Quantitative investigation of surface functionalization of cylindrical nanopores derived from polystyrene-poly(methylmethacrylate) diblock copolymers†
Feng
Li
,
Ruben
Diaz
and
Takashi
Ito
*
Department of Chemistry, Kansas State University, 213 CBC Building, Manhattan, Kansas 66506-0401, USA. E-mail: ito@ksu.edu; Fax: +1 785-532-6666; Tel: +1 785-532-1451
First published on 19th October 2011
Abstract
This paper reports quantitative measurements of surface modification efficiency on nanoporous films (20–35 nm in thickness; 14, 20 and 30 nm in pore diameter) derived from cylinder-forming polystyrene-poly(methylmethacrylate) diblock copolymers. The density of free –COOH groups on the nanopore surface was determined by measuring their probe counter cations released via cation exchange using spectrofluorometry and inductively coupled plasma mass spectrometry. The yields of aqueous-phase amidation mediated by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide in phosphate buffer (pH 6) were 20–30%. In contrast, organic-phase amidation and esterification using oxalyl chloride offered much higher yields (87–89%), providing efficient means for functionalizing the nanopore surface. Quantitative information on the surface modification will provide a basis for assessing the influences of the surface functionality on the efficiency of chemical separations with the nanoporous films.
Introduction
Nanoporous films derived from cylinder-forming block copolymers1–3 are promising materials for chemical and biological separations because of their high porosities, cylindrical pore shapes, and uniform and controllable pore diameters.4,5 Previous work has mainly focused on investigating their size-selective molecular permeability: Such nanoporous films were applied for virus filtration,6,7macromolecule separations8,9 and protein delivery.10Film permeability can be tailored based on chemical interactions by functionalizing the nanopore surface, as demonstrated with track-etched and anodic alumina membranes.11 However, the surface chemistry of block copolymer-derived nanoporous films has attracted limited attention. Novel block copolymers were synthesized to fabricate nanoporous films comprising cylindrical nanopores with predictable surface functional groups upon chemical degradation of the cylindrical domains.12–16 Chemical modification of the resulting nanopore surface was assessed using NMR and FTIR.12–14 Very recently, a polystyrene-poly(methylmethacrylate) block copolymer (PS-b-PMMA) with a di-COOH group at the PMMA terminal was synthesized to fabricate thin films comprising COOH-decorated nanopores.17 Nanoporous films were obtained by dissolving PMMA homopolymer from block copolymer-homopolymer blend films. The surface –COOH groups were employed to covalently immobilize single-stranded probe DNA.
We have investigated the surface chemistry of cylindrical nanopores derived from PS-b-PMMA.18–21 Nanoporous films were prepared by selective removal of the cylindrical PMMA domains in thermally-annealed thin films via UV-based degradation and subsequent acetic acid (AcOH) treatment.22,23Electrochemical methods were employed to assess the pH-dependence of film permeability for charged redox species, revealing the presence of surface –COOH groups.18 The presence of surface –COOH groups was further validated by scanning force microscopy studies on UV/AcOH-treated PS-b-PMMA films.21,24Electrochemical methods were also used to verify nanopore surface functionalization via aqueous-phase amidation of the surface –COOH groups mediated by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide chloride (EDC).19,20 However, the electrochemical and force microscopy approaches measure changes in film permeability and tip-surface force primarily based on electrostatic interactions.18,20,21 Because of surface charge screening involved in electrostatic effects, it is challenging to directly and quantitatively correlate electrochemical and surface force data to the density of surface –COOH groups.25PS-b-PMMA-derived nanoporous films cannot be characterized using solution-phase NMR as was done previously,12–14 because they are insoluble in organic solvent as a result of the UV-induced crosslinking of the PS matrix.7,26 The surface functionalization of these films cannot be assessed using FTIR spectroscopy, because the absorption bands of surface –COOH groups were not observed in the FTIR spectra of these films.18
Here, the surface functionalization efficiency of PS-b-PMMA-derived nanopores was quantitatively assessed by measuring monovalent probe cations released from the surface –COOH groupsvia cation-exchange processes. The nanopore surface was functionalized by aqueous-phase amidation mediated by EDC and organic-phase amidation/esterification through acid chloride. These reactions reduce free –COOH groups on nanopore surface, and thus their yields can be determined by comparing the surface –COOH densities before and after the reactions. The quantity of cations released from a 30-nm thick film was on the order of pmol, and thus was measured using highly-sensitive analytical techniques including spectrofluorometry and inductively coupled plasma mass spectrometry (ICP-MS). Similar approaches using spectrofluorometry have been employed previously to determine the modification yields of surface -COOH groups in track-etched membranes.27,28
Experimental section
Chemicals and materials
Three types of PS-b-PMMA (43 K PS-b-PMMA: Mn = 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 for PS and 11500 g mol−1 for PMMA, Mw/Mn = 1.06; 57 K PS-b-PMMA: Mn = 39800 g mol−1 for PS and 17000 g mol−1 for PMMA, Mw/Mn = 1.06; 82 K PS-b-PMMA: Mn = 57000 g mol−1 for PS and 25000 g mol−1 for PMMA, Mw/Mn = 1.07) were purchased from Polymer Source and used as received. Rhodamine 6G (Aldrich), cesium chloride (Aldrich), thionine acetate (Acros), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC; Chemimpex international), N-hydroxysuccinimide (Acros Organics), tetraethyleneglycol monoamine ((PEO)4NH2; Molecular Biosciences, Inc.), oxalyl chloride (Acros) and glacial acetic acid (AcOH) were used as received. Gold-coated Si wafers, which were prepared by sputtering 10 nm of Ti followed by 20 nm of Au onto Si (100) wafers, were purchased from LGA Thin Films (Foster City, CA). All aqueous solutions were prepared with water having a resistivity of 18 MΩ cm or higher (Barnstead Nanopure Systems). Detailed synthesis procedures for ferrocenylmethylamine (FcCH2NH2), 16-hydroxy-1-oxohexylferrocene (FcCO(CH2)5OH) and 16-hydroxy-1-oxohexadecylferrocene (FcCO(CH2)15OH) are given in the ESI†. The ferrocene derivatives were examined for electrochemical studies that would be reported elsewhere.
400 g mol−1 for PS and 11500 g mol−1 for PMMA, Mw/Mn = 1.06; 57 K PS-b-PMMA: Mn = 39800 g mol−1 for PS and 17000 g mol−1 for PMMA, Mw/Mn = 1.06; 82 K PS-b-PMMA: Mn = 57000 g mol−1 for PS and 25000 g mol−1 for PMMA, Mw/Mn = 1.07) were purchased from Polymer Source and used as received. Rhodamine 6G (Aldrich), cesium chloride (Aldrich), thionine acetate (Acros), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC; Chemimpex international), N-hydroxysuccinimide (Acros Organics), tetraethyleneglycol monoamine ((PEO)4NH2; Molecular Biosciences, Inc.), oxalyl chloride (Acros) and glacial acetic acid (AcOH) were used as received. Gold-coated Si wafers, which were prepared by sputtering 10 nm of Ti followed by 20 nm of Au onto Si (100) wafers, were purchased from LGA Thin Films (Foster City, CA). All aqueous solutions were prepared with water having a resistivity of 18 MΩ cm or higher (Barnstead Nanopure Systems). Detailed synthesis procedures for ferrocenylmethylamine (FcCH2NH2), 16-hydroxy-1-oxohexylferrocene (FcCO(CH2)5OH) and 16-hydroxy-1-oxohexadecylferrocene (FcCO(CH2)15OH) are given in the ESI†. The ferrocene derivatives were examined for electrochemical studies that would be reported elsewhere.
Preparation of PS-b-PMMA-derived nanoporous films
PS-b-PMMA-derived nanoporous films on gold electrodes were prepared according to procedures reported previously.18,19,23Gold-coated Si wafers were sonicated in ultra pure water and ethanol for 30 min, and then cleaned in a Novascan PSD-UVT UV-ozone system for 60 min prior to use. Thin films of PS-b-PMMA were prepared on cleaned gold substrates by spin-coating (2000 rpm) from its toluene solution (0.6% (w/w)) and annealed at 170 °C in vacuum (ca. 0.3 Torr) for 60 h. According to AFM images, annealed PS-b-PMMA films with thickness of 20–35 nm comprised cylindrical PMMA domains oriented perpendicularly to the film surface. The PMMA domains were removed by irradiation with UV light using a Novascan PSDUVT UV-ozone system (ca. 20 mW cm−2) under an Ar atmosphere and by subsequent immersing in AcOH. The thickness of annealed PS-b-PMMA films prior to the UV irradiation was measured using a J. A. Woollam alpha-SE spectroscopic ellipsometer. In addition, those before and after UV/acetic acid treatment were measured with a surface profiler (XP-2, Ambios Technology).Surface functionalization of PS-b-PMMA-derived nanopores
AFM measurements
AFM images were obtained by contact mode imaging in air, using a Picoscan SPM (Molecular Imaging). Contact mode tips from Vista Probes (CS-25) were employed. These tips were first rinsed with ethanol and then cleaned in a Novascan PSD-UVT UV-ozone system for 20 min prior to use.Determination of free surface –COOH density via cation exchange
A freshly-prepared nanoporous film was immobilized at the bottom of a cell similar to those employed for electrochemical measurements.18,20 The area of a film in contact with the solution was defined by an O–ring (0.80 cm in diameter). The film was immersed in an aqueous solution of a probe (Rhodamine 6G or CsCl; 1 mM) overnight. The cell was thoroughly washed with ultrapure water, disassembled, and then further washed with ultrapure water to remove physisorbed probe cations. Then, the sample was immersed in 0.01 M HCl solution (2.00 mL) for 2 h. The amount of the released probe molecules in the HCl solution was measured with spectrofluorometry (for Rhodamine 6G) or ICP-MS (for Cs+). For measurements with thionine acetate, the cell was soaked in an ethanol solution of thionine acetate (10 mM) overnight, thoroughly washed with ethanol, and then immersed in 0.01 M HCl (in a 1:1 mixture of ethanol and water).27,28
For spectrofluorometry, emission spectra at 525 nm (Rhodamine 6G) or 594 nm (thionine) as excitation wavelength were measured with an ISA-SPEX FluoroMax-2 spectrofluorometer. Fluorescence intensity at 550 nm (Rhodamine 6G) or 620 nm (thionine) was measured to determine the concentration of fluorescent probe cations released to the HCl solution (The fluorescence spectra of Rhodamine 6G and thionine are given in ESI). Standard probe solutions (1 nM, 5 nM, 10 nM, 50 nM and 100 nM) were prepared to obtain a calibration curve for each day's measurements.
For ICP-MS, Cs+ was employed because the influence of contamination was negligible. ICP-MS measurements were performed by Dr Javier Seravall (Redox Biology Center, Department of Biochemistry, University of Nebraska-Lincoln). A calibration curve was prepared by mixing 14 μM CsCl (0.0, 1.0, 2.0, 3.0 or 4.0 μL) with 200 μL of solution containing 10 mM HCl, 0.1% HNO3 and 50 ppb Ga3+ (as an internal standard). A Cs+ signal intensity measured at m/z = 133 was normalized against the Ga signal for each sample solution. The detection limit was 0.11 nM, which was significantly higher than the Cs concentration measured in the sample solutions (20–65 nM).
Results and discussion
The effective density of free –COOH groups on a nanoporous film was measured with ion-exchange processes as follows (Fig. 1): A nanoporous film was immersed in an aqueous solution of neutral pH or in an ethanol solution containing a base (acetate ion)29 to replace the proton of surface –COOH groups with monovalent probe cations (Q+ in Fig. 1). Subsequently, the probe-loaded film was soaked in a 0.01 M HCl solution (2.00 mL) to release the probe cations from the films. The amount of the released probes was determined from the probe concentration measured with spectrofluorometry or ICP-MS.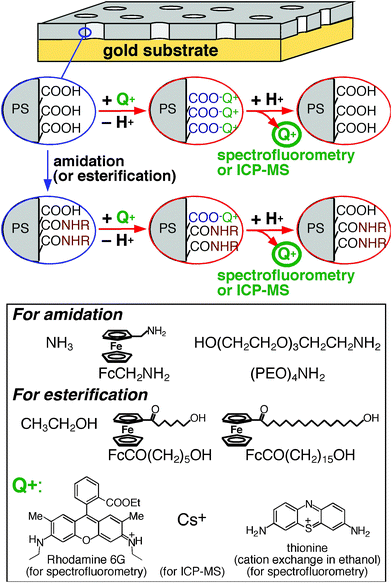 | ||
| Fig. 1 Experimental procedures for amidation/esterification of surface –COOH groups and measurements of surface free –COOH groups with monovalent probe cations (Q+) via cation exchange processes. Amine and alcohol reactants employed for nanopore surface modification were also shown. | ||
The –COOH density was calculated from the amount of probe cations released to 0.01 M HCl using the following equation:
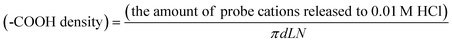 | (1) |
In this equation, d is pore diameter obtained from AFM images (Fig. 2), and L is pore length that corresponds to the thickness of the annealed PS-b-PMMA film. N is the total number of pores calculated from pore density obtained from AFM images (Fig. 2) and film area in contact with solution defined by an O–ring (0.8 cm in diameter). In this study, the ellipsometric thickness of an annealed film was used as L, the thickness of the nanoporous film obtained upon UV/AcOH treatment, because a change in film thickness was negligible for 20–35-nm thick films (Fig. 3): Profilometry data did not exhibit detectable changes in film thickness between UV-exposed and unexposed areas in a film (Fig. 3, top right). In addition, the thickness of a UV/AcOH-treated film, which was measured by dissolving the UV-unexposed region of the film with toluene, was almost the same as the ellipsometric thickness of the same film prior to the UV/AcOH treatment (Fig. 3, bottom right). The ratio of these thicknesses, i.e., the quotient of the profilometric thickness of a UV/AcOH-treated film divided by the ellipsometric thickness of the untreated film, measured on multiple different films was 0.99 ± 0.07.
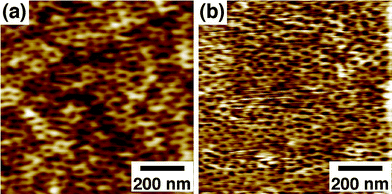 | ||
| Fig. 2 AFM images of a 57 K PS-b-PMMA-derived nanoporous film (31 nm thick) on a gold substrate (a) before and (b) after organic-phase esterification with FcCO(CH2)15OH. Δz = 6.4 nm. | ||
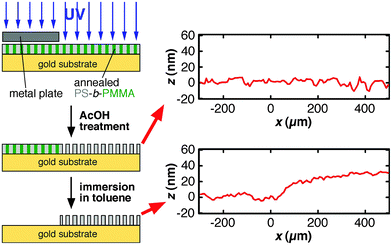 | ||
| Fig. 3 Sample preparation procedure for profilometry measurements to verify the changes in film thickness between UV-exposed and unexposed areas in a film (top right) and to determine the thickness of a UV/AcOH- treated film (bottom right). The ellipsometric thickness of the film prior to UV/AcOH treatment was 31 nm. In the profilometry data, the edge of the metal plate during the UV irradiation was located around x = 0. | ||
This equation can be employed under the following two assumptions: (1) Most –COOH groups (pKa∼4.5) were deprotonated at pH∼7 and formed 1:1 ion pairs with probe cations. This assumption was validated by comparing the –COOH density values measured with three different cation-exchange-based approaches (vide infra). (2) All the nanopores were oriented perpendicularly to film surface, and had cylindrical shapes with uniform lengths and diameters. In addition, based on the following results reported by us,21,24 it was hypothesized that –COOH groups were mainly present on the etched, nanoporous PMMA surface, and negligible on PS-based film surface: Friction force images obtained with chemically functionalized tips revealed that the etched PMMA surface was more hydrophilic. In addition, anionic protein molecules were preferentially adsorbed onto the PS-based surface, reflecting electrostatic repulsion from the negatively-charged, etched PMMA surface.
First, the effective density of free –COOH groups was determined for unmodified (native) nanoporous films. Three cation-exchange-based methods were employed for 57 K PS-b-PMMA-derived nanoporous films (Table 1): spectrofluorometric method in aqueous solution with Rhodamine 6G; spectrofluorometric method in ethanol solution with thionine acetate, and ICP-MS method in aqueous solution with Cs+. Among them, the second method was previously employed to measure the effective –COOH density,27,28 because the acetate ion leads to the deprotonation of a surface –COOH group as a base29 to form a 1:1 ion pair of thionine cation and surface –COO−group. As shown in Table 1, the –COOH density values measured in aqueous solution with Rhodamine 6G (0.79 ± 0.14/nm2) was very similar to that (0.86 ± 0.08/nm2) measured in ethanol solution with thionine acetate. This result indicates that most of the surface –COOH groups formed 1:1 ion pairs with Rhodamine 6G under the experimental condition. In addition, the density value obtained with Cs+ (0.81 ± 0.32/nm2) was almost identical to that with Rhodamine 6G, indicating that the density values were primarily determined by surface –COOH groups and were not limited by the probe cation size. Based on these results, it is concluded that the three cation-exchange-based methods can be similarly employed to determine the effective surface –COOH density on a nanoporous film. Because of the experimental simplicity, the spectrofluorometric method in aqueous solution with Rhodamine 6G was used for the further investigations.
| MW of polymer | Diameter (nm)a | Pore density (/μm2)a | –COOH density (/nm2) |
|---|---|---|---|
| a Measured from AFM images of three separate samples. b The average and standard deviation of surface –COOH density measured using Rhodamine 6G as a counter cation. The numbers of samples examined are shown in square brackets. c The average and standard deviation of surface –COOH density measured using Cs+ as a counter cation. The numbers of samples examined are shown in square brackets. d The average and standard deviation of surface –COOH density measured using thionine acetate (in ethanol) as a counter cation.27,28 The numbers of samples examined are shown in square brackets. | |||
| 82 000 (82 K) | 30 ± 3 | 570 ± 50 | 0.93 ± 0.12 [7]b |
| 57 000 (57 K) | 20 ± 3 | 890 ± 70 | 0.79 ± 0.14 [9]b |
| 0.81 ± 0.32 [4]c | |||
| 0.86 ± 0.08 [6]d | |||
| 43 000 (43 K) | 14 ± 1 | 1220 ± 70 | 0.78 ± 0.07 [6]b |
Next, the effective surface –COOH density was measured on nanoporous films derived from three types of PS-b-PMMA with different molecular weights (82 K, 57 K and 43 K) (Table 1). These polymers offer nanoporous films with different pore diameters, and thus were used to investigate the effect of pore diameter on surface –COOH density. The effective density values were very similar for the three types of films. This result indicates the involvement of the same reaction(s) in nanopore formation.
Subsequently, surface –COOH densities were measured on 57 K PS-b-PMMA-derived nanoporous films modified via amidation or esterification. EDC-mediated aqueous-phase amidation led to decreases in surface –COOH density (Table 2 (a)), reflecting the amidation of surface –COOH groups with amine reactants. However, the amidation yields were relatively low (20–30%) under the experimental condition examined (in 0.1 M phosphate buffer, pH 6), as inferred by previous electrochemical results.19 The low amidation yields were probably determined by the low efficiency of intermediate reaction with N-hydroxysuccinimide in phosphate buffer of pH 6.13,30
| Reactant | –COOH density (/nm2)a | Functionalization Yield (%)b |
|---|---|---|
| a The average and standard deviation of surface –COOH density determined using Rhodamine 6G as a counter cation. The numbers of samples examined are shown in square brackets. b The yield was calculated from the average surface –COOH density values for 57 K PS-b-PMMA-derived nanoporous films with and without surface modification. c Represents the yield of intermediate reaction with N-hydroxysuccinimide. d The chemical structures are shown in Fig. 1. e Tetraethyleneglycol monoamine. | ||
| (a) Aqueous-phase modification mediated by EDC | ||
| H2O c | 0.65 ± 0.03 [4] | 18 |
| NH3 | 0.56 ± 0.02 [3] | 29 |
| FcCH2NH2d | 0.63 ± 0.01 [3] | 20 |
| (PEO)4NH2d,e | 0.58 ± 0.14 [4] | 26 |
| (b) Organic-phase modification using oxalyl chloride | ||
| H2O | 0.82 ± 0.09 [7] | — |
| Ethanol (2 mM in CH2Cl2) | 0.10 ± 0.01 [4] | 88 |
| FcCO(CH2)5OH d | 0.09 ± 0.01 [6] | 88 |
| FcCO(CH2)15OH d | 0.10 ± 0.02 [12] | 87 |
| FcCH2NH2d | 0.10 ± 0.03 [4] | 88 |
| (PEO)4NH2d,e | 0.09 ± 0.02 [5] | 89 |
In addition, the nanopore surface was functionalized via organic-phase amidation/esterification using oxalyl chloride. The organic-phase reactions were applicable for PS-b-PMMA-derived nanopores because of their high stability under organic environments: As shown in Fig. 2, a PS-b-PMMA-derived nanoporous film retained the nanoporous structure after organic-phase esterification with FcCO(CH2)15OH. Very similar pore diameters for AFM images in Fig. 2a and 2b (20 ± 3 nm and 19 ± 2 nm, respectively) indicate the high solvent-resistance of the films due to the crosslinking of the PS matrix during UV irradiation.7,26,31 The surface –COOH density after the hydrolysis of acid chloride with water (0.82 ± 0.09/nm2; Table 2 (b)) was very similar to that of native films (0.79 ± 0.14/nm2; Table 1), supporting the high stability of the nanoporous structure. Importantly, as compared with aqueous-phase amidation, organic-phase reactions led to much larger decreases in surface –COOH density (Table 2 (b)). The surface modification yields were 87–89%, indicating that organic-phase reactions provide efficient means for modifying the nanopore surface. The modification yields were very similar for the different reactants examined, indicating that these reactants were sufficiently small as compared to the pore size and thus could easily access the nanopore surface.
Conclusions
In this paper, the density and modification of surface –COOH groups on PS-b-PMMA-derived nanoporous films were quantitatively assessed from the amount of probe cations released via cation exchange. Aqueous-phase amidation mediated by EDC is suitable to immobilize biological molecules that are insoluble or denatured in organic solvents. The yields measured in this study were not high, probably due to the presence of phosphate in the solutions.13,30 The yield of EDC-mediated aqueous phase amidation can be improved by using an appropriate buffer such as a 2-morpholinoethanesulfonic acid.13 In contrast, organic-phase amidation and esterification using oxalyl chloride are suitable for functionalizing the solvent-resistive nanoporous films with high modification yields. The high resistance of these films to organic solvents makes it possible to employ various organic-phase reactions for functionalizing the nanopore surface. An appropriate modification method should be chosen according to the applications of nanoporous films. Quantitative information on the surface functionalization will enable systematic studies of the influences of the surface modification efficiency on the performances of the nanoporous films as chemical separation membranes.Acknowledgements
The authors thank Prof. Royce Murray (Univ. North Carolina, Chapel Hill), Profs. Duy Hua and Daniel Higgins (Kansas State Univ.) for their suggestions on this work, Prof. Christopher Culbertson (Kansas State Univ.) for access to the surface profiler, and Dr Javier Seravalli (Dept. Biochem., Univ. Nebraska, Lincoln) for ICP-MS measurements. The authors gratefully acknowledge the US Department of Energy (DE-SC0002362), ACS Petroleum Research Funds (ACS PRF# 46192-G5), Terry C. Johnson Center for Basic Cancer Research and Targeted Excellence Funds of Kansas State University for financial support of this work. RD acknowledges the support of the Kansas State University Summer Undergraduate Research Opportunity Program.References
- M. A. Hillmyer, Adv. Polym. Sci., 2005, 190, 137–181 CrossRef CAS.
- D. A. Olson, L. Chen and M. A. Hillmyer, Chem. Mater., 2008, 20, 869–890 CrossRef CAS.
- J. Bang, U. Jeong, D. Y. Ryu, T. P. Russell and C. J. Hawker, Adv. Mater., 2009, 21, 4769–4792 CrossRef CAS.
- T. Ito and D. M. N. T. Perera, in Trace Analysis with Nanomaterials, ed. D. T. Pierce and J. X. Zhao, Wiley-VCH, Weinheim, Editon edn, 2010, pp. 341–358 Search PubMed.
- E. A. Jackson and M. A. Hillmyer, ACS Nano, 2010, 4, 3548–3553 CrossRef CAS.
- S. Y. Yang, I. Ryu, H. Y. Kim, J. K. Kim, S. K. Jang and T. P. Russell, Adv. Mater., 2006, 18, 709–712 CrossRef CAS.
- S. Y. Yang, J. Park, J. Yoon, M. Ree, S. K. Jang and J. K. Kim, Adv. Funct. Mater., 2008, 18, 1371–1377 CrossRef CAS.
- E. E. Nuxoll, M. A. Hillmyer, R. Wang, C. Leighton and R. A. Siegel, ACS Appl. Mater. Interfaces, 2009, 1, 888–893 CAS.
- W. A. Phillip, B. O'Neill, M. Rodwogin, M. A. Hillmyer and E. L. Cussler, ACS Appl. Mater. Interfaces, 2010, 2, 847–853 CAS.
- S. Y. Yang, J.-A. Yang, E.-S. Kim, G. Jeon, E. J. Oh, K. Y. Choi, S. K. Hahn and J. K. Kim, ACS Nano, 2010, 4, 3817–3822 CrossRef CAS.
- L. A. Baker, P. Jin and C. R. Martin, Crit. Rev. Solid State Mater. Sci., 2005, 30, 183–205 CrossRef CAS.
- A. S. Zalusky, R. Olayo-Valles, J. H. Wolf and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 12761–12773 CrossRef CAS.
- J. Rzayev and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 13373–13379 CrossRef CAS.
- T. S. Bailey, J. Rzayev and M. A. Hillmyer, Macromolecules, 2006, 39, 8772–8781 CrossRef CAS.
- A. Klaikherd, S. Ghosh and S. Thayumanavan, Macromolecules, 2007, 40, 8518–8520 CrossRef CAS.
- J.-H. Ryu, S. Park, B. Kim, A. Klaikherd, T. P. Russell and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 9870–9871 CrossRef CAS.
- S. Y. Yang, S. Son, S. Jang, H. Kim, G. Jeon, W. J. Kim and J. K. Kim, Nano Lett., 2011, 11, 1032–1035 CrossRef CAS.
- Y. Li, H. C. Maire and T. Ito, Langmuir, 2007, 23, 12771–12776 CrossRef CAS.
- Y. Li and T. Ito, Langmuir, 2008, 24, 8959–8963 CrossRef CAS.
- Y. Li and T. Ito, Anal. Chem., 2009, 81, 851–855 CrossRef CAS.
- S. Ibrahim and T. Ito, Langmuir, 2010, 26, 2119–2123 CrossRef CAS.
- T. Thurn-Albrecht, R. Steiner, J. DeRouchey, C. M. Stafford, E. Huang, M. Bal, M. Tuominen, C. J. Hawker and T. P. Russell, Adv. Mater., 2000, 12, 787–791 CrossRef CAS.
- H. C. Maire, S. Ibrahim, Y. Li and T. Ito, Polymer, 2009, 50, 2273–2280 CrossRef CAS.
- T. Ito, I. Grabowska and S. Ibrahim, TrAC, Trends Anal. Chem., 2010, 29, 225–233 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, 2nd Ed., Wiley, New York, 2001 Search PubMed.
- U. Jeong, D. Y. Ryu, J. K. Kim, D. H. Kim, T. P. Russell and C. J. Hawker, Adv. Mater., 2003, 15, 1247–1250 CrossRef CAS.
- A. Papra, H.-G. Hicke and D. Paul, J. Appl. Polym. Sci., 1999, 74, 1669–1674 CrossRef CAS.
- C. Geismann and M. Ulbricht, Macromol. Chem. Phys., 2005, 206, 268–281 CrossRef CAS.
- K. Odashima, T. Ito, K. Tohda and Y. Umezawa, Chem. Pharm. Bull., 1998, 46, 1248–1253 CAS.
- M. A. Gilles, A. Q. Hudson and C. L. Borders, Jr., Anal. Biochem., 1990, 184, 244–248 CrossRef CAS.
- D. M. N. T. Perera, B. Pandey and T. Ito, Langmuir, 2011, 27, 11111–11117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis procedures, 1H-NMR spectra, 13C-NMR spectra and FTIR spectra of ferrocenylmethylamine (FcCH2NH2), 16-hydroxy-1-oxohexylferrocene (FcCO(CH2)5OH) and 16-hydroxy-1-oxohexadecylferrocene (FcCO(CH2)15OH), and the fluorescence spectra of Rhodamine 6G and thionine. See DOI: 10.1039/c1ra00471a |
| This journal is © The Royal Society of Chemistry 2011 |