A reaction of formaldehyde with acetonitrile: understanding the preparation of RDX (I)†
Xiao-Fang
Chen
ab,
Bo-Zhou
Wang
b and
Ke-Li
Han
*a
aState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, 457 Zhongshan Road, Dalian, 116023, P. R. China. E-mail: klhan@dicp.ac.cn; Fax: (+86)-411-84675584; Tel: (+86)-411-84379293
bXi’an Modern Chemistry Research Institute, 168 Zhangba Road, Xi’an, 710065, P. R. China
First published on 15th September 2011
Abstract
The reaction of HCHO with CH3CN to yield a key RDX precursor (TRAT) was proposed to undergo four sequential stages. More attention was suggested to be paid to the mid and final terms. A parallel product could be controlled by using the concentrated sulfuric acid in almost drying conditions.
Explosives are defined as special compounds containing a large amount of chemical energy. More explosives have been used for the extensive mining of minerals and the exploration of outer space than for wars and conflicts.1 Their preparation is a complex process. It usually involves huge danger and risks personal security, and even the taking of lives and destruction of property. Some online apparatus are experimentally employed to detect the synthetic course of explosives, such as thermal controlling apparatus, pressure gauge, and spectrometry-based detectors. Comparatively, the computational chemistry is an independent predictable tool, effectively providing which element steps are potentially explosive in the preparation of the hazardous materials.
One of the most common oxidizer in the rocket propellant, the cyclic nitramine hexahydro-1,3,5-trinitro-s-triazine (RDX), was selected for a test, which is also used as a more powerful explosive than TNT.1–3RDX has been received considerable attention in the past several decades.2,4–10 Many theoretical studies have been directed toward its structure,7,9spectra,2,6 unimolecular decomposition,7,10 improved-performance in mixtures. They provide a great deal of valuable information on characterizing, detecting, and functioning RDX.
RDX is usually manufactured industrially by the nitrolysis of hexamethylenetetramine.1 This conventional process suffers from huge challenges in the separation and purification of products. It was reported that the nitrolysis reaction employing hexahydro-1,3,5-triacetyl-s-triazine (TRAT) (Scheme 1) was a preferentially alternative route to RDX, possessing higher yields and economic investment.1,8TRAT can be obtained from HCHO and CH3CN through a sulfuric acid-catalyzed reaction (eqn (1)).11–13
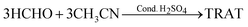 | (1) |
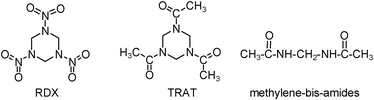 | ||
| Scheme 1 Hexahydro-1,3,5-trinitro-s-triazine (RDX), hexahydro-1,3,5-triacetyl-s-triazine (TRAT), and methylene-bis-amides. | ||
Initially, the concentrated sulfuric acid makes acetonitrile sulfated to become the salt-like adduct IM2 through electrophilic addition (Scheme 2). The C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond length in CH3CN is 1.148 Å, close to the experimentally observed value.17 When a CH3CN molecule approaches H2SO4, a van der Waals complex, IM1, is firstly formed, where the H⋯N bond is actually the hydrogen bond (1.755 Å). The H⋯N bond is gradually enhanced, and then the (S)O–C(R) bond is formed via a six-membered ring transition state (TS1), generating IM2. In the six-membered ring of TS1, the C–O, O–H, and N–H bond distances are 1.176, 1.631, and 1.063 Å, respectively. The C–NH bond in IM2 is 1.249 Å, among the C–N single and double bond distances. IM1 is slightly lying above two isolated reagents by 0.14 kcal mol−1 in the Gibbs free energy (ΔG(298 K, CCl4)) (Fig. 1). Forming sulfated imine surmounts an activation ΔG(298 K, CCl4) of 23.94 kcal mol−1, absorbing a net reaction free-energy of 14.04 kcal mol−1. The energy baseline in Fig. 1 is the energy sum of three isolated reaction molecules, H2SO4, CH3CN, and HCHO, for the sake of convenience. In TS1, the Mulliken charge sum of CH3CN counterpart is 0.22501e, and CH3CN serves as the nucleophilic reagent when the C–N π triple bond is opened.
N bond length in CH3CN is 1.148 Å, close to the experimentally observed value.17 When a CH3CN molecule approaches H2SO4, a van der Waals complex, IM1, is firstly formed, where the H⋯N bond is actually the hydrogen bond (1.755 Å). The H⋯N bond is gradually enhanced, and then the (S)O–C(R) bond is formed via a six-membered ring transition state (TS1), generating IM2. In the six-membered ring of TS1, the C–O, O–H, and N–H bond distances are 1.176, 1.631, and 1.063 Å, respectively. The C–NH bond in IM2 is 1.249 Å, among the C–N single and double bond distances. IM1 is slightly lying above two isolated reagents by 0.14 kcal mol−1 in the Gibbs free energy (ΔG(298 K, CCl4)) (Fig. 1). Forming sulfated imine surmounts an activation ΔG(298 K, CCl4) of 23.94 kcal mol−1, absorbing a net reaction free-energy of 14.04 kcal mol−1. The energy baseline in Fig. 1 is the energy sum of three isolated reaction molecules, H2SO4, CH3CN, and HCHO, for the sake of convenience. In TS1, the Mulliken charge sum of CH3CN counterpart is 0.22501e, and CH3CN serves as the nucleophilic reagent when the C–N π triple bond is opened.
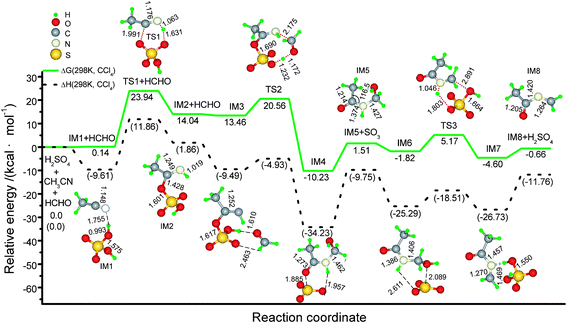 | ||
| Fig. 1 Schematic energy (in kcal mol−1) profile for the ketoimine intermediate (IM8) generated from the concentrated sulfuric acid catalyzed reaction of HCHO with CH3CN at the M06-2X(SMD,CCl4)/6-311+G** level, along with optimized geometry attributes. Notes: 1) bond lengths in Å and bond angels in degrees; 2) ΔG (298 K, CCl4) and ΔH (298 K, CCl4) designate the Gibbs free energy and enthalpy with the thermal correlations under the standard conditions. | ||
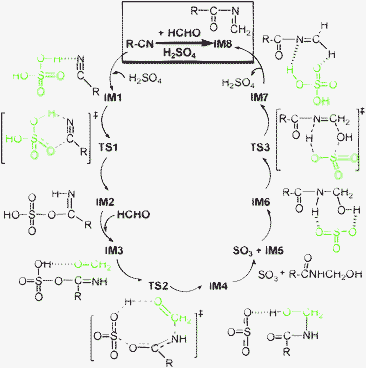 | ||
| Scheme 2 Proposed catalytic cycle for generating important ketoimine IM8 from HCHO and CH3CN (R = CH3–). Note that the blue-dashed lines denote hydrogen bonds. | ||
Next is assumedly the aminomethylation via an eight-membered ring TS2 when a HCHO molecule participates. A SO3 molecule is gradually avulsed with the cleavage of the O–SO3 and O2SO–H bonds, forming a counterpart of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)–NH–CH2OH. The necessary intrinsic reaction coordinates calculations on TS2 and TS3 (mentioned below) indicate that, another van der Waals complex, IM3, is pre-coordinated viahydrogen bonds (1.610 and 2.463 Å); and a SO3 molecule does not immediately escape, but it is coordinated to different sites (IM4 and IM6) of the OC(CH3)–NH–CH2OH moiety though hydrogen bonds when stirred. ΔG(298 K, CCl4) are 13.46, 20.56, −10.23, 1.51 and −1.82 kcal mol−1 for IM3, TS2, IM4, IM5 + SO3, and IM6, respectively.
C(CH3)–NH–CH2OH. The necessary intrinsic reaction coordinates calculations on TS2 and TS3 (mentioned below) indicate that, another van der Waals complex, IM3, is pre-coordinated viahydrogen bonds (1.610 and 2.463 Å); and a SO3 molecule does not immediately escape, but it is coordinated to different sites (IM4 and IM6) of the OC(CH3)–NH–CH2OH moiety though hydrogen bonds when stirred. ΔG(298 K, CCl4) are 13.46, 20.56, −10.23, 1.51 and −1.82 kcal mol−1 for IM3, TS2, IM4, IM5 + SO3, and IM6, respectively.
Sequentially, the dehydration makes IM6 liberate the catalyst molecule of H2SO4 when the SO3 moiety draws strongly on the H and OH groupsvia six-membered ring TS3. A van der Waals complex IM7 with the lower ΔG(298 K, CCl4) (−4.60 kcal mol−1) is formed between IM8 and H2SO4, prior to complete release of H2SO4. This dehydrolysis process readily occurs, only overcoming a much lower ΔG(298 K, CCl4) (5.17 kcal mol−1).
In a word, three sequential stages (i.e., forming imine → aminomethylation → dehydration) contribute to producing important ketoimine intermediate IM8, completing a catalytic cycle (Scheme 2). This process is similar to the course of a typical Mannich condensation reaction. Herein, acetonitrile serves as a new variant rather than an active hydrogen compound. It is derived from the most significant sulfated imine intermediate IM2, which is analogous to N-acylimino esters.18
Finally, the concerted trimerization of ketoimine gives rise to the desired product AAA conformation4,7TRATvia TS4 (Scheme 3), whose quasi H2C–N bond length is 2.789, 2.649, and 1.923 Å, respectively (Fig. 2). It is a strong exothermic process by −50.49 kcal mol−1 with an activation ΔG(298 K, CCl4) of 26.87 kcal mol1. These energies are higher than the corresponding internal energies (13.5/−59.4 kcal mol−1) for the reverse concerted decomposition of RDX in the gas phase.7 A relevant exothermic effect was also experimentally observed by Gradsten and Pollock.11
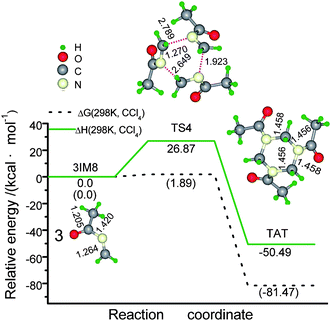 | ||
| Fig. 2 Schematic energy (in kcal mol−1) profile for the concerted trimerization of ketoimine IM8 to TRAT at the M06-2X(SMD,CCl4)/6-311+G** level. Notes: 1) bond lengths in Å and bond angels in degrees; 2) ΔG (298 K, CCl4) and ΔH (298 K, CCl4) designate the Gibbs free energy and enthalpy with the thermal correlations under the standard conditions. | ||
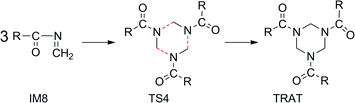 | ||
| Scheme 3 Concerted trimerization of ketoimine IM8 to TRAT (R = –CH3). | ||
In this catalytic synthesis of TRAT, forming sulfated imine is the rate-determining step (RDS) to generate ketoimine IM8, while its trimerization is the RDS in yielding TRAT. Considering the rate equation of k ∼ ln(−ΔG≠), we suggested that forming imine is a slower (ΔG(298 K, CCl4): 23.94 kcal mol−1) and endothermic process (14.04 kcal mol−1); dehydration is a fast (ΔG(298 K, CCl4)): 5.17 kcal mol−1) and almost iso-free energy process; both aminomethylation and trimerization are slow (ΔG(298 K, CCl4)): 20.56 and 26.87 kcal mol−1, respectively), but they proceed with the release of huge net free energy (−10.23 and −50.49 kcal mol−1, respectively), these two steps are likely to be full of potential hazards once they surmount their active barriers.
The mechanism above is different distinctly from a postulated carbonium ion mechanism on affording methylene-bis-amides ((CH3–CO–NH)2–CH2) from HCHO and CH3CN when an excess of strong H2SO4 solution employed.11,19 Obviously, the concentrated H2SO4 influences greatly the main product. Probable effects of water considered, one molecule of water reacted with one molecule of SO3 requires overcoming a higher activation ΔG(298 K, CCl4) of 24.65 kcal mol−1, via a strongly strained four-membered cyclic TS5 (Fig. 3); however, the activation free-energy barrier (1.89 kcal mol−1) is significantly reduced, where another water molecule acts as hydrogen bridges in a six-membered TS6. This facile reaction depletes SO3 molecules generated assumedly, and then retards IM6 being dehydrated. Therefore, the presence of water have disadvantage on forming ketoimine IM8, and essentially alters the course of the reaction between HCHO and CH3CN. The concentrated sulfuric acid functions as the catalyst and dehydrant in yielding TRAT. This Lewis acid enhances both the electrophilic and nucleophilic character of reagents to which they are bound.20
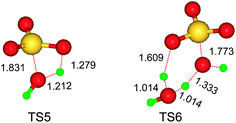 | ||
| Fig. 3 Calculated geometries of transition states for the reaction between SO3 and water molecule(s) at the M06-2X(SMD,CCl4)/6-311+G** level. | ||
In conclusion, the catalytic synthesis of a key RDX precursor (TRAT) from HCHO and CH3CN is interesting, and undergoes four sequential stages, i.e., forming imine (slower and endothermic), aminomethylation, dehydration (fast and almost iso-free energy) and trimerization. The second and fourth stages are slow, but they can release the huge net free energy. More attention is suggested to be paid to them in synthesizing TRAT. A parallel product, methylene-bis-amides could be controlled by using the concentrated sulfuric acid in drying conditions. A continuous study on the nitrolysis of TRAT to RDX is in progress.
Acknowledgements
The authors gracefully thank the National Key Basic Research Special Foundation of China (Grant No. 2007CB815202) and the National Natural Science Foundation of China (Grant No. 20833008) for financial support.References
- J. P. Agrawal and R. D. Hodgson, Organic Chemistry of Explosives, John Wiley & Sons, Ltd, 2007 Search PubMed.
- E. C. Meurer, H. Chen, L. Riter, I. Cotte-Rodriguez, M. N. Eberlin and R. G. Cooks, Chem. Commun., 2004, 40 RSC.
- (a) X. Zhao, Y. Cong, F. Lv, L. Li, X. Wang and T. Zhang, Chem. Commun., 2010, 46, 3028 RSC; (b) S. Zhu, X. Wang, A. Wang, Y. Cong and T. Zhang, Chem. Commun., 2007, 1695 RSC; (c) X. Chen, T. Zhang, M. Zheng, L. Xia, T. Li, W. Wu, X. Wang and C. Li, Ind. Eng. Chem. Res., 2004, 43, 6040 CrossRef CAS; (d) X. Chen, T. Zhang, M. Zheng, Z. Wu, W. Wu and C. Li, J. Catal., 2004, 224, 473 CrossRef CAS; (e) X. Chen, T. Zhang, P. Ying, M. Zheng, W. Wu, L. Xia, T. Li, X. Wang and C. Li, Chem. Commun., 2002, 288 RSC; (f) X.-F. Chen, J.-F. Liu, Z.-H. Meng and K.-L. Han, Theor. Chem. Acc., 2010, 127, 327 CrossRef CAS; (g) X.-F. Chen, C.-Y. Hou and K.-L. Han, J. Phys. Chem. A, 2010, 114, 1169 CrossRef CAS.
- D. I. A. Millar, I. D. H. Oswald, D. J. Francis, W. G. Marshall, C. R. Pulham and A. S. Cumming, Chem. Commun., 2009, 562 RSC.
- (a) C. J. McHugh, W. E. Smith, R. Lacey and D. Graham, Chem. Commun., 2002, 2514 RSC; (b) W.-B. Yi and C. Cai, J. Hazard. Mater., 2008, 150, 839 CrossRef CAS; (c) S. Radhakrishnan, M. B. Talawar, S. Venugopalan and V. L. Narasimhan, J. Hazard. Mater., 2008, 152, 1317 CrossRef CAS.
- D. G. Allis, J. A. Zeitler, P. F. Taday and T. M. Korter, Chem. Phys. Lett., 2008, 463, 84 CrossRef CAS.
- D. Chakrabory, R. P. Muller, S. Dasgupta and W. A. Goddard III, J. Phys. Chem. A, 2000, 104, 2261 CrossRef.
- E. E. Gilbert, J. R. Leccacorvi and M. Warman, Industrial and Laboratory Nitrations, ACS Symposium Series, 1976, 22, ch. 23,p. 327 Search PubMed.
- (a) R. Podeszwa, B. M. Rice and K. Szalewicz, Phys. Chem. Chem. Phys., 2009, 11, 5512 RSC; (b) R. Podeszwa, R. Bukowski, B. M. Rice and K. Szalewicz, Phys. Chem. Chem. Phys., 2007, 9, 5561 RSC.
- (a) A. Strachan, E. M. Kober, A. C. T. van Duin, J. Oxgaard and W. A. Goddard III, J. Chem. Phys., 2005, 122, 054502 CrossRef; (b) A. Strachan, A. C. T. van Duin, D. Chakraborty, S. Dasgupta and W. A. Goddard III, Phys. Rev. Lett., 2003, 91, 098301 CrossRef; (c) Y. Q. Guo, M. Greenfield and E. R. Bernstein, J. Chem. Phys., 2005, 122, 244310 CrossRef CAS.
- M. A. Gradsten and M. W. Pollock, J. Am. Chem. Soc., 1948, 70, 3079 CrossRef CAS.
- T. L. Gresham and T. R. Steadman, J. Am. Chem. Soc., 1949, 71, 1872 CrossRef CAS.
- W. D. Emmons, H. A. Rolewicz, W. N. Cannon and R. M. Ross, J. Am. Chem. Soc., 1952, 74, 5524 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef; (b) Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- (a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS; (b) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 4538 CrossRef CAS.
- D. R. Lide, Handbook of Chemistry & Physics, 84th ed., CRC press, 2003 Search PubMed.
- S. Kobayashi, R. Matsubara, Y. Nakamura, H. Kitagawa and M. Sugiura, J. Am. Chem. Soc., 2003, 125, 2507 CrossRef CAS.
- (a) C. C. Price and I. V. Krishnamurti, J. Am. Chem. Soc., 1950, 72, 5334 CrossRef CAS; (b) E. E. Magat, B. F. Faris, J. E. Reith and L. F. Salisbury, J. Am. Chem. Soc., 1951, 73, 1028 CrossRef CAS; (c) E. E. Magat, L. B. Chandler, B. F. Faris, J. E. Reith and L. F. Salisbury, J. Am. Chem. Soc., 1951, 73, 1031 CrossRef CAS.
- B. Alcaide and P. Almendros, Angew. Chem., Int. Ed., 2008, 47, 4632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full computational details and XYZ coordinates. See DOI: 10.1039/c1ra00239b |
| This journal is © The Royal Society of Chemistry 2011 |