An oxidative Hosomi-Sakurai strategy toward the synthesis of illioliganones B and C†
Wesley J.
Moran
* and
Arantxa
Rodríguez
Department of Chemical & Biological Sciences, University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK. E-mail: w.j.moran@hud.ac.uk; Tel: +44 (0)1484 473741; Fax: +44 (0)1484 472182
First published on 18th July 2011
Abstract
The core structures of illioliganones B and C have been made by an oxidative Hosomi-Sakurai reaction of a phenol derivative. The use of a silyl protecting group is shown to be crucial for this reaction to proceed.
Illioliganones B and C were isolated from the stem bark of Illicium oligandrum by Yu and co-workers and the structures reported in 2009.1 These compounds were shown to have mild anti-inflammatory properties. In order to develop a synthetic route toward these molecules, we were interested in utilising an intermolecular oxidative Hosomi-Sakurai reaction.2,3 This reaction has been shown to have promise but there are several drawbacks. In particular, Canesi and co-workers have shown that two ortho substituents and a para substituent are required for a successful reaction (Scheme 1a). Without two ortho substituents, a formal [2+3] cycloaddition occurs instead of the dearomatising allylation process (Scheme 1b).4
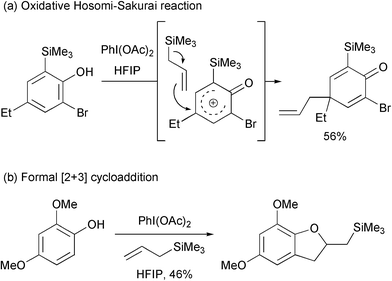 | ||
| Scheme 1 Known reactions of substituted phenols with allyltrimethylsilane in the presence of iodobenzene diacetate. | ||
Our retrosynthetic analysis of illioliganones B and C involved disconnection of the diols back to the alkenes1 and 2, which were envisaged to be produced from an oxidative Hosomi-Sakurai reaction of phenol derivative 3 (Scheme 2). Note that compound 3 does not have two ortho substituents to the phenol group, therefore a formal [2+3] cycloaddition, rather than the oxidative Hosomi-Sakurai, would appear to be more likely based on Canesi's findings.
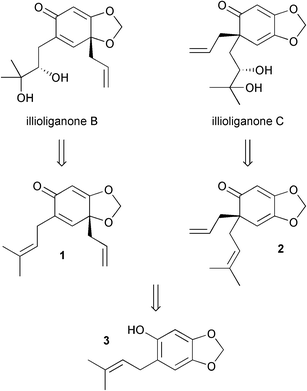 | ||
| Scheme 2 Retrosynthetic analysis. | ||
The synthesis began with a Pd-catalysed allylation of commercially available sesamol 4 which proceeded in excellent yield (Scheme 3).5Filtration, to remove the Pd salts, followed by concentration provided the crude material 5 which was heated neat at 125 °C for 3 h to effect a Claisen rearrangement. Purification provided the phenol derivative 3 in up to 90% yield over the two steps. Notably, the Claisen rearrangement was completely regioselective. Subsequent treatment with Canesi's oxidative Hosomi-Sakurai reaction conditions led only to decomposition with no identifiable products being formed. It is postulated that the reaction intermediates were too unstable.
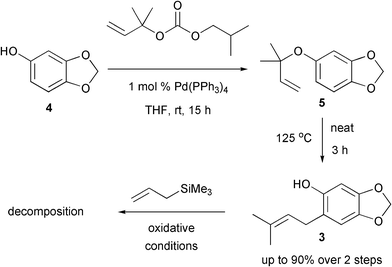 | ||
| Scheme 3 Attempted Hosomi-Sakurai reaction with 3. | ||
Felpin suggested that the conversion of a phenol into a silyl ether allows for a change in mechanism from radical to ionic for hypervalent iodine(III) mediated oxidative dearomatisation reactions.6 Thus, more stable reaction intermediates are present leading to increased yields of final product. Accordingly, phenol derivative 3 was silylated and then subjected to the oxidative allylation conditions (Scheme 4). In the event, we were pleased to obtain the desired compounds. However, the reaction proved to be somewhat capricious with the major product 2 being isolated in between 30 and 55% yields. The minor regioisomer 1 was generally found to be impossible to isolate in pure form from the reaction mixture. A third product 7 was also formed in up to 30% yield, and this proved to be a solvent addition product. Use of the less nucleophilic hexafluoroisopropanol did not lead to greater yields of the desired products however, possibly due to its increased acidity leading to silyl ether cleavage in situ. Nonetheless, a reasonable amount of one of the desired compounds, 2, could be isolated.
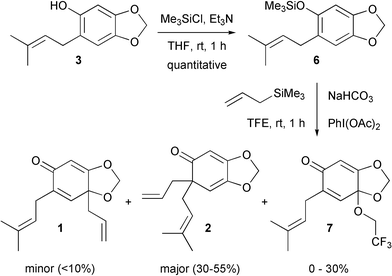 | ||
| Scheme 4 Successful Hosomi-Sakurai reaction with silyl ether6. | ||
Canesi reported that the regioselectivity of the Hosomi-Sakurai reaction is controlled by steric factors.2c However, it is difficult to rationalise the formation of products 1, 2 and 7 on this basis. In addition, the absence of any cycloaddition product is interesting and is in contrast with Canesi's work.4 As such, further work is required to delineate these issues.
At this stage, it was envisaged that the synthesis could be completed with a dihydroxylation of 2 using the Upjohn conditions.7 Indeed, illioliganone C was formed, however the crude reaction mixture showed that the dihydroxylation was completely non-selective (Scheme 5). Moreover, a clean sample of the natural product could only be isolated in small quantities after two successive flash chromatography columns.8 The use of other dihydroxylation conditions were investigated,9 but, unfortunately, either no reaction or decomposition of the starting material occurred in all cases.
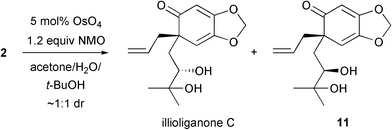 | ||
| Scheme 5 Non-selective dihydroxylation. | ||
A small sample of the minor regioisomer 1 was subjected to the Upjohn dihydroxylation conditions but again an essentially inseparable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of illioliganone B and its epimer was formed.
:1 mixture of illioliganone B and its epimer was formed.
In order to improve the end-game of the synthesis, an alternative strategy involving epoxidation followed by ring opening was devised. We were delighted to find that simply by treating the polyene2 with m-chloroperbenzoic acid led to completely chemoselective and diastereoselective epoxidation in 70% yield (Scheme 6). Subsequently, heating the epoxide in water at 60 °C overnight furnished one compound which was determined to be 11, the epimer of illioliganone C.10 Obviously, the undesired face of the alkene had been epoxidised.
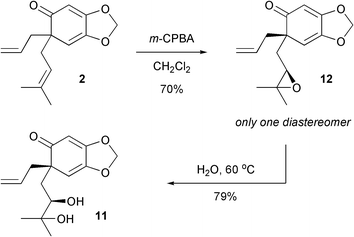 | ||
| Scheme 6 Preparation of the epimer of illioliganone C. | ||
Attempts to invert the alcohol stereocentre by a Mitsunobu reaction were unsuccessful. Subsequently, an inelegant oxidation/reduction sequence was conceived to provide the natural product, however carbon–carbon bond cleavage occurred to generate the corresponding aldehyde (and, presumably, acetone).
In conclusion, we have shown that the oxidative Hosomi-Sakurai reaction is successful on a silylated phenol derivative without two ortho substituents. This reaction is proposed to proceed by a purely ionic mechanism, unlike the analogous phenol process, and it demonstrates that this transformation has wider scope than was initially thought.
Acknowledgements
We thank the Leverhulme Trust for a Research Grant (F/01 582/D).References
- W.-Z. Tang, S.-G. Ma, S.-S. Yu, J. Qu, Y.-B. Liu and J. Liu, J. Nat. Prod., 2009, 72, 1017 CrossRef CAS.
- (a) K. C. Guérard, C. Sabot, M.-A. Beaulieu, M.-A. Giroux and S. Canesi, Tetrahedron, 2010, 66, 5893 CrossRef; (b) C. Sabot, K. C. Guérard and S. Canesi, Chem. Commun., 2009, 2941 RSC; (c) C. Sabot, B. Commare, S. Nahi, M. A. Duceppe, K. C. Guérard and S. Canesi, Synlett, 2008, 3226 CAS; (d) K. C. Nicolaou, D. J. Edmonds and G. S. Tria, Angew. Chem., Int. Ed., 2007, 46, 3942 CrossRef CAS; (e) S. Quideau, L. Pouységu, M. Oxoby and M. A. Looney, Tetrahedron, 2001, 57, 319 CrossRef CAS; (f) S. Quideau, M. A. Looney and L. Pouységu, Org. Lett., 1999, 1, 1651 CrossRef CAS.
- (a) V. V. Zhdankin, Arkivoc, 2009, 1 CAS; (b) T. Dohi and Y. Kita, Chem. Commun., 2009, 2073 RSC; (c) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS; (d) T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656 CrossRef CAS; (e) D. Magdziak, S. J. Meek and T. R. R. Pettus, Chem. Rev., 2004, 104, 1383 CrossRef CAS; (f) A. N. French, S. Bissmire and T. Wirth, Chem. Soc. Rev., 2004, 33, 354 RSC; (g) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS; (h) G. F. Koser, Aldrichimica Acta, 2001, 34, 89 CAS; (i) V. V. Zhdankin and P. J. Stang, Chem. Rev., 1996, 96, 1123 CrossRef.
- (a) D. Bérard, M.-A. Giroux, L. Racicot, C. Sabot and S. Canesi, Tetrahedron, 2008, 64, 7537 CrossRef; (b) D. Bérard, L. Racicot, C. Sabot and S. Canesi, Synlett, 2008, 1076 Search PubMed.
- H. Jiang and Y. Hamada, Org. Biomol. Chem., 2009, 7, 4173 CAS.
- F.-X. Felpin, Tetrahedron Lett., 2007, 48, 409 CrossRef CAS.
- V. VanRheenan, R. C. Kelly and D. Y. Cha, Tetrahedron Lett., 1976, 17, 1973 CrossRef.
- The analytical data obtained were in close agreement with the literature values: see ESI† for details.
- Other dihydroxylation conditions tested included (a) the Prevost-Woodward reaction: L Emmanuvel, T. M. A. Shaikh and A. Sudulai, Org. Lett., 2005, 7, 5071 CrossRef CAS; (b) a RuO4-catalysed procedure: B. Pleitker and M. Niggemann, Org. Lett., 2003, 5, 3353 CrossRef.
- Z. Wang, Y.-C. Cui, Z.-B. Zu and J. Qu, J. Org. Chem., 2008, 73, 2270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, characterisation data and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c1ra00095k |
| This journal is © The Royal Society of Chemistry 2011 |