | 50200 | 1.03 |
| G-3 | 20 | 64100 | 68900 | 64500 | 1.07 |
| G-4 a | 20 | 87600 | 96400 | 70600 | 1.10 |
To better understand initiation efficiency, the experiment was repeated using an average arm length of five (L)-lactide units (DP = 5). By shortening the polymer arms, chemical shifts attributed to the polymer chains were greatly reduced while allowing more than sufficient arm length to effect complete initiation. Heteronuclear multiple quantum coherence (HMQC) experiments were performed to obtain 2D-NMR spectra. This experiment was selected over traditional 13C NMR because of the ease of peak identification as well as increased signal amplification compared to 13C NMR.50Fig. 2 demonstrates an overlayed 2D-NMR spectra of the G-3 bis-MPA dendrimer (red) and G-3 bis-MPA dendrimer with an average of 5 L-lactide repeat units initiated off each arm (blue). Strong signals from both b and c (red) can be clearly seen prior to polymerization. After the polymerization of (L)-lactide (blue), b completely disappears while signal c shows very little movement indicating quantitative initiation.
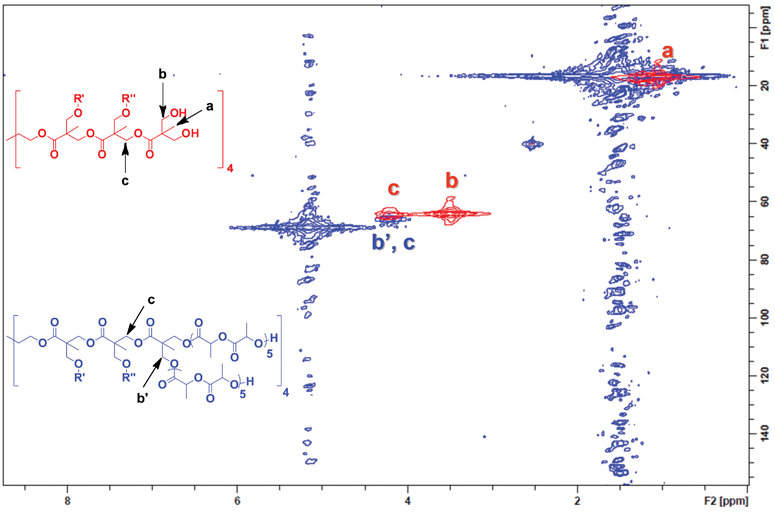 | ||
| Fig. 2 Complete initiation of the bis-MPA G-3 dendrimer was confirmed by 2D-NMR. The G-3 initiator (red spectra) shows the terminal methylene proton/carbon coupling b, which completely transforms into a new chemical shift b′ on the star polymer spectra (blue). | ||
The bis-MPA dendrimers provided an ideal system for evaluating the effects of increasing star polymer arms on initiation and resulting materials. Through increasing the dendrimer generation the number of polymer arms could be sequentially doubled. Again, using 2D-NMR quantitative initiation was found for all (L)-lactide polymerizations from bis-MPA dendrimer generations 1–4 (8, 16, 32, 64 arms respectively, remaining spectra in supporting information). These results suggest that the initiating hydroxyl groups were not adversely affected by the densely packed initiation sites found on higher generation dendrimers. However, the GPC trace found for the G-4 dendrimer produced an oddly shaped peak (ESI,† Fig. 9). The observed front loading of the peak was assumed to be a product of the extremely bulky nature of the star system.
The ability to form block stars is an essential feature for synthesizing delivery vehicles. Since it was previously determined that bis-MPA dendrimers are capable of forming homo-star polymers from (L)-lactide, we turned our attention to forming a second block from TMC. The formation of block stars can be accomplished in two different ways - polymerization from a macroinitiator or by sequential addition of a second monomer at near full first block conversion. Both methods were evaluated and compared using the G-3 bis-MPA dendrimer. A macroinitiator was produced using the same polymerization procedure (DP = 20) and fully characterized after precipitation into cold isopropanol. It was then subjected to chain extension with TMC (DP = 20) using DBU/TU as the polymerization catalyst in 2M DCM. It should be noted that DMSO was no longer required because of improved solubility and DBU was chosen due to its more favorable kinetics when polymerizing TMC. Despite the macroinitiator having 2° alcohols no initiation issues were observed from any dendrimer generation.
Utilizing the sequential addition method proved more difficult than forming block stars from macroinitiators. As with the macroinitiator method the first block was synthesized as previously described. However, upon >90% conversion of (L)-lactide, a 4M DCM solution of TMC was immediately added for installation of the second block. Unfortunately, the presence of DMSO and continued catalyst dilution drastically reduced polymerization kinetics. This problem was addressed by adding 5 mol% DBU catalyst following TMC addition. The DBU facilitated the 2nd block polymerization in ∼2.5 h whilst keeping excellent reaction control (Fig. 3). Sparteine is generally a more selective catalyst to use despite its slower polymerization kinetics because it lacks the ability to epimerize (L)-lactide and shows little to no unwanted transterification.44 However, when increased reaction rates are desired DBU provides an excellent alternative assuming the reaction is stopped promptly prior to or upon complete conversion. Regardless, the sequentially formed block stars produced material closely matching that of the macroinitiator method (Table 2).
| Catalyst | Rxn Time | Mn | PDI | |
|---|---|---|---|---|
| Macro Initiator | ||||
| Bis MPA G-3 | ||||
| LAC20 | SP | 30 min | 70735 | 1.05 |
| LAC20 TMC16 | SP | 24 h | 106906 | 1.08 |
| LAC20 TMC15 | DBU | 2 h | 93000 | 1.07 |
| Boltorn | ||||
| LAC20 | SP | 30 min | 45415 | 1.15 |
| LAC20 TMC18 | DBU | 24 h | 55998 | 1.25 |
| B Cyclodextrin | ||||
| LAC20 | SP | 30 min | 59400 | 1.05 |
| LAC20 TMC14 | DBU | 2 h | 70200 | 1.05 |
| Sequential Addition | ||||
| Bis MPA G-3 | ||||
| LAC20 | SP | 30 min | 67168 | 1.04 |
| LAC20 TMC16 | SP | 14 days | 112221 | 1.07 |
| LAC20 TMC14 | DBU | 2.25 h | 94526 | 1.08 |
| PPI G-3 | ||||
| LAC20 | SP | 30 min | 64108 | 1.07 |
| LAC20 TMC18 | SP | 15 h | 100714 | 1.06 |
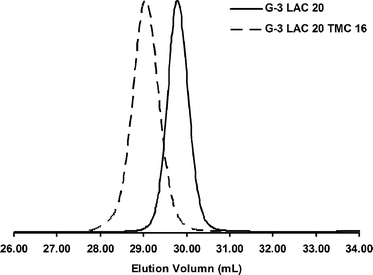 | ||
| Fig. 3 THF GPC traces of bis-MPA G-3 dendrimer initiated star polymers. | ||
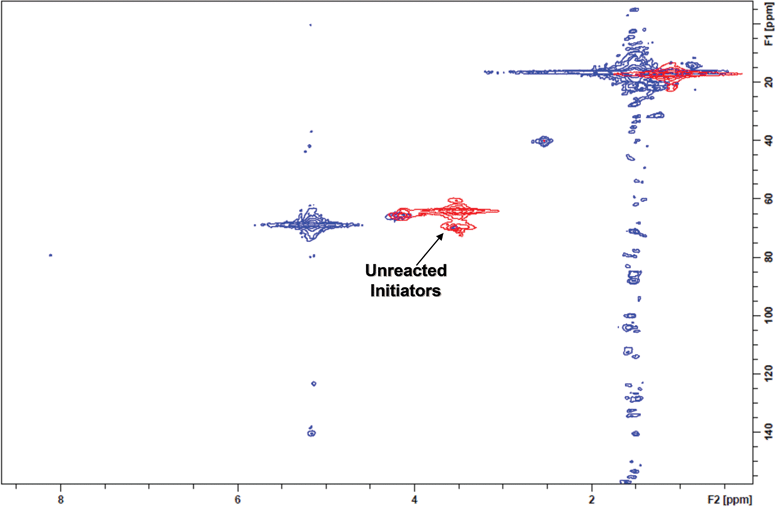 | ||
| Fig. 4 The 2D-NMR of Boltorn H20® (red) was unable to grow polymer from all initiation sites (blue) as determined by the incomplete shift of all terminal methylene/carbon coupling signals. | ||
An alternative to using discrete dendrimers was found in Boltorn H20® a commercially available condensation product of 2,2-bis(hydroxymethyl) propionic acid (bis-MPA). This material provided an inexpensive multifunctional initiator having an average of 16 hydroxyl groups per initiator molecule.51–53 Analogous to the monodisperse bis-MPA dendrimers, the Boltorn H20®dendrimers were essentially insoluble in all appropriate ROP solvents; therefore DMSO was again used to promote a homogenous polymerization. Using this method, reaction homogeneity was maintained and permitted full conversion of (L)-lactide as determined by proton NMR. By GPC the molecular weight and PDI were determined to be 45,400 Da and 1.15, respectively. Despite having a relatively narrow GPC trace, the shape of the curve was not completely Gaussian and consistently tails in the lower molecular weight region. This tailing in the lower molecular weight region despite the relatively narrow PDI was found to be intrinsic to the initiator. Since the Boltorn® material was formed through a poly-condensation reaction of bis-MPA, primary hydroxyl groups are theoretically dispersed throughout the generation layers. This dispersion of hydroxyl groups translates into sterically hindered interior primary alcohols that cannot initiate at the same efficiency as ones found on the periphery. The incomplete initiation was readily observed using 2D-NMR (Fig. 4). Additionally, even though Boltorn H20® has an average of 16 hydroxyl groups per molecule there is still a distribution of individual functionality amongst molecules, contributing to the further broadening of polydispersity.
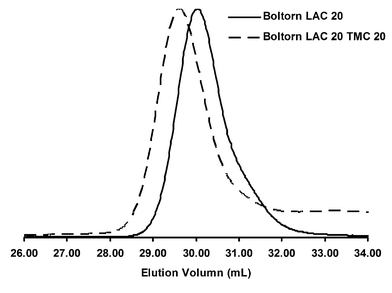 | ||
| Fig. 5 GPC traces of Boltorn H20® initiated star polymers. | ||
Although there was not complete initiation of the Boltorn H20®, we were able to add a TMC block with relatively high success. Formation of the second block proceeded uniformly suggesting that any unreacted hydroxyl groups remained unreacted post second block formation (Fig. 5). Even though Boltorn H20® was not able to form star polymers with the same fidelity as the previous dendrimer system, it did prove to be a readily available option for the synthesis of inexpensive star polymers.
Amine functionalized dendrimers
In addition to alcohols, primary amines have also been found to make outstanding star polymer initiators. Poly(propyleneimine) (PPI) dendrimers provided a readily available and highly regular multifunctional amine initiator.54,55 An advantage of PPI dendrimers when compared to polyamidoamine (PAMAM) dendrimers, and the aforementioned poly(alcohol)s, was their complete solubility in DCM eliminating the need for a solubilizing agent such as DMSO. The trial polymerization of (L)-lactide from PPI G-1 (8 amine functional groups) using (-)-sparteine/TU was found to give experimentally identical results to the G-1 bis-MPA initiator with the minor exception of amide solubility (Scheme 3). This change in solubility was believed to be a manifestation of the increased intermolecular hydrogen bonding interactions of the newly formed amide bonds. The temporary haze (∼30 s) quickly subsided during the initial stages of the reaction, returning the polymerization solution to full homogeneity. As expected narrow PDI values for the PPI initiators were obtained (Table 1).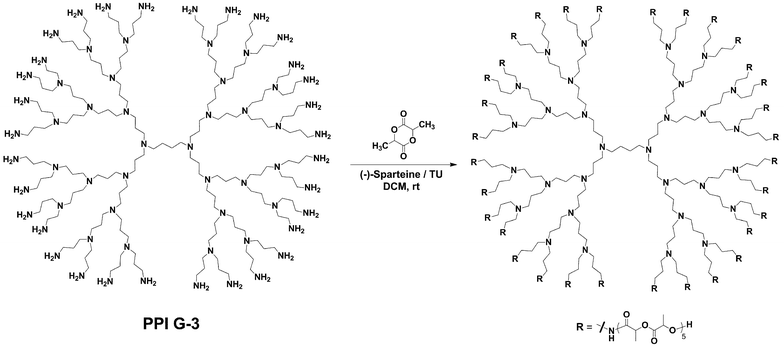 | ||
| Scheme 3 Synthesis of a lactide star polymer from the PPI G-3 initiator. | ||
The PPI dendrimer series provided an ideal system for direct comparison of hydroxyl compared to amine initiation whilst keeping the respective amounts of polymer arms constant. Both the PPI and bis-MPA systems presented an initiator series having 8, 16, 32, and 64 polymer arms which produced stars having comparable Mn and PDI values (Table 1). As seen with the G-4 bis-MPA system, the G-4 PPI initiators produced similar front loading on their GPC traces. (Table 1 and ESI,† Fig. 9).
To confirm quantitative initiation of the PPI system, both 1H-NMR and 2D-NMR characterization were conducted on G-(1–4) star polymers (arm DP = 5) (Fig. 6). The proton NMR spectra proved very useful because the terminal (L)-lactide methine proton could be integrated against the other poly(L-lactide) methine peaks to corroborate the theoretical average of five repeat units per arm.‡ Using 2D-NMR it was observed that all signals attributed to the methylene carbon adjacent to the amine initiator disappeared upon formation of amide (see ESI†).
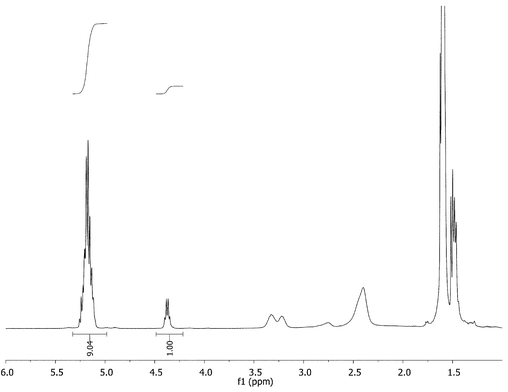 | ||
| Fig. 6 Complete initiation of the PPI G-3 dendrimer was confirmed by 1H-NMR. | ||
The formation of block stars was found to proceed nearly identically to the previous systems with the exception that DBU could not be used as a catalyst. When polymerizing from amines the newly formed amide could be deprotonated by strong bases creating unwanted and inefficient initiation sites. If only the (-)-sparteine/TU system was used monomodal block stars were readily formed. Consequently, because DMSO was not used the TMC polymerization times with (-)-sparteine were significantly reduced (<24 h, Table 2).
β-Cyclodextrin
The success of dendritic initiators lead to our exploration of polysaccharides as star polymer initiators. A prime candidate for this evaluation was β-cyclodextrin (BCD) because of its high purity, low cost, and great utility within the greater scientific community.56,57 Comparable to the bis-MPA dendrimers, BCD was also poorly soluble in DCM requiring DMSO to homogenize the reaction mixture. Using the previously mentioned protocol we performed an experiment to determine whether the cyclic sugar was able to form a star polymer (arm DP = 20). Comparable to the dendritic system a narrow PDI (1.06) was obtained with Mn = 54,300 Da. Cyclic sugars contain a mixture of primary and secondary alcohols in a 1:2 ratio, respectively. It was feared that the corresponding difference in sterics may affect uniform initiation thus creating non-uniform polymer arms. To identify any initiation problems presented by the different alcohols 2D-NMR was used to monitor initiation efficiency for a (L)-lactide polymerization (arm DP = 5). The results conclusively showed that within five (L)-lactide units both the primary and secondary alcohols were quantitatively initiated when using (-)-sparteine (NMR spectra can be found in the ESI†). Formation of a second TMC block proceeded as before showing no dependence on block forming method providing narrow PDI values (Table 2).
Conclusion
Advancements in synthetic techniques and the commercial availability of dendritic and other multifunctional initiators has allowed the production of nearly monodispersed star polymers. Considering the wide variety of initiating moieties, it is possible to incorportate additional functionality that is independent of monomer selection (i.e. inclusion complexes with cyclodextrins, pH responsiveness of PPI, etc.)The current and rising demand for therapeutics has engendered the need for new and creative delivery platforms. Such vectors can be readily produced via the combination of judiciously chosen initiators with generic and functionalized monomers. The use of cyclic esters and carbonates have been shown to provide additional benefits because their inherent stability can be balanced with a natural biodegradability. The literature has also repeatedly shown that such monomers are non-toxic and compatible within the body facilitating use in vivo. Furthermore, star polymers form globular 3D-structures that can mimic dendritic systems while being synthesized by rapid chain extension polymerizations negating the need for extra protection/deprotection steps. Finally, the development of organocatalysis has greatly aided the utility of ROP within the healthcare fields. Its high fidelity, fast kinetics and easy catalyst removal make it an ideal choice for polymerizing delivery systems.
Experimental
Materials
Reagents were available commercially from Aldrich or Perstorp and used as received unless otherwise noted. 2,2-Bis(hydroxymethyl)propanoic acid (bis-MPA) based dendrimers were synthesized by a modified literature procedure,49 and poly(propyleneimine) dendrimers were purchased from Symo-Chem. All multifunctional initiators were either azeotropically dried from toluene (β-cyclodextrin and Bis-MPA dendrimers) or heated at 80 °C for 48 h under HV (Boltorn®H20 and PPI dendrimers). (L)-lactide and trimethylene carbonate (TMC) were purified by recrystallization from dry toluene and stored in a N2 filled drybox. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (98%) and (-)-sparteine were stirred with CaH2, vacuum distilled, and stored over molecular sieves (3 Å). N′-(3,5-bis(trifluoromethyl)phenyl)-N-cyclohexyl-thiourea (TU) was synthesized according to literature procedure58 and recrystallized from dry toluene. Dry tetrahydrofuran (THF), dichloromethane (DCM) and toluene were obtained by using a solvent drying system from Innovative Technology employing a 5% slurry of activated alumina. Dry dimethylsulfoxide (DMSO) was obtained viavacuum distillation from CaH2.Methods
All polymerizations were carried out in a nitrogen atmosphere glove box. 1H-NMR spectra were obtained on a Bruker Avance 400 instrument using CDCl3 or d6DMSO solutions. Gel permeation chromatography (GPC) was performed in THF at 30 °C using a Waters chromatograph equipped with four 5 μm Waters columns (300 mm × 7.7 mm) connected in series with increasing pore size (100, 1000, 105, 106 Å), a Waters 410 differential refractometer for refractive index (RI) detection and a 996 photodiode array detector, calibrated with polystyrene standards (750 − (2 × 106) g mol−1).Acknowledgements
The authors acknowledge Perstorp (Sweden) for the generous supply of Boltorn H20® and Bert Meijer for the supply of PPI dendrimer. We thank Jane Frommer and Kristina Nilsson for obtaining the AFM image and Delores Miller for her helpful discussion on 2D NMR.Notes and references
- BLS Spotlight on Statistics: Health Care, http://www.bls.gov/spotlight/2009/health_care/, Accessed June 6, 2011, 2011.
- Advanced Drug Delivery Systems: New Developments, New Technologies, http://www.bccresearch.com/report/drug-delivery-systems-phm006g.html, Accessed June 6, 2011, 2011.
- M. Goldberg, R. Langer and J. Xinqiao, J. Biomater. Sci., Polym. Ed., 2007, 18, 241–268 CrossRef CAS
.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS
- N. Kumar, M. N. V. Ravikumar and A. J. Domb, Adv. Drug Delivery Rev., 2001, 53, 23–44 CrossRef CAS
- S. F. van Dongen, H. P. de Hoog, R. J. Peters, M. Nallani, R. J. Nolte and J. C. van Hest, Chem. Rev., 2009, 109, 6212–6274 CrossRef CAS
-
K. Osada and K. Kataoka, in Peptide Hybrid Polymers, Springer-Verlag Berlin, Berlin, 2006, vol. 202, pp. 113–153 Search PubMed
- Z. L. Tyrrell, Y. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128–1143 CrossRef CAS
- S. Stolnik, L. Illum and S. S. Davis, Adv. Drug Delivery Rev., 1995, 16, 195–214 CrossRef CAS
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS
- H. Gao, W. Shi and L. B. Freund, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9469–9474 CrossRef CAS
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC
- T. Tang, D. J. Coady, A. J. Boydston, O. L. Dykhno and C. W. Bielawski, Adv. Mater., 2008, 20, 3096–3099 CrossRef CAS
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS
- A. D'Emanuele and D. Attwood, Adv. Drug Delivery Rev., 2005, 57, 2147–2162 CrossRef CAS
- P. Antoni, M. J. Robb, L. Campos, M. Montanez, A. Hult, E. Malmström, M. Malkoch and C. J. Hawker, Macromolecules, 2010, 43, 6625–6631 CrossRef CAS
- L. Röglin, E. H. M. Lempens and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 102–112 CrossRef
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS
- A.-M. Zorn, M. Malkoch, A. Carlmark and C. Barner-Kowollik, Polym. Chem., 2011, 2, 1163–1173 RSC
- M. I. Montañez, L. M. Campos, P. Antoni, Y. Hed, M. V. Walter, B. T. Krull, A. Khan, A. Hult, C. J. Hawker and M. Malkoch, Macromolecules, 2010, 43, 6004–6013 CrossRef
- Z. Poon, S. Chen, A. C. Engler, H.-i. Lee, E. Atas, G. von Maltzahn, S. N. Bhatia and P. T. Hammond, Angew. Chem., Int. Ed., 2010, 49, 7266–7270 CrossRef CAS
- W. Cao and L. Zhu, Macromolecules, 2011, 44, 1500–1512 CrossRef CAS
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171–1185 RSC
- O. Altintas, U. Tunca and C. Barner-Kowollik, Polym. Chem., 2011, 2, 1146–1155 RSC
- D. J. A. Cameron and M. P. Shaver, Chem. Soc. Rev., 2011, 40, 1761–1776 RSC
- J. T. Wiltshire and G. G. Qiao, Aust. J. Chem., 2007, 60, 699–705 CrossRef CAS
-
G. Odian, Principles of Polymerization, Wiley-Interscience, Staten Island, New York, 2004 Search PubMed
- F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS
- R. Fu and G.-D. Fu, Polym. Chem., 2011, 2, 465–475 RSC
- A. W. Bosman, R. Vestberg, A. Heumann, J. M. J. Fréchet and C. J. Hawker, J. Am. Chem. Soc., 2002, 125, 715–728 CrossRef
- K.-i. Fukukawa, R. Rossin, A. Hagooly, E. D. Pressly, J. N. Hunt, B. W. Messmore, K. L. Wooley, M. J. Welch and C. J. Hawker, Biomacromolecules, 2008, 9, 1329–1339 CrossRef CAS
- H. Gao and K. Matyjaszewski, Macromolecules, 2008, 41, 1118–1125 CrossRef CAS
- Z. Li, J. Ma, N. S. Lee and K. L. Wooley, J. Am. Chem. Soc., 2011, 133, 1228–1231 CrossRef CAS
- N. S. Ieong, M. Redhead, C. Bosquillon, C. Alexander, M. Kelland and R. K. O'Reilly, Macromolecules, 2011, 44, 886–893 CrossRef CAS
- J. M. Stukel, R. C. Li, H. D. Maynard and M. R. Caplan, Biomacromolecules, 2009, 11, 160–167 CrossRef
- P. M. Wright, G. Mantovani and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7376–7385 CrossRef CAS
- F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois, Polym. Chem., 2011, 2, 528–533 RSC
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC
- J. Watanabe, H. Kotera and M. Akashi, Macromolecules, 2007, 40, 8731–8736 CrossRef CAS
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS
- A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749–6754 CrossRef CAS
- D. J. Coady, K. Fukushima, H. W. Horn, J. E. Rice and J. L. Hedrick, Chem. Commun., 2011, 47, 3105–3107 RSC
- S. Csihony, D. A. Culkin, A. C. Sentman, A. P. Dove, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 9079–9084 CrossRef CAS
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS
- H. Ihre, O. L. Padilla De Jesús and J. M. J. Fréchet, J. Am. Chem. Soc., 2001, 123, 5908–5917 CrossRef CAS
- M. Trollsås, B. Atthof, A. Würsch, J. L. Hedrick, J. A. Pople and A. P. Gast, Macromolecules, 2000, 33, 6423–6438 CrossRef
- S. Aryal, M. Prabaharan, S. Pilla and S. Gong, Int. J. Biol. Macromol., 2009, 44, 346–352 CrossRef CAS
- G. Kreutzer, C. Ternat, T. Q. Nguyen, C. J. G. Plummer, J.-A. E. Månson, V. Castelletto, I. W. Hamley, F. Sun, S. S. Sheiko, A. Herrmann, L. Ouali, H. Sommer, W. Fieber, M. I. Velazco and H.-A. Klok, Macromolecules, 2006, 39, 4507–4516 CrossRef CAS
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Macromol. Biosci., 2009, 9, 515–524 CrossRef CAS
- M. R. Radowski, A. Shukla, H. von Berlepsch, C. Böttcher, G. Pickaert, H. Rehage and R. Haag, Angew. Chem., Int. Ed., 2007, 46, 1189–1189 CrossRef
- E. M. M. de Brabander-van den Berg and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1308–1311 CrossRef
- Y. Chen and Y. Liu, Chem. Soc. Rev., 2010, 39, 495–505 RSC
- L. X. Song, L. Bai, X. M. Xu, J. He and S. Z. Pan, Coord. Chem. Rev., 2009, 253, 1276–1284 CrossRef CAS
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS
Footnotes |
| † Electronic Supplementary Information (ESI) available: 2D NMR spectra and GPC traces. See DOI: 10.1039/c1py00272d/ |
| ‡ It should be noted that a repeat unit is defined as being one ring opened lactide monomer. |
| This journal is © The Royal Society of Chemistry 2011 |