Fluorinated contrast agents for magnetic resonance imaging; a review of recent developments
James C.
Knight
*a,
Peter G.
Edwards
b and
Stephen J.
Paisey
c
aDepartment of Oncology, University of Alberta, Edmonton, AB, T6G 1Z2, Canada. E-mail: jknight@ualberta.ca; Tel: (780) 432 8932
bDepartment of Chemistry, Cardiff University, PO Box 912, Cardiff CF10 3TB. E-mail: edwardspg@cf.ac.uk; Tel: +44 (0)29 208 74083; Fax: +44 (0)29 208 74030
cWales Research and Diagnostic Positron Emission Tomography Imaging Centre, Cardiff University, School of Medicine, Heath Park, Cardiff, CF14 4XN. E-mail: paiseysj@cf.ac.uk; Tel: +44 (0)29 207 43676; Fax: +44 (0)29 207 46879
First published on 26th October 2011
Abstract
The development of medical imaging probes for magnetic resonance imaging (MRI) is a particularly dynamic area of research. At present, many prominent groups are dedicating significant resources to tailoring and optimising the performance of potential contrast agents. Whilst 1H MRI has become an indispensable tool for the imaging of disease states, it frequently suffers from low contrast owing to background signal from intrinsic 1H. As a result, increasing attention is being directed at compounds containing 19F as this nucleus has a similar NMR sensitivity to 1H and, importantly, intrinsic 19F signals are virtually undetectable in vivo. For several decades, perfluorinated molecules (in which all of the C–H bonds in the parent molecule have been replaced with C–F bonds) and highly fluorous gases such as SF6 have traditionally been used for these kinds of investigations and there have been some excellent reviews of these compounds and their applications. However, recently 19F imaging is showing signs of evolution, particularly as there have been several reports of fluorinated responsive (smart) agents, micelles, dendrimers and hyperbranched polymers being investigated as targets for 19F-MRI. Furthermore, examples of multimodal contrast agents containing 19F nuclei are also starting to emerge. In this review we aim to summarise these exciting recent chemical developments.
 James C. Knight | Dr James C. Knight obtained a BSc in chemistry from Cardiff University in 2005 and subsequently a PhD in 2009 under the supervision of Dr Angelo J. Amoroso and Prof. Peter G. Edwards. He subsequently spent two years working between the Cardiff School of Biosciences and Chemistry departments developing multimodal imaging agents under the supervision of Dr Richard W. E. Clarkson, Dr Stephen J. Paisey and Prof. Peter G. Edwards. Currently, he is a postdoctoral fellow based at the University of Alberta with Dr Frank Wuest. His work now focuses on the development and testing of novel cancer-targeting imaging agents. |
 Peter G. Edwards | Prof. Peter G. Edwards received his BSc degree in Chemistry from Imperial College in 1976 where he continued to a PhD in 1979 under the supervision of Sir Geoffrey Wilkinson. This was followed by postdoctoral fellowships with Professors Robert Bau at the University of Southern California, Los Angeles (1979–80), Richard Andersen at The University of California at Berkeley (1980–83) and Sir Geoffrey Wilkinson (1983–84) before being appointed as Lecturer in Chemistry at Cardiff University in 1984. He was awarded his Chair position in 1995. His research has focused on ligand design and functional complexes in coordination, organometallic, catalysis chemistry and in bio-medical imaging. |
 Stephen J. Paisey | Dr Stephen J. Paisey, born in Southampton in 1975, studied at Cardiff University obtaining a (BSc(Hons) in chemistry, 1995) and a PhD supervised by Prof. Peter G. Edwards. He has subsequently worked in medicinal-inorganinc chemistry under Prof. Peter Sadler at the University of Edinburgh, NMR protein structure assignment under Prof. Steve Homans at the University of Leeds and as MRI experimental liaison officer at Cardiff University's Experimental MRI Centre (2007–2011). He is currently a research fellow at the Wales Research and Diagnostic Positron Emission Tomography Imaging Centre. |
1. Introduction and scope
1.1 Advantages of magnetic resonance imaging
There are many imaging techniques, each of which has its own advantages and disadvantages. For example, the optical-based imaging techniques such as fluorescence imaging, coherent anti-Stokes Raman spectroscopy (CARS) and optical coherence tomography can provide sub-cellular resolution images with sub-second time resolution. However, these techniques are limited to surface investigations as they typically only have shallow depth penetration of samples.Ultrasound is a rapid, real time, soft tissue imaging technique which has a much better depth penetration than optical imaging (∼20 cm) but at the expense of resolution (mm–cm). In fact, the resolution of ultrasound images is variable depending upon the depth penetration required. Also, ultrasound does not penetrate air gaps or bone well, making it unsuitable for lung imaging and adult brain imaging. Computed tomography (CT) and positron emission tomography (PET) imaging, commonly used in tandem, do not have issues with depth penetration but rely upon ionising radiation and so the number of scans a patient can receive are limited for safety reasons. CT provides mm–cm resolution and provides good contrast to between hard and soft tissues with a typical scan taking 3–4 min to acquire. PET imaging measures the localisation of biologically active short lived radioactive isotopes at pM concentrations and mm–cm resolution typically with a 30–40 min scan time. As a result PET provides metabolic and functional information but needs to be coupled to an anatomical imaging method such as CT or MRI. The requirement for radioactive chemicals freshly prepared by skilled scientists makes PET an extremely expensive imaging technique.
MRI is an extremely versatile anatomical and functional imaging technique which excels at deep, soft tissue imaging. It can provide finer distinctions between soft tissues at slightly higher resolution (mm for human scanners and 50–100 μm for ultra high-field animal scanners) than CT/PET without the need for ionising radiation. MRI has no practical limitations due to sample penetration and the contrast between tissues can be manipulated by altering the scan acquisition parameters to best highlight the tissues of interest. MRI is a fairly slow imaging technique with scans times being highly dependent upon the resolution required and the field of view size. Typical anatomical scans take between 10 min and an hour to acquire.
Standard MRI relies upon the detection of differences in relaxation rates of the protons of water molecules within tissue types. However, there are occasions where the relaxation rates of protons do not differ sufficiently between tissue types to produce useful contrast. One way around this is to introduce paramagnetic metal ion containing contrast agents which cause a localised alteration of endogenous proton relaxation times.
A detailed discussion of this strategy is beyond the scope of this review, however the subject is well represented in the literature.1–4 An alternative approach to highlighting particular tissues is to introduce an additional biologically localising tracer MRI signal to the system.
1.2 Usefulness of fluorine-19 in MR applications
In principle, any NMR active nucleus can be imaged by MRI, subject to the availability of appropriately tuned amplifiers and transceiver coils. However, in practice the choice of useful imaging nuclei is severely limited by a combination of the inherent physical, chemical and biological properties of each nucleus. The relative natural abundance of an NMR active nucleus and its response to a magnetic field vary from element to element meaning that the NMR signal per mole of compound varies from element to element. The overall NMR sensitivity of a nucleus at natural abundance can be expressed as its NMR receptivity. 1H has the highest receptivity of any nucleus, so for comparison sake the receptivity of a nucleus is often quoted relative to 1H (where 1H has a receptivity of 1).5 For example in samples of naturally abundant isotopes, 13C has a receptivity relative to 1H of just 1.7 × 10−4 and 15N has a receptivity of 3.84 × 10−6, largely due to the low natural abundance of their NMR active nuclei. By comparison 19F, which has a natural abundance of 100%, has much more useful receptivity of 0.834 relative to 1H.5Fluorine also has a large chemical shift range (>300 ppm).Fluorine also is an attractive element from a chemical/biological perspective as the substitution of a hydrogen for a fluorine within a molecule often has very little effect on the chemical and biological properties of the molecule. Thus novel tracers can be easily created by the fluorination of compounds of known biological activity. Also, unlike the radioactive isotope 18F used for PET imaging, there is no need for specialist synthetic methodologies to introduce hot fluorine into tracers for MRI as 19F is a stable isotope.
In addition, 19F containing MRI tracers are more stable than lanthanide based coordination compounds used as contrast agents since the MRI active part of a 19F containing MRI tracer is covalently bound to the part of the molecule conferring biological selectivity. This increased stability reduces the risk of false positives due to compound degradation and completely negates the risk of toxicity due to unbound metal ions. Finally, there is virtually no background 19F-MRI signal in biological samples as the endogenous fluorine concentrations are below MRI detection limits.6,7
1.3 A brief history of 19F imaging
Literature reports of 19F imaging date back to 1977, just 4 years after the development of 1H MRI,8 when Holland et al. described images of sodium fluoride and perfluorotributylamine phantoms.9 However, it was not until 1985 that McFarland and co-workers presented in vivo19F images of rat abdomens.10 Since then, there have been several hundred publications produced reporting upon advances in 19F-MRI/MRS and its efficacy in a broad range of applications such as pO2 sensing,11 hypoxia,12pH sensing,13drug metabolism,14cell tracking15 and plaque detection.16 Much of this work has been described in a comprehensive review of the subject area by Yu et al. in 2005.17 There have been reviews since this one that largely focus on the use of perfluorocarbons in biological applications.18,19 The aim of this review is to provide a summary (particularly from a chemical perspective) of the exciting developments in contrast agent design that have occurred in this area since 2005.2. Micelles
Micelles are lipid-based compounds composed of amphiphilic molecules which undergo spontaneous self-assembly.20 In aqueous solution these amphiphiles typically form micelles in which the hydrophobic regions associate to form a central core and the hydrophilic groups remain in contact with the water (Fig. 1). The application of micelles for in vivo imaging has attracted much attention over the last two decades, and many groups have constructed micelles consisting of amphiphiles which incorporate a hydrophilic Gd(III) chelate.21–23 The slow tumbling rate of these large species has been shown to be crucial in shortening the T1 and T2 relaxation times, particularly at clinically relevant field strengths. Furthermore, the ability to conjugate groups which bind to specific biological targets and, also, the potential to incorporate multimodal functionality (for example, by further integrating optical and/or nuclear imaging probes) has led to a wave of interest in these structures. The use of this technology for applications in 19F-MRI however has only recently begun to be explored.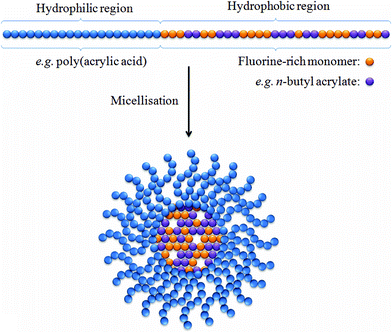 | ||
| Fig. 1 A simplified representation of a micelle containing a hydrophobic core with a large population of fluorine nuclei. The incorporation of n-butyl acrylate (purple spheres) into the hydrophobic block viastatistical copolymerisation has been shown by Peng et al. to enable longer T2 relaxation times by lowering the glass transition temperature.24 | ||
Recently, Peng et al.24 reported a series of amphiphilic diblock copolymers in which the hydrophilic region was composed of poly(acrylic acid) and the hydrophobic region consisted of n-butyl acrylate copolymerised with partly fluorinated acrylate or methacrylate. These diblock copolymers were shown to successfully self-assemble into stable micelles, containing a high density of fluorine within the hydrophobic core, ranging in size between 20 and 45 nm. 19F NMR experiments revealed that for each of the samples under investigation only a single 19F resonance frequency was observable. This is a desirable characteristic as multiple resonance signals can often necessitate the use of selective MRI pulse sequences to prevent the acquisition of ghost images. In all samples, a single T1 relaxation time was recorded between ca. 500–600 ms. The authors investigated the role of plasticising organic solvents and found a favourable effect upon imaging performance, resulting from an increase in segmental mobility within the hydrophobic core of the micelles. This flexibility helps to prevent incomplete averaging of the dipolar and chemical shift interactions The favourable NMR and MRI properties of these micelles highlighted the potential of this approach.
A similar study reported by Du et al. looked at the properties of another new series of hyperbranched-star amphiphilic fluoropolymers.25 These polymers were also shown to undergo micelle formation and were designed as potential targets for 19F-MRI. In this example, stable micelles were formed which ranged in size between 20 and 30 nm. 19F NMR experiments revealed a single narrow 19F signal and good T1 and T2 relaxation times of ca. 500 and 50 ms, respectively. These relaxation properties remained largely unaffected by the size and composition of the micelles. 19F-MRI phantom images were collected and high signal-to-noise ratios were obtained which demonstrates the potential usefulness of these micelles for a range of biomedical applications. However, it was noted that due to the long scan times which were required, these micelles were not suitable as blood pool imaging agents.
A bispherical molecule called 19FIT was developed by Jiang et al. for application as a 19F imaging tracer (Fig. 2).26 This water-soluble molecule has 27 fluorine atoms which generate a single 19F signal. 19F-NMR experiments revealed that when dissolved in phosphate buffered saline (PBS), 19FIT forms micelles with a critical micelle concentration of ca. 7 mM. The longitudinal relaxation time of 19FIT was reported to be 163 ms which is significantly shorter on comparison to the macrocyclic compound perfluoro-15-crown-5-ether (T1 = 1069 ms). This short T1 relaxation time is beneficial as it results in a shorter data collection time. Whole-body 1H and 19F images of a mouse injected with a PBS solution of 19FIT were obtained and revealed that at 1-2 h post-injection the 19F signal decreased quickly to unobservable levels in all organs. Based on the rate of decrease in 19F signal in whole-body spectra and in urine samples, the in vivo residence half-life of 19FIT was estimated at ca. 0.5 days which is substantially less than perfluorocarbons which have been reported to persist in the organs and tissues for several months. Importantly, high-performance liquid chromatography profiles of urine samples prior to and post-injection, revealed the presence of only one fluorinated compound with a molecular mass equal to that of 19FIT.
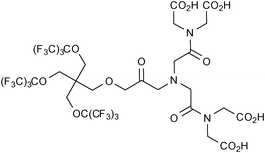 | ||
| Fig. 2 The bispherical molecule ‘19FIT’ developed by Jiang et al.26 for application as a 19F imaging tracer. | ||
The authors suggested that this demonstrates the in vivo stability of this compound and eliminates complications involving degradation products. Furthermore, after being injected with the compound, mice were monitored for a period of 45 days and experienced no weight loss or showed any signs of acute toxicity.
These studies hint at the potential of micelles to act as effective platforms for the delivery of large amounts of 19F nuclei and to function as intravascular 19F-MRI probes.
Furthermore, several groups have demonstrated the ability to functionalise the periphery of micelles with a variety of groups capable of targeting biological markers of disease.27–30 Such modifications can be realistically applied to micelles containing large populations of 19F nuclei, thus enabling the directed accumulation of these potential contrast agents at selected targets. In addition, attaching chains of poly(ethylene) oxide to micelles has previously been shown to extend blood residence times by enabling these so-called ‘stealth micelles’ to evade immune recognition.22 Such an approach might also prove favourable in some 19F-MRI applications involving micelles.
3. Dendrimers and hyperbranched polymers
Dendrimers are a class of nanoscale macromolecules which have well-defined, repeating branched polymeric chains which emanate from a central core (For an example, see Fig. 3).31–36 The ability to selectively functionalise these materials has facilitated advances in several fields, perhaps most notably in biomedical applications. There have been a host of examples over the last two decades of dendritic MRI contrast agents comprising multiple Gd(III) moieties.37 Such dendrimers frequently exhibit substantial increases in rotational correlation times which leads to strong enhancements in T1 relaxation rates compared to lower molecular weight chelates. Several in vivo studies have demonstrated that dendrimers typically exhibit prolonged retention times within the vascular system compared to lower molecular weight chelates which usually undergo rapid renal clearance.38,39 Furthermore, the conjugation of targeting groups to the periphery of dendrimers has been successfully employed to bind specific biological markers of disease.40–42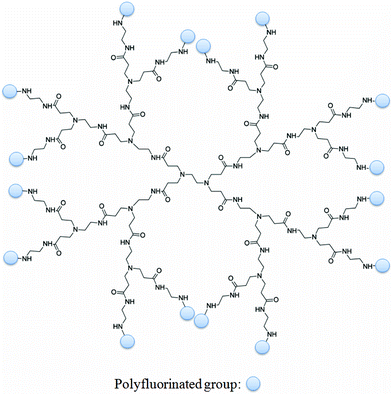 | ||
| Fig. 3 A representation of a dendrimer (in this case, PAMAM-G2) functionalised with multiple polyfluorinated groups. It is easy to theorise bi-functionalised dendrimers in which targeting moieties capable of binding markers of disease are also incorporated. | ||
Such materials are attractive in 19F MR probe design due to large number of surface functional groups which can be reacted with polyfluorinated precursors, leading to materials with potentially very high populations of fluorine nuclei. The number of surface groups present on a dendrimer increases geometrically with the generation number. For example, a conventional PAMAM dendrimer will have the following number of end groups: G0 = 4, G1 = 8, G2 = 16, G3 = 32, G4 = 64, G5 = 128, G6 = 256, etc. Therefore, a G6-PAMAM dendrimer reacted with, for example, heptafluorobutyric acid could in theory carry up to 1,792 fluorine nuclei. Furthermore, bi-functionalising the dendrimers to include targeting groups could facilitate the site-directed delivery of these large payloads of fluorine. Recently, examples of dendrimers and hyperbranched polymers incorporating large quantities of 19F nuclei have started to emerge which is leading to interest in these structures as potential 19F MR imaging agents.
An example of this type of study was recently reported by Thurecht et al. who synthesised a series of fluorinated (trifluoroethylacrylate) hyperbranched polymers having acid, alkyne and mannose end-groupsvia a slightly modified reversible addition-fragmentation chain transfer methodology (RAFT).43 The size of each of these polymers in pure water was determined by dynamic light scattering (DLS) and was typically ca. 10 nm. The polymer containing acid end-groups which formed the basis of the study displayed favourable in vitro imaging properties, yielding a T2 relaxation time of 88 ms at 16.4 T (20 mg mL−1 in pure water). A T1 relaxation time of 480 ms was also recorded. The non-targeting hyperbranched copolymer was examined in vivo using a mouse model. A high-resolution 1H MRI scan of the abdominal cavity 2 h post injection was recorded and quickly followed by a 19F-targeted scan of the same area. These images were superimposed (Fig. 4) and reveal a clearly observable 19F signal, largely present within the bladder. This indicates that the polymer can be removed from the blood and excreted via the kidneys.
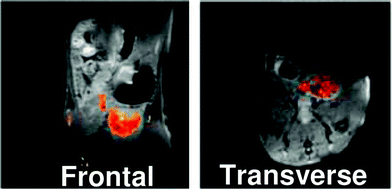 | ||
| Fig. 4 MR images of mouse abdominal region 2 h after tail vein injection of polymer with acid end groups (P1) into mouse. 1H image is shown in greyscale and 19F image is overlaid. P1 can be seen to accumulate in the bladder region. Reprinted with permission from ref. 43. Copyright 2010 American Chemical Society. | ||
This work also showed that by utilising Huisgen-type click reactions, these hyperbranched polymers can also be readily functionalised with potentially a large array of targeting moieties. This could lead to the site-specific delivery of high payloads of fluorine nuclei to, for example, markers of disease.
In 2009, a series of poly(amidoamine) (PAMAM) starburst dendrimers were reported by Criscione et al. and were shown to form complex nanoscopic and microscopic particulates via a spontaneous self-assembly process.44 These dendrimers were modified via the covalent attachment of perfluoroalkyl groups to the constituent dendrimer subunits. The synthesis of these fluorinated particulates involved reacting PAMAM(G3) dendrimers with heptafluorobutyric acid in the presence of triethylamine, leading to dendrimers with 15 covalently attached heptafluoroacyl groups. These fluorinated dendrimers, which were then further modified with PEG in order to improve their transportation properties within the cardiovascular system, were evaluated in vivo (Fig. 5). The dendrimeric probes were shown to stay within the vasculatory system before being removed by the liver. An interesting feature of these particulates is their capacity to gradually and irreversibly break up upon exposure to acidic conditions. The authors postulated that this behaviour could perhaps be useful in enabling the controlled release of encapsulated materials, e.g. therapeutic agents, and demonstrated this by inducing the release of the small molecule fluorescent dye Rhodamine B.
 | ||
| Fig. 5 19F MR images taken following intravenous injection of PEGylated, fluorinated PAMAM(G3) particulates. (a) 19F CSI image and (b) 1H image reveal the presence of PEGylated particulates in the mouse kidney vasculature. These images are merged in (c). (d) 19F CSI image and (e) 1H image showing the filtration of the PEGylated particulates in the mouse liver. These images are merged in (f). Reprinted from Biomaterials, Vol. 30, Criscione et al., Self-assembly of pH-responsive fluorinated dendrimers-based particulates for drug delivery and non-invasive imaging, p. 3946–3955, Copyright 2009, with permission from Elsevier. | ||
More recently, Ogawaet al. examined the imaging potential of two novel polymer nanoparticles (PNPs) which were composed of fluorinated methacrylate monomers grafted onto PAMAM.45 These PNPs were PAMAM-g-PTFPMA (TFPMA = 2,2,3,3-tetrafluoropropyl methacrylate) and PAMAM-g-PTFEMA (TFEMA = 2,2,2-trifluoroethyl methacrylate). DLS experiments performed on PAMAM-g-PTFPMA at different time points indicated that these nanoparticles were spherical with hydrodynamic diameters reaching 24.3 nm after 490 min. Furthermore, these PNPs were also shown by GPC to have a small polydispersity index of 1.5. Micrographs obtained from transmission electron microscopy (TEM) revealed spherical PAMAM-g-PTFPMA nanoparticles with an average diameter of 19.5 nm (σ = 1.5 nm). This value is slightly smaller than that obtained from the DLS experiments due to the lack of solvent which causes the nanoparticles to swell. The T1 and T2 relaxation times of both PNPs were investigated by NMR techniques to establish their imaging potential. An inversion recovery sequence was used to determine T1 values for both PAMAM-g-PTFEMA and PAMAM-g-PTFPMA which were 0.6 and 0.3 s, respectively. These relaxation times are short in comparison to low-molecular weight fluorinated agents such as trifluorotoluene (2 s). A multiecho spin-echo NMR sequence was used to establish the corresponding T2 values of 0.4 and 0.1 s, respectively. The authors noted that these values, while shorter than small molecules such as TFT (2 s), were significantly longer compared to neat perfluorocarbons such as perfluoro-2,2,2′,2′-tetramethyl-4,4′-bis(1,3-dioxolane) (6–20 ms).
The same group also reported a similar study into the synthesis and in vivo performance of related PNPs with a high fluorine content.46 The fluorinated polymer nanoparticles (PAMAM-g-PTFPMA-b-PCMB) were highly soluble in aqueous conditions. DLS measurements and TEM revealed that the PNPs were spherical in appearance and had hydrodynamic diameters in the range of 15–80 nm in ultrapure water. Four samples of PAMAM-g-PTFPMA-b-PCMB nanoparticles exhibited T1 and T2 relaxation times of <250 ms and ca. 10 ms, respectively. Despite the short T2 which leads to a reduction in signal, the fast T1 allowed for a shorter repetition time and therefore the rapid collection of imaging signals. In vitro19F-MRI experiments indicated that the T2 relaxation time was influenced largely by the molecular weight of the polymer, with the largest PNPs exhibiting the lowest mobility and therefore the shortest T2 relaxation times. Despite this, the rapid signal acquisition meant that signal was observed at particle concentrations in the sub-micromolar concentration level.
A series of PEGylated nanogels containing a cross-linked poly[2-(N,N-diethylamino)ethyl methacrylate]-copoly(2,2,2-trifluoroethyl methacrylate) gel core were generated by Oishi et al.47,48 These nanoprobes contain tethered poly(ethylene glycol) (PEG) chains with acetal terminal groups which could allow for the conjugation of tumour-targeting moieties. The authors demonstrated that the 19F-MRI signal intensities and signal-to-noise ratios of these materials could be controlled by adjusting the extracellular pH (from pH 7.4 to 6.5). It was suggested that the pH-dependent behaviour of these nanoprobes could be a result of the gel core ‘swelling’ in acidic environments as the amino groups of the PEAMA segments become protonated. The consequent enhancement in the molecular motion would expectedly increase the T2 relaxation time. Indeed, the hydrodynamic volume of these nanoprobes was shown to increase proportionally as the pH was decreased from 7.4 to 7.0. At pH 6.5, the hydrodynamic volume was 8.7-fold larger when compared to the original size at physiological pH (7.4). The MR signal intensity of the fluorinated nanoprobes was effectively ‘switched off’ at pH 7.4 due to the decrease in molecular motion as the hydrodynamic volume is reduced, leading to extremely short T2 values (ca. 100 μs). Conversely, the MR signal was effectively switched on at pH 6.5 as the gel core swelled. As solid tumours often exhibit low extracellular pH due to the Warburg effect,49,50 this ‘smart’ agent therefore demonstrates the potential to selectively image these disease sites.
Clearly, these studies highlight the great potential of dendrimers and hyperbranched polymers to generate highly fluorinated probes with favourable imaging properties. Dendrimeric precursors with a range of terminal functional groups (e.g. –NH2, –OH, –COO−, Si(OCH3)3, alkyl, succinamic acid, and 3-carbomethoxypyrrolidinone) are commercially available which allows for a number of different synthetic strategies with a range of polyfluorinated precursors. Furthermore, the existence of bi- and multi-functional dendrimers40–42 makes it easy to imagine such polyfluorinated dendrimers also being co-modified with, for example, targeting moieties to enable site-specific delivery to areas of biological interest, and also other moieties capable of ‘tuning’ their in vivo properties, e.g. solubility, circulation times, biodistribution.
4. Smart 19F-containing contrast agents
An imaging agent which can either be switched on or off upon being activated by a certain stimulus can be given a number of appropriate terms, e.g. activated, responsive, or smart. This class of probes offers the ability to sense a wide range of phenomena, including (i) the presence of certain biological markers (cellular receptors, proteinsetc.), (ii) metal ion concentration, (iii) pO2 (oxygen concentration), and (iv) pH (the nanogels described in Section 3 which undergo pH responsive swelling are excellent examples). As the smart imaging probe will remain effectively invisible until the particular stimulus in encountered, these agents have the potential to afford extremely specific and useful information. There have consequently been numerous reports in recent years of agents designed for this purpose; one archetypal example of an enzyme-activated smart agent is the compound (4,7,10-triacetic acid)-1-(2-β-galactopyranosylethoxy)-1,4,7,10-tetraazacyclododecane)gadolinium (EGad) developed by Meade and co-workers.51 This Gd-DOTA derivative incorporates a galactopyranose ‘cap’ which is selectively cleaved upon interaction with the enzymeβ-galactosidase, allowing water to access the inner coordination sphere of the gadolinium ion and effectively switching the probe ‘on’. Despite the sustained interest that such smart agents have received, reports of fluorinated probes which activate in such a manner have been comparatively sparse.The long T1 of fluorine has been a determining factor in its limited application as a target for MRI as it necessitates extended repetition times which can lead to prohibitively long scan times. By utilising the well-documented ability of paramagnetic metals to enhance the relaxation rates of nuclei in close proximity, some groups have applied paramagnetic metals in order to shorten the T1 relaxation times of 19F nuclei. This shortening of T1 allows more scans to be recorded within a certain timeframe and therefore leads to a better signal-to-noise ratio. Conversely, paramagnetic metals will also reduce the T2 relaxation time which in some cases can lead to line broadening so severe that it becomes very difficult to obtain a meaningful signal at all. Therefore, when designing an imaging agent of this type it is clearly important to tailor the structure of the compound (e.g. distance of 19F nuclei to the metal) and optimise the scan acquisition parameters to best suit the aims of the study. Some groups have started to utilise these phenomena in tandem with innovative chemical design in order to develop fluorinated smart agents capable responding to external stimuli.
In 2008, Mizukami et al. developed a novel probe capable of detecting the activity of the protease caspase-3.52Caspase-3 is a marker enzyme of apoptosis which recognises the tetrapeptide motif Asp-X-X-Asp (where X is optional). Their first report was based on the development of Gd-DOTA-DEVD-Tfb (Tfb = para-trifluoromethoxybenzyl), where DEVD is a tetrapeptide (Asp-Glu-Val-Asp) which undergoes selective cleavage of the C-terminal peptide bond by caspase-3. This probe is composed of three components which are arranged in a linear sequence: (i) a Gd(III) ion chelated by the macrocylic ligand DOTA, (ii) the enzyme substrate DEVD, and (iii) a moiety containing three fluorine atoms. This agent operates in a ‘smart’ fashion and switches from an OFF to an ON status upon interaction with the target enzyme. In its initial form, the highly paramagnetic Gd(III) ion conveys a intramolecular paramagnetic relaxation enhancement (PRE) effect upon the fluorine nuclei, causing T2 to become so short that it is essentially unable to be measured accurately. However, in the presence of caspase-3, the peptide sequence is selectively cleaved and the intramolecular PRE upon the 19F nuclei is effectively negated which leads to a subsequent lengthening of T2 and therefore a significant enhancement of signal. This was demonstrated by both 2D-NMR and 19F-MRI phantom imaging experiments which showed an increase in signal over time following the addition of caspase-3.
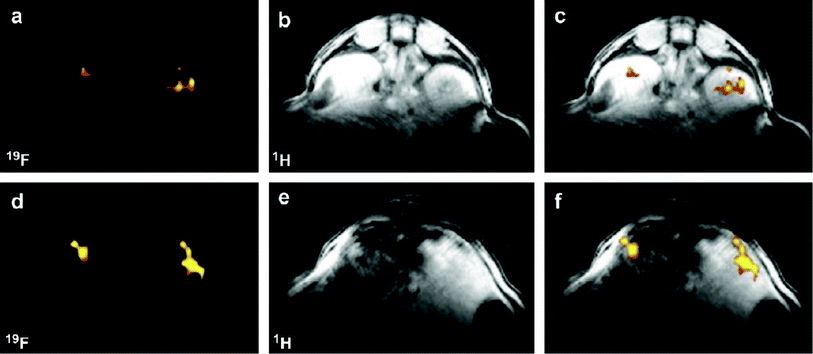
The following year, the same group reported a similar probe in which Tfb was replaced with the fluorescent group 7-amino-4-trifluoromethylcoumarin (AFC), thus generating a novel dual-functional (fluorescence and 19F-MRI) probe also intended for imaging caspase-3 activity (Fig. 6).53
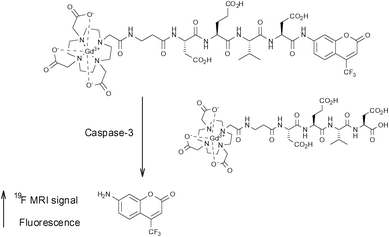 | ||
| Fig. 6 The smart bimodal contrast agent reported by Mizukami et al.53 switches ‘on’ upon interaction with the protease caspase-3. | ||
Generating a bimodal imaging agent via the incorporation of an optically active component provides some advantages over the Tfb analogue as it offers a choice of methods depending on the nature of the experiment being performed. The fluorescence properties of AFC are dependent upon the electron-donating ability of the 7-amino group and upon cleavage from the peptide by caspase-3, a quantifiable increase in fluorescence intensity and a marked blue to yellow-green (500 nm) shift in emission wavelength occurs when excited at 400 nm. Indeed, 19F-NMR and 19F-MR phantom imaging studies each showed an enhancement of signal following incubation with caspase-3 at 37 °C as a function of time (Fig. 7; lower). A corresponding increase in fluorescence intensity was also observed (Fig. 7; upper). These two reports clearly demonstrate the exciting potential of enzyme-activated 19F-MRI contrast agents for imaging biological activity.
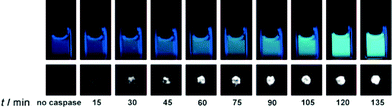 | ||
| Fig. 7 Time-dependent fluorescence images (upper, 10 μM) and 19F MR phantom images (lower, 1 mM) of Gd-DOTA-DEVD-AFC incubated with caspase-3. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced from ref. 53 with permission. | ||
In the last few years, there have also been a number of reports of paramagnetic fluorine-labelled lanthanide complexes,54–58 many of which can be considered as smart imaging probes due to their response to changes in pH. Such probes are potentially very useful, particularly in oncological imaging as solid tumours often exhibit low extracellular pH.49,50 In 2007, Senanayake et al. reported a series of pH-responsive probes containing CF3 groups in close proximity to paramagnetic lanthanide(III) ions.54 These compounds were designed with the intention of reducing the T1 relaxation time of the fluorine nuclei in order to facilitate shorter scan acquisition times. The ligands L1 and L3 (Fig. 8) were shown to form charge neutral mono-aqua complexes with a series of lanthanides whilst the sulfonamide-containing framework, L2 (Fig. 8), revealed reversible co-ordination of the sulfonamide N-donor in response to changes in pH. This behaviour also led to a corresponding change in the hydration of the metal centre. NMR experiments designed to measure the relaxation properties of these lanthanide complexes revealed that the CF3 groups in ortho-positions consistently experienced the greatest increase in R1 relaxation rate. Notably, the ortho-CF3 group of [DyL2]− exhibited a relaxation rate of 93 s−1 (at 188 MHz, 298 K, pH 5.5) and also a sizeable shift in resonance frequency of 100 ppm to lower frequency compared to the free ligand. The lanthanide complexes of L3 revealed a significant shift in resonance frequency of the CF3 group depending upon cleavage of the ester (ΔδF = 10 and 5.5 ppm for Tb and Tm complexes, respectively). This feature offers a potentially useful means of examining the activity of an esterase which is capable of catalysing the hydrolysis of the ester. This work successfully demonstrated the ability to enhance the R1 relaxation rate of 19F nuclei by incorporating a paramagnetic lanthanide ion within the molecular framework, facilitating shortened scan acquisition times and enhancing chemical shift non-equivalence in reporter resonances. This principle has since been successfully applied to perfluorocarbon nanoparticles by incorporating gadolinium ions within the surrounding lipid monolayer.59
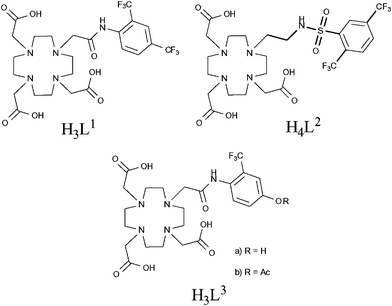 | ||
| Fig. 8 Senanayake et al. have effectively reduced T1 relaxation times by incorporating paramagnetic metals in close proximity to fluorine nuclei, enabling faster acquisition and therefore shorter scan times.54 | ||
This work was followed by similar studies,55–57 one of which examined the pH-responsive behaviour of similar lanthanide complexes (Fig. 9).55 In this example, the acidity of the amide hydrogen was tuned by incorporating either –NO2, –NH2 or –OH functional groups in the para-position of the aromatic ring. Of particular note were the holmium(III) complexes which, in the case of L5 and L6, revealed changes in chemical shift of the 19F (–CF3) resonance frequency of 18.3 and 40 ppm, respectively. This study again demonstrates the potential of these types of compound to be utilised in 19F MRS/MRI and, in particular, chemical shift imaging studies.
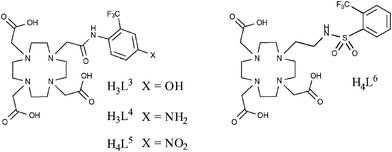 | ||
| Fig. 9 Kenwright et al. later looked at the pH-responsive behaviour of lanthanide complexes based on these ligands and for some metals, notably Holmium, observed chemical shift of the 19F signal, making these compounds potentially useful for chemical shift imaging studies.52 | ||
5. Multi-chromic 19F imaging tracers
A similar approach to that of the preceding two examples has recently been employed by Jiang et al. in their development of a ligand framework which contains 19F nuclei in proximity to a series of metal ions (Fig. 10).60 Whilst not strictly a smart agent, the frequency of the 19F signal and the T1 and T2 relaxation times were shown to be highly dependent upon the identity of the chelated metal. The largest shift in frequency was measured when the ligand (the fluorinated chelator) was complexed with Tb3+. The paramagnetic metals Fe3+, Gd3+, Er3+, Dy3+, and Ho3+ also resulted in large shifts. The T1 and T2 relaxation times were most significantly reduced when the ligand was chelated with Gd3+, however Fe3+, Ni2+ and Cu2+ also caused dramatic reductions compared to the free ligand. This compound could feasibly be modified to facilitate its conjugation to a variety of therapeutic agents and, by using different metals, could yield a multi-chromic (or multi-spectral) tracer capable of being tracked at specific frequencies.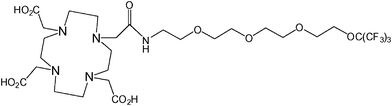 | ||
| Fig. 10 A fluorinated chelator developed by Jiang et al. yields a single fluorine resonance signal which can be shifted by changing the identity of the chelated metal.60 | ||
Another example of a multi-chromic 19F-based tracer has recently been developed and patented by Yu et al.61 This patent focuses on the modification of the 19FIT framework (Fig. 2) by incorporating paramagnetic metal ions via covalently attached chelating groups. The modulation of the 19F resonance frequency in this case can be accomplished by a variety of means. For example, the choice of metal ion will induce a unique and diagnostic shift in the frequency of the 19F signal. Furthermore, altering the 19FIT framework by (i) changing the proximity of the fluorine nuclei to the paramagnetic species, and/or (ii) changing the ratio of 19F nuclei to paramagnetic species would also have an effect upon signal modulation. This patent covers a range of possibilities along this theme and presents an attractive approach to multi-chromic MR imaging.
6. Targeted 19F-containing contrast agents
6.1 Amyloid plaques
The imaging of neurological diseases by 19F-MRI is also an emerging area of research which is attracting significant interest over recent years. In the last decade, benzoxazole compounds and many other frameworks incorporating PET-active 18F nuclei have been successfully employed to detect the early stages Alzheimer's disease and monitor its progression. This is generally accomplished by targeting amyloid plaques which are associated with the disease.In 2009, Amatsubo et al.62 investigated two benzoxazole derivatives, TFMB-2Et and TFMB-3Et (Fig. 11; upper and lower, respectively) containing trifluoromethoxy groups which were designed to image these amyloids using 19F-MRI. These compounds were shown to successfully cross the blood brain barrier (BBB) and selectively bind to amyloid plaques. This study highlighted the influence that the hydrophilic character of this series of compounds can have upon the 19F-NMR signal intensity in various tissues including the brain. Whilst these compounds produced sharp 19F NMR signals in PBS, the signal intensities acquired from homogenated tissue samples were consistently lower with the homogenated brain sample yielding the weakest and broadest 19F signal. Of the two compounds tested, the slightly more hydrophilic compound TFMB-3Et exhibited the highest signal-to-noise ratio. The in vitro19F-NMR observations were in good agreement with single pulse MR imaging measurements of mice which, following cervical dislocation, had been infused with either TFMB-2Et or TFMB-3Et directly into the brain and temporal muscle. These measurements revealed that TFMB-3Et afforded superior 19F signal intensity. Furthermore, chemical shift images revealed that while both compounds were visible in muscle tissue, only TFMB-3ET gave meaningful signal at the injection site in the brain. As brain tissue contains a higher concentration of membrane lipids compared to other tissues, more hydrophilic compounds maintain a higher degree of molecular mobility on comparison to more hydrophobic compounds. Therefore, based on these observations, it seems likely that increasing the hydrophilic character of neurological imaging agents of this type is beneficial in providing higher 19F signal intensity. However, the authors also pointed out that the continual increase of hydrophilic character of these compounds would eventually inhibit the ability to pass through the BBB. Therefore, it is clear that when designing a neurological 19F-MR imaging probe, each of these factors should be taken into consideration.
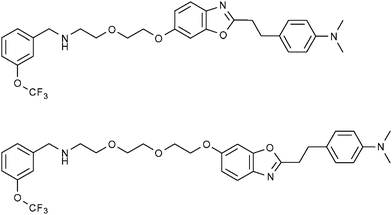 | ||
| Fig. 11 The fluorinated benzoxazole derivatives, TFMB-2Et (upper) and TFMB-3Et (lower) have been investigated by Amatsubo et al. as potential markers of amyloid plaques which are associated with Alzheimer's disease.62 | ||
Over the last decade, another class of compounds based on bis-styrylbenzene has also been shown to bind strongly to amyloid plaques, providing the opportunity to generate 19F-MRI contrast agents by incorporating 19F nuclei within the molecular framework of these compounds. In 2004, Sato et al. established that the mono-fluorinated amyloidophilic probe (E,E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB; Fig. 12) was capable of staining amyloid plaques and neurofibrillary tangles with high sensitivity and selectivity.63 The authors retained maximum variability by performing the low yielding fluorine labelling step at the first stage in the synthesis of this compound. Starting with the precursor 2,5-dimethylaniline, the amine was converted to a diazonium tetrafluoroborate salt that spontaneously decomposed yielding a crude fluorinated product which was then purified by column chromatography on SiO2 using hexane as the mobile phase (yield = 15%) prior to the remainder of the synthesis. The favourable binding properties of this compound made it an attractive multimodal (19F/1H MRI and fluorescence imaging) probe for the early stage detection and monitoring of Alzhemier's disease.
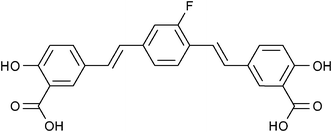 | ||
| Fig. 12 A fluorinated derivative of bis-styrylbenzene, FSB, has been shown to selectively target amyloid plaques.63 | ||
This potential was confirmed when Higuchi et al.64 demonstrated the ability to visualise brain plaques using FSB by 19F and 1H MRI in living mice (Fig. 13).
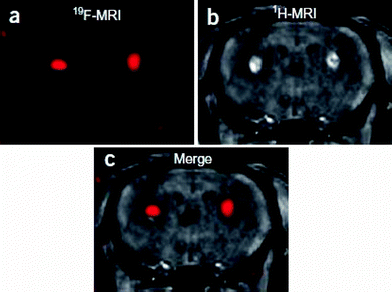 | ||
| Fig. 13 The fluorinated probe FSB was introduced by stereotaxic injection into the bilateral basal ganglia of 6-month-old wild-type mice. (a) 3D RARE coronal 19F MR image, (b) T1-weighted gradient-echo coronal 1H MR image, (c) merged 19F and 1H MR images. Reprinted by permission from Macmillan Publishers Ltd: Nature Neuroscience (ref. 64), copyright 2005. Only relevant frames a, b and c have been extracted from the original figure. | ||
More recently, Flaherty et al.65 reported a series of compounds (Fig. 14) based on the same bis-styrylbenzene framework which incorporated multiple symmetrically equivalent fluorine atoms in order to generate maximum 19F signal. These modifications were made either in the form of a tetrafluoro-substituted phenyl core or by incorporating trifluoromethyl or trifluoromethoxy substituents. Despite these modifications, the molecules were shown by a fluorescence-based assay to maintain high binding affinity and selectively to amyloid plaques. The molecules with a tetrafluorinated phenyl core exhibited the highest binding affinities overall. Furthermore, the neutral compounds revealed higher affinities compared to those with carboxylate functionalities. A selection of compounds (including 3 and 6) were shown to be able to cross the BBB. While MRI experiments on these compounds have not yet been reported, they show promise as imaging agents for Alzheimer's disease.
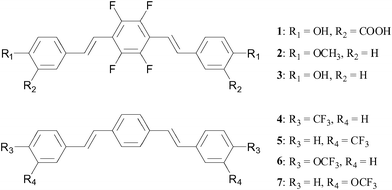 | ||
| Fig. 14 In an effort to maximise fluorine signal, Flaherty et al.65 have incorporated multiple fluorine nuclei within a series of bis-styrylbenzene derivatives. | ||
6.2 Tumours
In addition to the highlighted examples of ‘smart’ tumour-targeted imaging probes in the Section 4, there have also been other interesting examples of potential ‘theranostic’ (combined therapeutic/diagnostic) agents designed to target tumours. Hattori et al. investigated two such agents, 10Bpa(2,6F2) and 10Bpa(2,6F2)–ol (Fig. 15; left and right, respectively), which were designed both as 19F MR diagnostics and as potential therapeutics against cancer by utilising boron-neutron capture therapy (BNCT).66 These molecular frameworks were chosen in the knowledge that dipeptides containing 3-(4-fluorophenyl)alanine moieties are carried into certain types of tumour cellsvia the oligopeptide transporter. Previous experiments performed by this group indicated that 3-(pentafluorophenyl)alanine was detectable by 19F-MRI down to micromolar concentrations.67 Using both of the compounds shown in Fig. 15 and also the parent non-fluorinated compound 10Bpa, experiments which were designed to examine cellular uptake were performed on three types of cancer cell lines. Specifically, these cell lines were C6 (rat glioma), HeLa (human epithelioma) and KB (human squamous cell carcinoma). Following incubation with each of the compounds, the concentration of 10B was determined viainductively coupled plasma atomic emission spectroscopy (ICP-AES). The results showed that the concentrations of both 10Bpa(2,6F2) and 10Bpa(2,6F2)-ol in C6 cells were approximately half that of the non-fluorinated analogue, 10Bpa. However, in the two human cancer lines (HeLa and KB) the concentration of 10B was not considerably different between all three compounds tested. Further studies examining the cytotoxicity of these compounds revealed a lack of toxicity in the range 1–20 mM. These results indicate that both 10Bpa(2,6F2) and 10Bpa(2,6F2)-ol each show promise as 19F-MRI diagnostic agents as well as carriers of 10B for BNCT.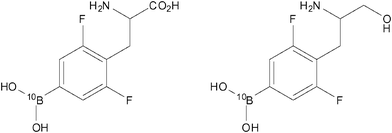 | ||
| Fig. 15 10Bpa(2,6F2) (left) and 10Bpa(2,6F2)-ol (right) have been designed to perform as both 19F MR contrast agents and potential therapeutics against cancer.66 | ||
In another interesting study, Brix et al. reported the synthesis of a fluorinated derivative (Fig. 16; 3-ABA-TFE) of 3-aminobenzamide (3-ABA) which is a known effective radiosensitiser and prevents DNA strand breaks from undergoing repair.68 The purpose of this investigation was to examine the biodistribution and pharmacokinetics of this compound in vivo using 19F-MRI. The 19F-labelling of this compound was accomplished via a highly efficient 2 step synthesis involving the trifluoroethylation of the starting material 3-nitrobenzoic acid chloride in dioxane to yield the corresponding N-alkylamide. The nitro group was then reduced to an amine either by RANEY®-nickel catalysis in the presence of hydrogen (70 bar) (86% yield) or by using SnCl2 dihydrate (97% yield). The authors demonstrated that fluorinating 3-ABA resulted in only marginally higher cytotoxicity relative to the parent compound. 3-ABA-TFE was injected into the intraperitoneal cavity of rats with prostate adenocarcinoma. Whole-body MR images were subsequently obtained at various time intervals and showed only a slight and heterogeneous signal arising from the adenocarcinoma. An optimal imaging time of approximately 45 h revealed the highest uptake of 3-ABA-TFE in liver, tumour and muscle tissue, yielding muscle-to-liver and tumour-to-liver signal ratios of 0.31 ± 0.07 and 0.11 ± 0.04, respectively. These observations suggest that, due to tumour-specific factors, the accumulation of 3-ABA-TFE in tumours may be impeded and in fact inferior to the uptake in healthy tissue. This demonstrates the requirement for developing superior systems to enable the delivery of compounds selectively to tumours.
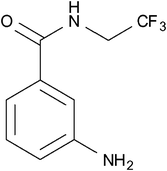 | ||
| Fig. 16 Brix et al. studied the biodistribution and pharmacokinetics of the fluorinated compound, 3- ABA-TFE.68 | ||
7. Summary and future perspectives
A recent surge in the number of reports based on fluorine-containing compounds intended as targets for 19F-MRI has served to highlight the enormous potential this technique has to offer for the visualisation of disease states.Of particular note has been the growing interest in the generation of nanoparticles and micellar assemblies with high fluorine content. These materials are demonstrating great promise for a variety of biomedical applications as they are capable of accommodating high concentrations of fluorine nuclei and also targeting specific biological sites and markers of disease.
The development of ‘smart’ probes which activate upon interaction with specific biological stimuli has been an area of intense multi-disciplinary investigation for several years. Whilst many 1H MRI and nuclear imaging (PET/SPECT) smart agents have been utilised as a means of investigating specific biochemical and metabolic processes, the relative sparsity of smart agents containing an 19F-MR probe indicates this is an open field for investigation. Some particularly innovative research has led to the development of multi-modal (19F-MR and fluorescence) smart agents containing 19F nuclei, which experience plural signal enhancement upon interaction with enzymes. It is easy to imagine that similar probes will be developed in the near future.
Whilst 19F-MRI presents many potential advantages over more conventional imaging modalities, current issues with sensitivity and specificity indicate a need to develop superior technology. Such improvements are necessary in order to support the next generation of 19F-MRI contrast agents that will surely come.
Acknowledgements
We thank the Welsh Assembly Government for funding this work. We also thank Dr Angelo J. Amoroso for his helpful insight and comments.References
- M. Bottrill, L. Kwok and N. J. Long, Chem. Soc. Rev., 2006, 35, 557–571 RSC.
- J. Klostergaard, K. Parga and R. G. Raptis, P. R. Health Sci. J., 2010, 29, 223–231 Search PubMed.
- A. M. Mohs and Z. R. Lu, Expert Opin. Drug Delivery, 2007, 4, 149–164 CrossRef CAS.
- S. Zhang, C. R. Malloy and A. D. Sherry, J. Am. Chem. Soc., 2005, 127, 17572–17573 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS.
- A. M. Morawski, P. M. Winter, X. Yu, R. W. Fuhrhop, M. J. Scott, F. Hockett, J. D. Robertson, P. J. Gaffney, G. M. Lanza and S. A. Wickline, Magn. Reson. Med., 2004, 52, 1255–1262 CrossRef CAS.
- D. G. Reid and P. S. Murphy, Drug Discovery Today, 2008, 13, 473 CrossRef CAS.
- P. C. Lauterbur, Nature, 1973, 242, 190–191 CrossRef CAS.
- G. N. Holland, P. A. Bottomley and W. S. Hinshaw, J. Magn. Reson., 1977, 28, 133–136 CAS.
- E. McFarland, J. A. Koutcher, B. R. Rosen, B. Teicher and T. J. Brady, J. Comput.-Assisted Tomogr., 1985, 9, 8–15 CrossRef CAS.
- D. Zhao, A. Constantinescu, L. Jiang, E. W. Hahn and R. P. Mason, Am. J. Clin. Oncol., 2001, 24, 462–466 CrossRef CAS.
- S. P. Robinson and J. R. Griffiths, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 987–996 CrossRef CAS.
- V. D. Mehta, P. V. Kulkarni, R. P. Mason, A. Constantinescu and P. P. Antich, Bioconjugate Chem., 1994, 5, 257–261 CrossRef CAS.
- P. Bachert, Prog. Nucl. Magn. Reson. Spectrosc., 1998, 33, 1–56 CrossRef CAS.
- M. Srinivas, A. Heerschap, E. T. Ahrens, C. G. Figdor and I. J. M. de Vries, Trends Biotechnol., 2010, 28, 363–370 CrossRef CAS.
- G. M. Lanza, X. Yu, P. M. Winter, D. R. Abendschein, K. K. Karukstis, M. J. Scott, L. K. Chinen, R. W. Fuhrhop, D. E. Scherrer and S. A. Wickline, Circulation, 2002, 106, 2842–2847 CrossRef CAS.
- J. Yu, V. D. Kodibagkar, W. Cui and R. P. Mason, Curr. Med. Chem., 2005, 12, 819–848 CrossRef CAS.
- J. Chen, G. M. Lanza and S. A. Wickline, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 431–440 CrossRef CAS.
- J. Ruiz-Cabello, B. P. Barnett, P. A. Bottomley and J. W. M. Bulte, NMR in Biomedicine, 2011, 24, 114–129 CrossRef CAS.
- W. J. M. Mulder, G. J. Strikers, G. A. F. van Tilborg, A. W. Griffioen and K. Nicolay, NMR Biomed., 2006, 19, 142–164 CrossRef CAS.
- J. P. André, É. Tóth, H. Fischer, A. Seelig, H. R. Mäcke and A. E. Merbach, Chem.–Eur. J., 1999, 5, 2977–2983 CrossRef.
- M. Grogna, R. Cloots, A. Luxen, C. Jérôme, C. Passirani, N. Lautram, J.-F. Desreux and C. Detrembleur, Polym. Chem., 2010, 1, 1485–1490 RSC.
- G. Zhang, R. Zhang, X. Wen, L. Li and C. Li, Biomacromolecules, 2008, 9, 36–42 CrossRef CAS.
- H. Peng, I. Blakey, B. Dargaville, F. Rasoul, S. Rose and A. K. Whittaker, Biomacromolecules, 2009, 10, 374–381 CrossRef CAS.
- W. Du, A. M. Nystrom, L. Zhang, K. T. Powell, Y. Li, C. Cheng, S. A. Wickline and K. L. Wooley, Biomacromolecules, 2008, 9, 2826–2833 CrossRef CAS.
- Z.-X. Jiang, X. Liu, E.-K. Jeong and Y. B. Yu, Angew. Chem., Int. Ed., 2009, 48, 4755–4758 CrossRef CAS.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S.-F. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS.
- B. C. M. te Boekhorst, S. M. Bovens, J. Rodrigues-Feo, H. M. H. F. Sanders, C. W. A. van de Kolk, A. I. P. M. de Kroon, M.-J. M. Cramer, P. A. F. M. Doevendans, M. ten Hove, G. Pasterkamp and C. J. A. van Echteld, Mol. Imaging Biol., 2010, 12, 635–651 CrossRef.
- G. A. F. van Tilborg, E. Vucic, G. J. Strijkers, D. P. Cormode, V. Mani, T. Skajaa, C. P. M. Reutelingsperger, Z. A. Fayad, W. J. M. Mulder and K. Nicolay, Bioconjugate Chem., 2010, 21, 1794–1803 CrossRef CAS.
- C. W. Kessinger, C. Khemtong, O. Togao, M. Takahashi, B. D. Sumer and J. Gao, Exp. Biol. Med., 2010, 235, 957–965 CrossRef CAS.
- S. Svenson and D. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129 CrossRef CAS.
- R. Esfand and D. Tomalia, Drug Discovery Today, 2001, 6, 427–436 CrossRef CAS.
- O. A. Matthews, A. N. Shipway and J. F. Stoddart, Prog. Polym. Sci., 1998, 23, 1–56 CrossRef CAS.
- J. M. Oliveira, A. J. Salgado, N. Sousa and J. F. Mano, Prog. Polym. Sci., 2010, 35, 1163–1194 CrossRef CAS.
- F. Vögtle, S. Gestermann, R. Hesse, H. Schwierz and B. Windisch, Prog. Polym. Sci., 2000, 25, 987–1041 CrossRef.
- K. Inoue, Prog. Polym. Sci., 2000, 25, 453–571 CrossRef CAS.
- M. Longmire, P. L. Choyke and H. Kobayashi, Curr. Top. Med. Chem., 2008, 8, 1180–1186 CrossRef CAS.
- S. Langereis, Q. G. d. Lussanet, M. H. P. van Genderen, E. W. Meijer, R. G. H. Beets-Tan, A. W. Griffioen, J. M. A. van Engelshoven and W. H. Backes, NMR Biomed., 2006, 19, 133–141 CrossRef CAS.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS.
- W. T. Chen, D. Thirumalai, T. T. Shih, R. C. Chen, S. Y. Tu, C. I. Lin and P. C. Yang, Mol. Imaging Biol., 2010, 12, 145 CrossRef.
- R. S. Navath, A. R. Menjoge, B. Wang, R. Romero, S. Kannan and R. M. Kannan, Biomacromolecules, 2010, 11, 1544–1563 CrossRef CAS.
- A. Quintana, E. Raczka, L. Piehler, I. Lee, A. Myc, I. Majoros, A. Patri, T. Thomas, J. Mulé and J. Baker, Pharm. Res., 2002, 19, 1310–1316 CrossRef CAS.
- K. J. Thurect, I. Blakey, H. Peng, O. Squires, S. Hsu, C. Alexander and A. K. Whittaker, J. Am. Chem. Soc., 2010, 132, 5336–5337 CrossRef.
- J. M. Criscione, B. L. Le, E. Stern, M. Brennan, C. Rahner, X. Papademetris and T. M. Fahmy, Biomaterials, 2009, 30, 3946–3955 CrossRef CAS.
- M. Ogawa, S. Nitahara, H. Aoki, S. Ito, M. Narazaki and T. Matsuda, Macromol. Chem. Phys., 2010, 211, 1369–1376 CrossRef CAS.
- M. Ogawa, S. Nitahara, H. Aoki, S. Ito, M. Narazaki and T. Matsuda, Macromol. Chem. Phys., 2010, 211, 1602–1609 CrossRef CAS.
- M. Oishi, S. Sumitani, T. K. Bronich, A. V. Kabanov, M. D. Boska and Y. Nagasaki, Chem. Lett., 2009, 38, 128–129 CrossRef CAS.
- M. Oishi, S. Sumitani and Y. Nagasaki, Bioconjugate Chem., 2007, 18, 1379–1382 CrossRef CAS.
- O. Warburg, Science, 1956, 123, 309–314 CAS.
- J.-w. Kim and C. V. Dang, Cancer Res., 2006, 66, 8927–8930 CrossRef CAS.
- R. A. Moats, S. E. Fraser and T. J. Meade, Angew. Chem., Int. Ed. Engl., 1997, 36, 726–728 CrossRef CAS.
- S. Mizukami, R. Takikawa, F. Sugihara, Y. Hori, H. Tochio, M. Walchli, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2008, 130, 794–795 CrossRef CAS.
- S. Mizukami, R. Takikawa, F. Sugihara, M. Shirakawa and K. Kikuchi, Angew. Chem., Int. Ed., 2009, 48, 3641–3643 CrossRef CAS.
- P. K. Senanayake, A. M. Kenwright, D. Parker and S. K. van der Hoom, Chem. Commun., 2007, 2923–2925 RSC.
- A. M. Kenwright, I. Kuprov, E. De Luca, D. Parker, S. U. Pandya, P. K. Senanayake and D. G. Smith, Chem. Commun., 2008, 2514–2516 RSC.
- K. H. Chalmers, M. Botta and D. Parker, Dalton Trans., 2011, 40, 904–913 RSC.
- K. H. Chalmers, E. De Luca, N. H. M. Hogg, A. M. Kenwright, I. Kuprov, D. Parker, M. Botta, J. I. Wilson and A. M. Blamire, Chem.–Eur. J., 2011, 16, 134–148 CrossRef.
- K. H. Chalmers, A. M. Kenwright, D. Parker and A. M. Blamire, Magn. Reson. Med., 2011 Search PubMed in press.
- A. M. Neubauer, J. Myerson, S. D. Caruthers, F. D. Hockett, P. M. Winter, J. Chen, P. J. Gaffney, J. D. Robertson, G. M. Lanza and S. A. Wickline, Magn. Reson. Med., 2008, 60, 1066–1072 CrossRef CAS.
- Z.-X. Jiang, Y. Feng and Y. B. Yu, Chem. Commun., 2011, 47, 7233–7235 RSC.
- United States Pat., 2011/0217241, 2011 Search PubMed.
- T. Amatsubo, S. Morikawa, T. Inubushi, M. Urushitani, H. Taguchi, N. Shirai, K. Hirao, M. Kato, K. Morino, H. Kimura, I. Nakano, C. Yoshida, T. Okada, M. Sano and I. Tooyama, Neurosci. Res., 2009, 63, 76–81 CrossRef CAS.
- K. Sato, M. Higuchi, N. Iwata, T. Saido and K. Sasamoto, Eur. J. Med. Chem., 2004, 39, 573–578 CrossRef CAS.
- M. Higuchi, N. Iwata, Y. Matsuba, K. Sato, K. Sasamoto and T. Saido, Nat. Neurosci., 2005, 8, 527–533 CrossRef CAS.
- D. P. Flaherty, S. M. Walsh, T. Kiyota, Y. Dong, T. Ikezu and J. L. Vennerstrom, J. Med. Chem., 2007, 50, 4986–4992 CrossRef CAS.
- Y. Hattori, T. Asano, Y. Niki, H. Kondoh, M. Kirihata, Y. Yamaguchi and T. Wakamiya, Bioorg. Med. Chem., 2006, 14, 3258–3262 CrossRef CAS.
- Y. Hattori, Y. Yamaguchi, H. Yamamoto, T. Asano, M. Kirihata, M. Takagaki and T. Wakamiya, Peptide Sci., 2004, 2005, 411 Search PubMed.
- G. Brix, A. Schlicker, W. Mier, P. Peschke and M. E. Bellemann, Magn. Reson. Imaging, 2005, 23, 967–976 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |