Dialkylbiaryl phosphines in Pd-catalyzed amination: a user's guide
David S.
Surry
and
Stephen L.
Buchwald
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: sbuchwal@mit.edu; Fax: +1-617-253-3297; Tel: +1-617-253-1885
First published on 9th September 2010
Abstract
Dialkylbiaryl phosphines are a valuable class of ligand for Pd-catalyzed amination reactions and have been applied in a range of contexts. This perspective attempts to aid the reader in the selection of the best choice of reaction conditions and ligand of this class for the most commonly encountered and practically important substrate combinations.
 David S. Surry | David S. Surry was born in London and carried out his undergraduate studies at the University of Cambridge, UK. During this time he performed research in the laboratories of Prof. I. Fleming. He then stayed on at Cambridge to complete a PhD with Dr D. R. Spring and spent a period at GlaxoSmithKline, UK. He is currently conducting postdoctoral research at the Massachusetts Institute of Technology with Prof. S. L. Buchwald. |
 Stephen L. Buchwald | Stephen L. Buchwald is the Camille Dreyfus Professor of Chemistry at MIT where he has been on the faculty since 1984. He has received numerous honors, the latest being the Gustavus J. Esselen Award for Chemistry in the Public Interest. During the period January 1999–June 2009, he was the most cited chemist in the world (per paper). He lives in Newton, Massachusetts with his wife, Susan Haber, and their two children, Sara (age 10) and Nathan (age 13), along with their three cats, Trixie, Rocket and the famous orange tabby, Rufus. |
1. Introduction
Palladium-catalyzed amination of aryl, vinyl and heteroaryl halides and pseudohalides has rapidly emerged as a valuable tool in the synthesis of pharmaceuticals, natural products and novel materials.1–4 The development of Pd-catalyzed C–N coupling has significantly contributed to the streamlining of the synthesis of small molecule pharmaceutical agents, allowing more efficient syntheses and facilitating a modular approach to analogue synthesis.5–7 The significance of this methodology in this regard stems from the prevalence of aromatic amines in biologically active molecules;8 important classes include kinase inhibitors,9,10antibiotics11,12 and CNS active agents.13Breakthroughs in this area have typically been driven by the implementation of new classes of ligands. Notable examples include chelating diphenylphosphino ligands such as BINAP,14,15dppf16 and Xantphos,17 more electron-rich chelating phosphines such as Josiphos,18N-heterocyclic carbenes19 and trialkylphosphines20,21 that have served to continually increase the substrate scope and to render the reactions more efficient.22,23 Despite the plethora of systems currently available for Pd-catalyzed C–N coupling, only a relatively limited group has seen extensive practical application. This reflects on a combination of the ease of use of a catalyst system, its robustness, availability of ligands and substrate scope. Catalysts based on dialkylbiaryl phosphines compare favorably with other systems in this regard and have been extensively applied in the synthesis of biologically active molecules.4 These ligands were first described by Buchwald for Pd-catalyzed cross-coupling in 1998.24 Since then further work25–37 has led to the development of a versatile family of structurally related ligands (Section 2.1) that have been shown to generate highly active catalysts for a range of reactions, notably Pd-catalyzed amination4 and etherification of aryl halides,38,39arylation of enolates,40 and Suzuki–Miyaura cross-coupling.41,42 A key advantage of these ligands over some others is that the reactions can typically be performed without a dry-box in standard laboratory glassware.
Progress in this field has been brisk and reactions with these ligand systems can now be applied to a diverse array of substrates. The optimal ligand and other reaction parameters (such as Pd source, base, solvent and temperature) can vary for different substrate combinations. Part of the reason for this disparity stems from the wide variation in the electronic and steric properties of the nitrogen-based nucleophiles when compared to other cross-coupling processes such as the Suzuki–Miyaura reaction. The amine and amides can differ in nucleophilicity and pKa which means that the rate determining step of the catalytic cycle can vary with substrate,43 contributing to the difficulty in selecting the best conditions. It is the goal of this perspective to provide a practical guide to the use of these catalysts that will enable the practitioner to more easily select the most efficacious conditions for a given substrate combination. As a result, mechanistic details will only be discussed where they directly impinge on the choice of reaction conditions. A generalized catalytic cycle is illustrated in order to aid later discussion (Scheme 1).
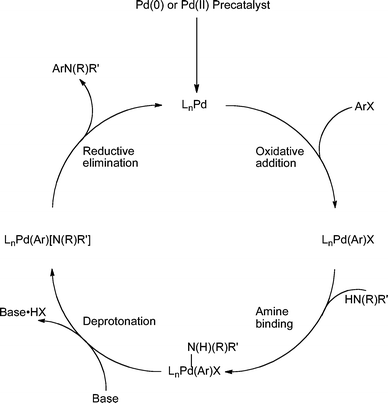 | ||
| Scheme 1 Generalized catalytic cycle for Pd-catalyzed amination with dialkylbiaryl phosphines. | ||
There are a number of other thorough reviews on Pd-catalyzed C–N bond formation that the reader is guided to for descriptions of the advancement of the field and applications,22,23,44–57 for mechanistic aspects58–62 or applications on the process development scale.2,63,64 There are also reviews specifically focused on applications of dialkylbiaryl phosphine ligands in Pd-catalyzed amination and Suzuki cross-coupling reactions.4,41
2. Key variables
The key reaction parameters of ligand, Pd source, base and solvent are discussed in detail below (Scheme 2). The selection is typically determined by both the structure of the amine and electrophile (see Section 3).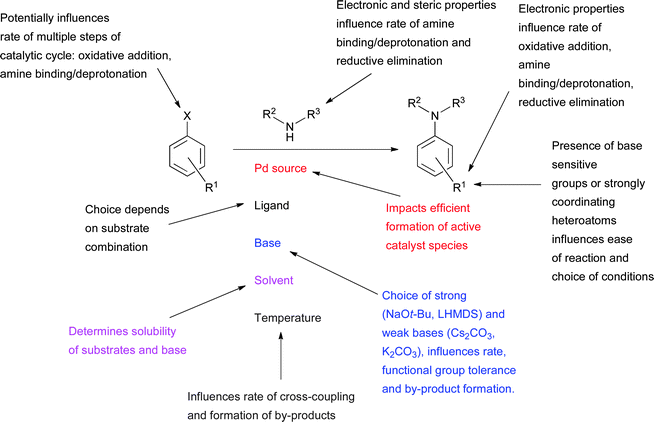 | ||
| Scheme 2 Factors influencing the outcome of a Pd-catalyzed amination reaction. | ||
It is worth noting that there is often an interaction between these variables, for example, the best base for a reaction may change depending on the solvent employed. Therefore the optimization of reaction conditions can often be most efficiently achieved by a statistical DOE (Design of Experiment) approach.65,66 Some of the considerations in the optimization of Pd-catalyzed amination reactions with dialkylbiaryl phosphine ligands may also apply to other ligand systems, however, the reader should be aware that many observations will not be directly transferable.
2.1 Ligand
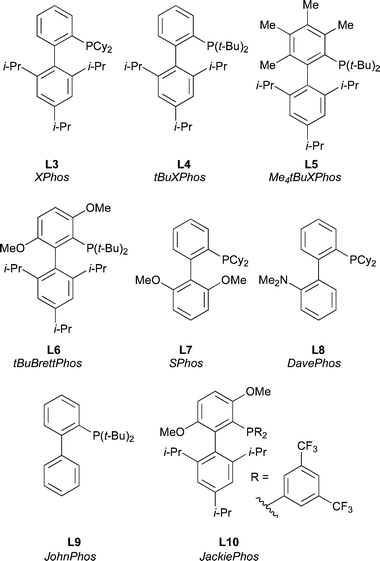
One of the most important determinants of the success of a given amination reaction with dialkylbiaryl phosphine-based catalysts is the structure of the ligand. These ligands can typically be made in a one step procedure via addition of an aryl lithium or Grignard reagent to an appropriate aryne followed by quenching with a chlorophosphine.67,68 As a result of the modularity afforded by this synthetic route numerous derivatives have been described and this has allowed fine-tuning of ligand structure for each application (Fig. 1).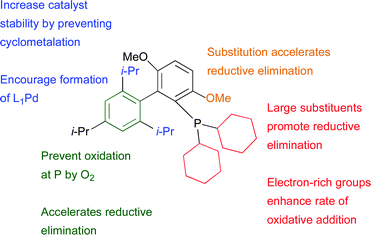 | ||
| Fig. 1 Important structural features of dialkylbiaryl phosphine ligands. | ||
These ligands are air stable,69 easily handled crystalline solids and a number are now commercially available. The original studies made use of DavePhos (L8) and JohnPhos (L9) for amination reactions24,70 and these ligands have found on-going application in the synthesis of natural products5,71–74 and pharmaceuticals,75–79 including examples in process development.80,81 Subsequently, several structural variants have been reported.82–88 A major breakthrough came with the discovery of XPhos (L3)83 and RuPhos (L2),84 which supply improved reactivity in a diversity of amination reactions. More recently, the ligand BrettPhos (L1)85 was disclosed which confers the most active dialkylbiaryl-based phosphine system for the selective arylation of 1° amines. To date, the two most generally useful of these ligands for amination are L185,89 for the arylation of 1° amines and L2 for 2° amines (Fig. 2).89,90
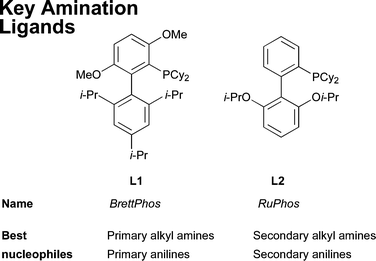
A number of other ligands of this class are also commercially available and are valuable in Pd-catalyzed amination; their particular applications are discussed below (Fig. 3). It should be noted that several closely related ligands were later developed by others and these can also provide active catalysts for Pd-catalyzed amination.26,28,31,33,34,91
The recent realization of the high reactivity of monoligated Pd(0) complexes towards oxidative addition has been decisive in the utilization of aryl chlorides in cross-coupling reactions.92 This might lead the reader to the conclusion that the ideal ratio of metal to ligand in these reactions is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. For simple substrates this may be true, but an extra equivalent of ligand relative to Pd is often needed in order to stabilize the catalyst in difficult cases in which long reaction times are required or when a high TON at the metal center is desired.90,93 This tactic is effective when using dialkylbiaryl phosphine ligands because in the presence of an extra equivalent of ligand the L1Pd complex can still be the dominant species in solution.94 It is also worth mentioning that an extra equivalent of ligand could be necessary for catalyst activation. This will be discussed in more detail in the following section.
:1. For simple substrates this may be true, but an extra equivalent of ligand relative to Pd is often needed in order to stabilize the catalyst in difficult cases in which long reaction times are required or when a high TON at the metal center is desired.90,93 This tactic is effective when using dialkylbiaryl phosphine ligands because in the presence of an extra equivalent of ligand the L1Pd complex can still be the dominant species in solution.94 It is also worth mentioning that an extra equivalent of ligand could be necessary for catalyst activation. This will be discussed in more detail in the following section.
2.2 Catalyst activation
The efficiency with which the catalytically active, monoligated Pd(0) complex is formed before entry into the catalytic cycle is a key factor in the selection of reaction conditions for Pd-catalyzed amination reactions. If a Pd(II) salt such as Pd(OAc)2 is used, reduction of Pd(II) to Pd(0) must occur before the cross-coupling reaction can take place. This can be brought about by the amine component in an amination reaction if the amine possesses hydrogen atoms α to the nitrogen atom as this enables the Pd(II) amine complex to undergo β-hydride elimination.95 The efficiency of this process varies between amines and it may take a significant amount of time for all of the Pd(OAc)2 to be reduced and enter into the catalytic cycle.96,97 Furthermore, for the arylation of nucleophiles lacking a β-hydrogen such as 1° amides, 1° anilines or ammonia, another reductant must be present. Pd(OAc)2, however, remains attractive on an industrial scale, although its source and morphology can have a strong impact on reactivity.98 In some instances, reduction of Pd(II) to Pd(0) has been expedited by the addition of a tertiary amine (typically NEt3).32,99 Inclusion of phenylboronic acid as a reducing agent for Pd in the reaction mixture has been beneficial in some contexts.83,100 This method is not always effective, however, perhaps because when the boronic acid is present stoichiometrically with respect to Pd the concentration of the boronic acid in the reaction medium is low which may have a deleterious effect on the efficacy and rate of the reduction step.The phosphine ligand may also affect the reduction of Pd(II), however this process is likely slow with bulky, electron-rich dialkylbiaryl phosphines.101 In order to address this issue and to provide a convenient means of ensuring efficient reduction of Pd(II) a protocol has been developed whereby water mediates reduction of Pd(OAc)2 by the phosphine.102 This procedure rapidly produces a highly active Pd catalyst that gives superior results to Pd2(dba)3, Pd(OAc)2/PhB(OH)2 or [(allyl)PdCl]2 for the arylation of both amides103 and anilines under the conditions examined. Using this method a number of demanding transformations can be efficiently accomplished, including the arylation of anilines with aryl chlorides at low catalyst loadings and the arylation of electron-deficient anilines in the presence of the weak base K2CO3 (Scheme 3).
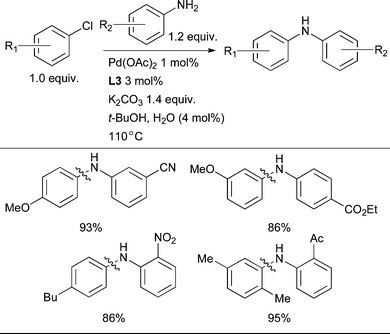 | ||
| Scheme 3 Water-mediated reduction of Pd(II) salts permits efficient amination of electron-deficient anilines. | ||
An active catalyst can also be produced using [(allyl)PdCl]2; here the catalytically active Pd(0) is generated by the attack of a nucleophile upon the allyl group.19 Unfortunately this complex is applicable only in a limited range of amination reactions with dialkylbiaryl phosphines.33,88,104
The need for a reduction step to form Pd(0) can be avoided by the use of a stable Pd(0) complex as the Pd source. In conjunction with dialkylbiaryl phosphines the air-stable complexes Pd2(dba)3 or Pd(dba)2 are suitable in a variety of situations.82–84,87,105–109 Coordination of dba to the metal can, however, attenuate the activity of the Pd catalyst.110,111 Preheating Pd2(dba)3, ligand and base in solvent prior to the introduction of substrates can have beneficial effects on reproducibility,81,112,113 perhaps highlighting the importance of efficient formation of the L1Pd complex.
As a result of the issues faced by these various Pd sources, efforts were made to devise a stable precatalyst19,114,115 containing the dialkylbiaryl phosphine ligand that would give direct access to a Pd complex within the catalytic cycle. Such a complex would also potentially be advantageous in providing a single component source of both Pd and ligand. Earlier efforts attempted to address this goal still required an exogenous reductant be present.83,99 The intramolecularly coordinated amine complexes 1–5 have proven to be an ideal Pd source that can form the catalytically active L1Pd complex under the reaction conditions simply by mixing with the reagents (i.e., no need to add a reducing agent) and are air and moisture stable.93 These precatalysts can be made with a variety of biaryl phosphine ligands in three high yielding steps. Precatalysts with the ligands BrettPhos 1, XPhos 2, SPhos 3, RuPhos 4 and tBuXPhos 5 are commercially available (Scheme 4).
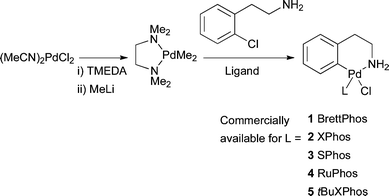 | ||
| Scheme 4 Synthesis of amine bound oxidative addition precatalysts. | ||
Under the reaction conditions the amine nitrogen is deprotonated and the resulting Pd amido undergoes rapid reductive elimination to generate L1Pd and indoline (this small amount of indoline is readily removed during work-up and purification at the end of the reaction) (Scheme 5).
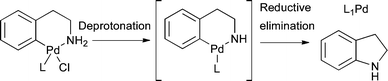 | ||
| Scheme 5 Activation of intramolecularly coordinated amine oxidative addition precatalyst. | ||
The active catalyst is fully generated in around 3 min at room temperature in the presence of a strong base such as NaOt-Bu. Activation can also be achieved with a weak base such as K2CO3 at 80 °C. These air-stable precatalysts provide extremely active catalysts in a variety of amination reactions, notably allowing the arylation of anilines with aryl chlorides to be performed with both low catalyst loadings and short reaction times (Scheme 6).89,90,116,117
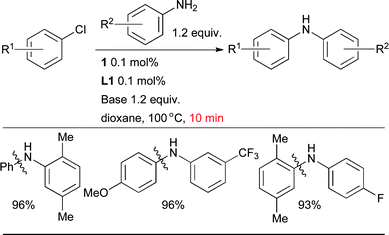 | ||
| Scheme 6 The application of precatalyst 1 allows arylation of anilines with low catalyst loading and short reaction times. | ||
The efficiency of these precatalysts is evidenced by the mild conditions under which these reactions can be performed. Indeed the oxidative addition of Pd(0) to an aryl chloride occurs at −40 °C with these precatalysts, illustrating the virtue of not having inhibitory additives such as dba present in the reaction mixture.93
In summary, Pd(OAc)2 and Pd2(dba)3 have been the most commonly utilized sources of Pd in amination reactions, however, the precatalysts 1–5 present a number of advantages (Scheme 7), ensuring that the active catalyst is formed. Many of these benefits are particularly felt in the case of difficult substrates or when low catalyst loadings or short reaction times are needed.
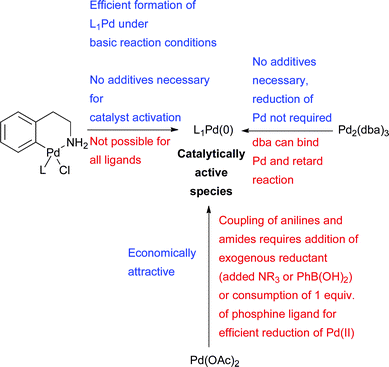 | ||
| Scheme 7 Considerations for choice of Pd source for amination reactions. | ||
2.3 Solvent
Pd-catalyzed amination reactions with dialkylbiaryl phosphine ligands can be performed in a wide variety of solvents. Toluene and 1,4-dioxane are most commonly employed, although 1,4-dioxane has an unfavorable toxicity profile and can typically be replaced with Bu2O.85 Other ethereal solvents including THF,106,1182-MeTHF81 and DME24 can also be used. Toluene is particularly advantageous in the coupling of aryl iodides due its weak ability to solubilize the inorganic iodide salts formed during the course of the reaction (Section 3.1).90t-BuOH is an appropriate solvent in numerous instances and when a higher reaction temperature is needed t-AmOH may be substituted.98 These solvents have the beneficial property of aiding in the solubilization of inorganic bases, such as K3PO4, K2CO3 or KOH,83 leading to rate enhancement of the desired cross-coupling reactions. Furthermore, mixing t-BuOH with less polar solvents such as toluene can have a significant accelerating effect on amination reactions.83 These solvents can also be effective in the dissolution of polar substrates. Similarly, polar, aprotic solvents such as DMSO, DMF and DMA have been successfully used in some examples,75,119–122 both alone and in mixtures with other solvents.In some instances Pd-catalyzed amination reactions are faster in DMF or DMA than in less polar solvents with K3PO4 as base,123 however other studies have indicated lower yields124 and increases in side reactions such as aryl halide reduction.125 These polar solvents have also been found to be advantageous for amination reactions with these ligands in conjunction with microwave heating.126,127
Water can be an attractive reaction medium for performing cross-coupling reactions128,129 due to its lack of toxicity or flammability. In certain cases Pd-catalyzed amination can be brought about in water with unmodified ligands such as L3.32,83 If water immiscible, non-polar substrates are used the active metal catalyst presumably dissolves in micelles of the substrate. Hydrophilic derivatives of dialkylbiaryl phosphines with improved water solubility130 have also been prepared,131–133 however, they have not yet been applied in Pd-catalyzed amination reactions. Biphasic reactions with water and a non-polar solvent such as toluene can result in enhanced functional group tolerance of bases such as KOH compared to monophasic systems.134–136
Another solvent worthy of particular mention is α,α,α-trifluorotoluene.137 Studies of Pd-catalyzed amination with dialkylbiaryl phosphine ligands in this medium or mixtures have shown it to possess certain advantages over toluene, including reduced foaming in biphasic reactions135 and better heating under microwave irradiation.138,139 (For more specific recommendations of the choice of solvent, see Fig. 6–8).
2.4 Base
The choice of base has a large bearing on the functional groups that may be present in amination substrates and thus has been the subject of considerable interest, including detailed mechanistic studies with some ligand systems.140 Unfortunately, it is not possible to select the base purely on the grounds of the pKa of the free N nucleophile as the pKa is changed significantly by binding to Pd, which typically occurs before deprotonation (Scheme 1). Furthermore, inorganic bases such as Cs2CO3 can act heterogeneously in non-polar solvents where they can behave as much stronger bases than might be predicted from their solution phase pKa value.141Early studies of the Pd-catalyzed coupling of amines with aryl halides utilized NaOt-Bu as base in toluene.142 This remains the most versatile base for Pd-catalyzed amination reactions with dialkylbiaryl phosphine ligands, often giving the highest reaction rates and enabling the lowest catalyst loadings. Unfortunately, because NaOt-Bu is a relatively strong base (pKa = 17.0) it can participate in undesirable side reactions with various electrophilic functional groups and some aromatic heterocycles and cause epimerization at acidic centers. KOt-Bu has also seen some application77 but suffers from similar limitations and is generally less satisfactory. These observations have prompted the search for alternative bases. NaOMe is somewhat less basic (pKa = 15.5) and can give better functional group tolerance than NaOt-Bu.99,143 It should be noted that studies with other ligand systems for Pd-catalyzed amination have revealed NaOPh to be a useful base for the arylation of heteroaryl amines, perhaps as a result of its good solubility in dioxane.144
LHMDS is another valuable strong base in Pd-catalyzed amination145 with dialkylbiaryl phosphine ligands. In particular, LHMDS allows amination of aryl halides to be performed in the presence of protic functional groups such as phenols, aliphatic alcohols and amides (Scheme 8).86,89,106
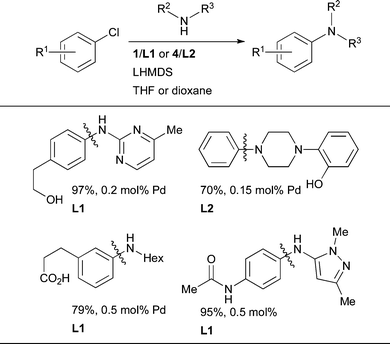 | ||
| Scheme 8 Use of LHMDS as base permits the cross-coupling of substrates bearing protic functional groups. | ||
This base is especially convenient because of the commercial availability of solutions in both toluene and THF, removing the necessity of storage and handling of the hygroscopic solid base. LHMDS also allows the amination of haloheterocycles possessing a free NH group.84,86,89
Hydroxide bases such as KOH or NaOH98 are attractive on scale from an economic standpoint and these can be used in conjunction with dialkylbiaryl phosphines in Pd-catalyzed amination reactions, although they generally give slower reactions than alkoxides.83,99 If powdered, hydroxide bases can be employed in toluene,98 although aqueous phase reactions of water immiscible substrates have also been demonstrated without the need for a phase transfer catalyst.83,146 It has been noted that the absence of a phase transfer catalyst can result in improved functional group tolerance.136Ba(OH)2 can also mediate amination under very mild, biphasic conditions, rendering it applicable to substrates which are prone to base-mediated epimerization.135
Weak inorganic bases such as Cs2CO3, K3PO4 or K2CO3 in place of alkoxides can bring significant benefits in the functional group tolerance of Pd-catalyzed amination reactions.147 These bases provide more general conditions for substrates containing electrophilic functional groups such as ketones, esters and nitro aromatics.147 Weak bases also facilitate the exploitation of aryl sulfonates as electrophiles, allowing these substrates to be cross-coupled without the requirement for slow addition of the electrophile.148 They have also been instrumental in allowing the arylation of amides (Section 3.3). In non-polar solvents such as toluene, in which these inorganic bases have very low solubility, the deprotonation of the Pd-bound amine is thought to occur at the solid–liquid boundary.149 In this situation, the particle size and shape of the inorganic base can be important in determining the rate and ultimate success of a cross-coupling reaction.150,151 This topic has seen the most detailed discussion by Maes in the use of Cs2CO3 in Pd-catalyzed aminations with BINAP as ligand,149 although the findings are almost certainly applicable to biaryl phosphine ligands. Cs2CO3 from different suppliers exhibited varying reactivity and SEM imaging of the base showed a correlation between the particle properties and the activity of the base. In point of fact, pregrinding the Cs2CO3 or K3PO4 before application in amination reactions has sometimes been recommended.112,113,149 Such particle size effects have also been invoked in a recent study at Merck to explain the difference in outcome between reactions carried out with a magnetic stir bar and with an overhead stirrer.81 Reactions performed with the overhead stirrer took up to five times longer to reach completion, presumably because the magnetic stirrer facilitated the reaction by grinding the base in situ. Studies at Pfizer have revealed that the rate of agitation can severely impact the rate of these amination reactions as the high density of Cs2CO3 can lead it to sink to the bottom of the reaction vessel.100 Scientists at LEO Pharma have found that inclusion of Celite in a reaction with Cs2CO3 as base in a Pd-catalyzed amination with L3 as ligand had a significant beneficial effect on the yield; it was hypothesized that this effect results from the Celite preventing clumping of the Cs2CO3.152 Aggregation effects may also be responsible for the large excess of base that some authors have shown to be necessary in certain amination reactions.153
On the other hand, in some reactions with weak inorganic bases it is the solubilized base that is important in bringing about the reaction,154 as evidenced by the rate accelerations that have been observed with more polar solvents such as DMA,123 the presence of water155,156 or additives such as 18-crown-6157 when used in conjunction with these weaker inorganic bases. Furthermore, it frequently turns out that Cs2CO3 is a more efficient base for amination reactions than K2CO3, which may relate to its greater solubility in organic solvents.158
Typically, alkali metal bases are moderately hygroscopic but they can usually be weighed into the reaction mixture in air without special precautions. If a substrate is moisture sensitive, however, (for example a hydrolyzable electrophile such as an aryl triflate) rigorous drying of the base159 and/or the addition of molecular sieves87,88 may be desirable. On the other hand, in some situations a beneficial effect of added water has been noted,155,156 perhaps as a result of improved solubilization of the base or by aiding in catalyst activation.102
A potential alternative solution to improving the functional group tolerance is provided by soluble organic bases, such as DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) or MTBD (7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene). These bases are, however, relatively expensive and have so far only proven effective in the amination of aryl nonaflates with anilines under microwave irradiation.160
The advantages and disadvantages of the most commonly used bases are summarized in Fig. 4.
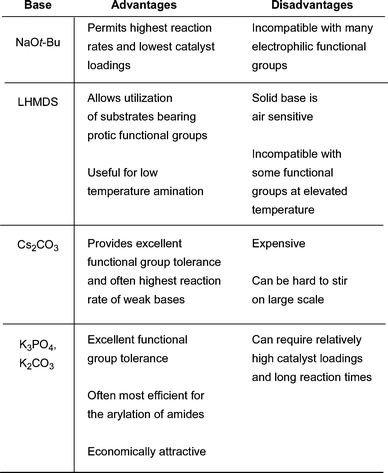 | ||
| Fig. 4 Comparison of bases typically used in Pd-catalyzed amination. | ||
3. Substrate
The structures of both the nucleophile and electrophile weigh substantially on the choice of reaction conditions. Aryl chlorides, bromides, iodides and sulfonates have distinct properties with respect to ease of oxidative addition. Furthermore, the halide or pseudohalide anion produced during the course of the reaction can also be significant.161–163 The presence of electron-donating or -withdrawing substituents on the aromatic ring or of heteroatoms within the ring also impinges on the rate of all steps of the catalytic cycle. The presence of substituents ortho to the aryl halide can be critical in determining the rate of reaction; such substitution can facilitate some steps of the catalytic cycle (for example, reductive elimination), while potentially retarding others (for example, oxidative addition).Similarly, N nucleophiles (e.g., aliphatic amines, anilines, amides, NH heterocycles) can possess widely differing nucleophilicity and pKa values as well as variable steric properties. These can also affect the rates of various steps on the catalytic cycle such as amine binding,43 deprotonation and reductive elimination, therefore necessitating different ligands or reaction conditions. Hence the origin of the variation in optimal reaction conditions for different substrate combinations.
3.1 Electrophile
Initial studies on Pd-catalyzed aminations were carried out predominantly with aryl bromides as electrophiles. Aryl chlorides, however, are typically more attractive substrates due to their lower cost and wider availability.164,165 The discovery of dialkylbiaryl phosphine ligands, among others, allowed these substrates to be engaged in amination reactions under milder conditions than had previously been reported.20,166–168 The implementation of L8 as ligand permitted deactivated aryl chlorides to be used24 (this may now be effected with a variety of ligands).21–23,26 With more modern ligand systems oxidative addition of L1Pd(0) is facile at or below room temperature (Scheme 9).93 The amination of aryl chlorides is now a routine procedure and in some cases aryl chlorides give more efficient reactions than aryl bromides.109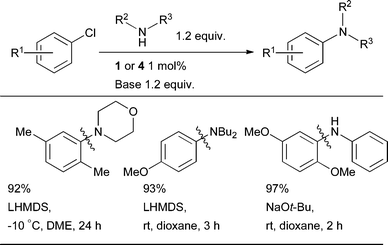 | ||
| Scheme 9 Amination of aryl chlorides at or below room temperature. | ||
Catalyst systems based on L1 and L2 allow the amination of electron-poor, -rich and -neutral aryl bromides and chlorides both with and without ortho-substitution, to be achieved with high efficiency and low catalyst loadings for a broad range of 1° and 2° amines. For unfunctionalized substrates this is best brought about by using NaOt-Bu in combination with an ethereal solvent or toluene. For substrates bearing protic functional groups the combination of LHMDS and THF or dioxane is most useful. Weak bases can also give good results; a typical starting point for optimization is K2CO3/t-BuOH.
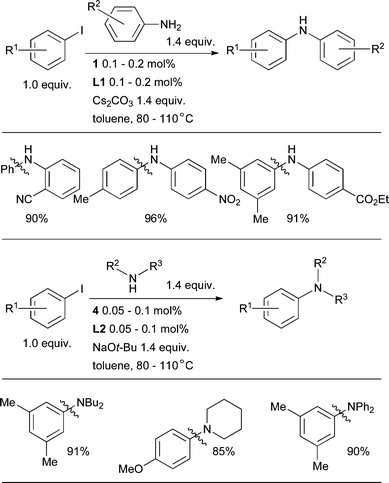
Intriguingly, aryl iodides, which are typically the easiest class of electrophile for C–C cross-coupling reactions, are relatively challenging substrates in Pd-catalyzed amination reactions.18,169 Mechanistic studies have suggested that this results from the formation of unreactive Pd dimers bridged by iodide anions.170L1, however, which produces monomeric oxidative addition complexes in solution, allows aryl and heteroaryl iodides to be efficiently aminated with 1° amines (and L2 may be used for 2° amines) (Scheme 10).90 The choice of solvent has a strong influence on these reactions: non-polar solvents such as toluene give the best results due to the low solubility of the iodide salts formed during the course of the reaction, although t-BuOH can also be used successfully when dealing with polar substrates. At 100 °C a range of solvents can be used, although this can reduce the efficiency of the reactions. By using 1, it is possible to accomplish the arylation of anilines with as little as 0.1 mol% Pd with Cs2CO3 as base, the lowest catalyst loadings that have yet been attained with a weak base.
A range of aromatic sulfonates are suitable as electrophiles for the Pd-catalyzed amination. The applicability of these substrates expands the range of available synthetic building blocks for cross-coupling as these compounds are readily accessible from phenols. Aryl triflates are the most reactive towards oxidative addition of this class.171 Unfortunately, they are also the most readily hydrolyzed by adventitious water, which can necessitate the addition of powdered molecular sieves to the reactions in order to improve yields.88 Strong bases such as NaOt-Bu can also mediate triflate hydrolysis, particularly of electron-deficient substrates.172 This problem can be partly resolved by slow introduction of the aryl triflate to the reaction mixture173 or by using LiOt-Bu which causes slower hydrolysis of the sulfonate.174 The most convenient solution is provided by employing milder inorganic bases such as Cs2CO3.148
Alternatively, aryl nonaflates175 as electrophiles give similar reactivity in Pd-catalyzed amination with ligands including L3 and L4,88,107,160 but undergo hydrolysis more slowly.176 For these substrates implementation of soluble organic bases such as DBU, or for more difficult examples MTBD, is beneficial in conjunction with microwave heating (Scheme 11).160
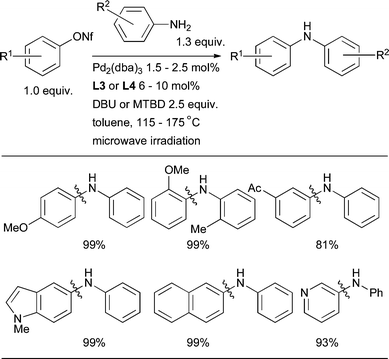 | ||
| Scheme 11 Pd-catalyzed coupling of anilines and aryl nonaflates under microwave irradiation. | ||
Aryl tosylates and benzenesulfonates are much more demanding substrates for Pd-catalyzed amination177 due to their lower propensity to undergo oxidative addition.163 They are, however, attractive from an economic standpoint due to their lower cost than aryl triflates. Dialkylbiaryl phosphine ligands can be successfully used for this transformation with a range of nitrogen nucleophiles, although the exact nature of the ligand has a strong influence on the outcome of the reaction, with L3 providing much higher yields and conversion than less bulky ligands such as L8.83 The most useful results are obtained with t-BuOH as the solvent in conjunction with a weak base such as Cs2CO3 or K2CO3. L3 also allows the amidation of aryl tosylates to be realized.
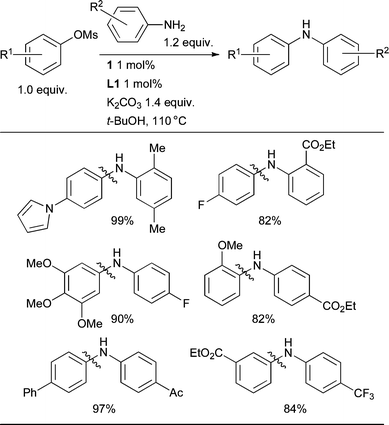
Aryl mesylates present a more exacting class of substrates due to the even greater difficulty of oxidative addition, however, both L185 and Kwong's indole-based dialkylbiaryl phosphine ligands32 have been found capable of aminating these compounds. The weak base K2CO3 is critical to the favorable outcome of these reactions (Scheme 12); stronger bases give lower yields due to competing cleavage of the sulfonate to the corresponding phenol.
By using L6 it is also possible to effect the amidation of aryl mesylates (Scheme 13).104 The combination of Cs2CO3 in t-BuOH is important in ensuring a successful outcome. The reaction is suitable for aryl mesylates with a range of steric and electronic properties, however, heteroaryl mesylates bearing a nucleophilic nitrogen atom provide lower yields, perhaps because these substrates act as nucleophilic catalysts for the undesired desulfonylation of the mesylates.
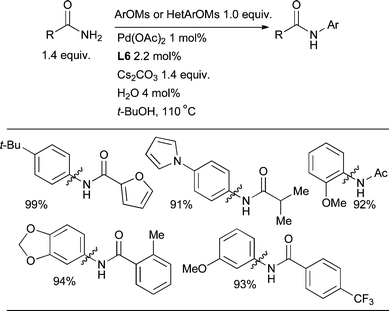 | ||
| Scheme 13 Amidation of aryl mesylates employing L6 as ligand. | ||
It should be noted that vinyl bromides, chlorides and triflates also constitute effective substrates under similar reaction conditions to the corresponding aryl halides,121,159,178–181 a feature that has been exploited in several innovative procedures to access heterocycles182–187 as well as in natural product synthesis.188
Heteroaryl halides are a particularly important class of electrophile for C–N cross-coupling reactions because of the appearance of heteroaryl amines in a range of valuable pharmaceutical agents, notably kinase inhibitors.9,10 Unfortunately these substrates are especially troublesome in metal-catalyzed C–N cross-coupling reactions; indeed even heteroaryl halides which are serviceable for Pd-catalyzed carbon–carbon bond forming reactions can be recalcitrant in amination reactions.189 Such electrophiles can be envisaged to produce a number of difficulties. First, they display a wide spectrum of electronic properties relative to aryl halides and can thus present a more stringent test of the metal to undergo the steps of the catalytic cycle such as oxidative addition190,191 or reductive elimination. Furthermore, if the heterocycles contain heteroatoms capable of coordination, for example pyridines, displacement of the phosphine ligand can occur, resulting in catalyst deactivation.192 Finally, these problems are compounded by the low solubility of these polar substrates in the solvents typically recommended for Pd-catalyzed amination such as toluene and dioxane. In these situations other solvents such as t-BuOH and DMF can be advantageous.89,90
The combination of the utility and challenge of these coupling partners has spurred research in this area. The coupling of halopyridines, -quinolines and -pyrimidines has received particular attention and some highly efficient catalyst systems are now available.18,193 Early studies in this area with dialkylbiaryl phosphine ligands showed L9 to constitute an effective catalyst system for the coupling of simple 1° and 2° amines with chloropyridines.82 These conditions were soon adopted by others in various synthetic applications.5,75,194–197 Following later developments in ligand design, synthetic work demonstrated that both L7198,199 and L3 can be very useful ligands in this context. More recent studies have shown that the most advantageous catalyst systems for the Pd-catalyzed amination of halogenated 6-membered ring heterocycles are comprised of L1 for 1° amines and L2 for 2° amines (Scheme 14).90
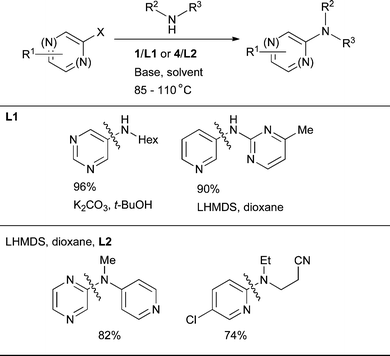 | ||
| Scheme 14 Pd-catalyzed amination of 6-membered ring heteroaryl halides using L1 and L2. | ||
5-Membered ring heteroaryl halides are recalcitrant substrates for Pd-catalyzed amination and low yields have often been encountered.200 Mechanistic studies have been directed towards understanding this effect with other ligand systems such as dppf201 and P(t-Bu)3,202 however, these results do not seem to be directly transferable to biaryl phosphines. Early efforts to engage these electrophiles in amination with dialkylbiaryl phosphine ligands showed that the successful outcome of the reactions is dependent on the correct choice of ligand. For the amination of halogenated thiophenes, furans, benzoxazoles and benzothiazoles with simple nucleophiles, L3, L7 and L2 were the ligands of choice.84 Once again, L1 and L2 are the best ligands at the present time for these substrates (Scheme 15), however, it must be noted that these ligands do not represent a general solution for this class of substrate.89 Furthermore, catalyst loadings are typically higher and reaction times longer than for simple aryl halides.
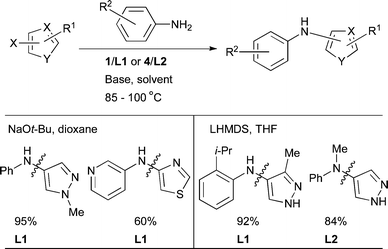
3.2 Nucleophile: anilines
The coupling of simple 1° anilines has typically been one of the easier classes of amination reactions due to the absence of β hydrogen atoms capable of undergoing undesirable elimination of Pd–H. Furthermore the importance of diarylamines as a structural unit of numerous pharmaceutical agents9,10 has prompted interest in this reaction. The coupling of anilines has also attracted study because of the attenuated nucleophilicity of anilines relative to aliphatic amines, which limits the applicability of SNAr reactions in the construction of diarylamines. A number of examples of the coupling of anilines using dialkylbiaryl phosphines in the synthesis of pharmaceuticals have appeared.6,120,122,197,203Catalysts based on L1 are the most active for the arylation of these substrates and provide excellent selectivity for mono-:diarylated products (i.e. for formation of diarylamine rather than triarylamine). A precatalyst that can activate under the reaction conditions is markedly advantageous for this class of substrate as a result of the inability of anilines to mediate reduction of Pd(II). By using the combination of precatalyst 1 and L1 it is possible to bring about the arylation of 1° anilines with aryl chlorides with both low catalyst loadings (0.01 mol% Pd) and short reaction times (1 h), a considerable improvement over other currently available catalyst systems (Scheme 16). Note in this case, the use of NaOt-Bu in conjunction with Bu2O, a solvent that presents advantages over other ethereal solvents such as dioxane when used on scale.
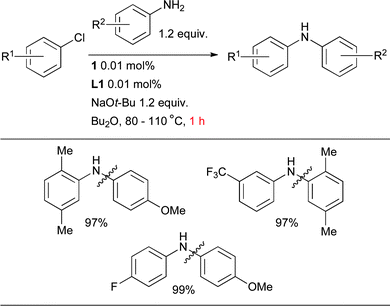
Indeed, with L1 it is possible to achieve the selective N-arylation of aminophenols (Scheme 17), providing complementary selectivity to that observed with Cu-based catalysts.117
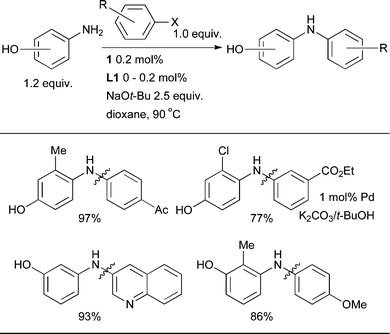 | ||
| Scheme 17 L1-based catalyst systems permit the selective N-arylation of aminophenols. | ||
The coupling of anilines in the presence of primary amides can be problematic. Use of catalyst based on L3 effectively overcomes this issue and allows for the selective arylation of anilines in the presence of primary amides (Scheme 18).83 Further, analogous to the results presented in Scheme 19, this provides complementary selectivity to what is observed when using a Cu-based catalyst.
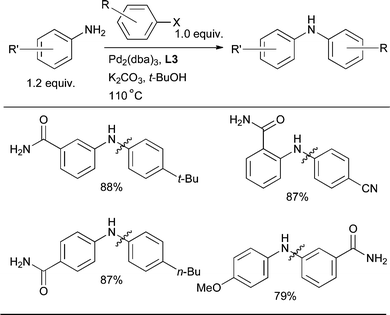 | ||
| Scheme 18 L3-based catalysts allow the selective arylation of an aniline in the presence of a primary amide.83 | ||
The coupling of heteroarylamines can be notably demanding, perhaps as a result of their low nucleophilicity and potential difficulty in undergoing reductive elimination. In this context the ligand L4 is especially efficacious, allowing the arylation of a range of pyridyl and pyrazoyl amines (Scheme 19).86
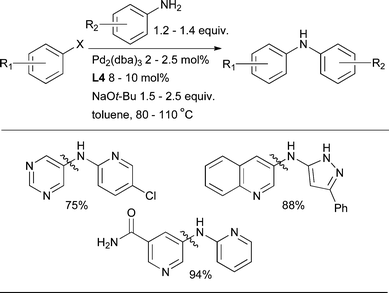 | ||
| Scheme 19 L4 can be a useful ligand for the arylation of electron-deficient heteroarylamines. | ||
L1 is also an effective ligand for this class of nucleophile under a variety of conditions, serving to further broaden the substrate scope (Scheme 20).89
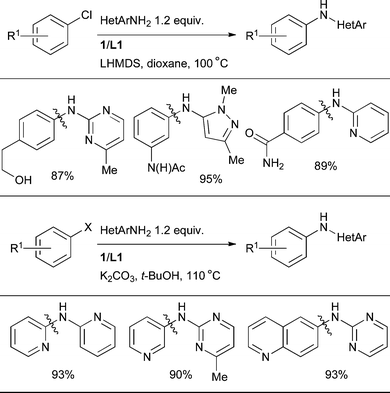 | ||
| Scheme 20 L1 provides an efficient catalyst system for the amination of 1° heteroarylamines under various reaction conditions. | ||
For the coupling of N-alkyl anilines, L2 is generally the ligand of choice, the base/solvent combinations of NaOt-Bu/THF or Cs2CO3/t-BuOH providing a broad substrate scope (Scheme 21).89
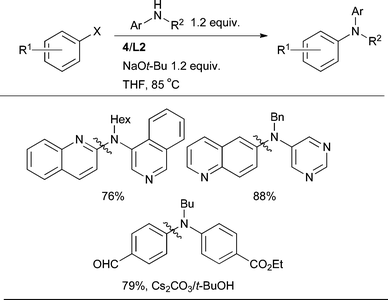 | ||
| Scheme 21 L2 is the best dialkylbiaryl phosphine for the arylation of 2° anilines. | ||
Cyclic amines such as indoline and N,N-diarylanilines are typically better coupling partners than N-alkyl anilines. An exception is N-methyl aniline which appears to be a privileged substrate for Pd-catalyzed amination. This amine readily undergoes arylation with a wide variety of ligand systems, often at lower catalyst loadings than are possible with other nucleophiles.
The synthesis of triarylamines has also been investigated. Using a catalyst based on L2 diarylamines can be arylated to afford the corresponding triarylamines in excellent yields (Scheme 22).89,90
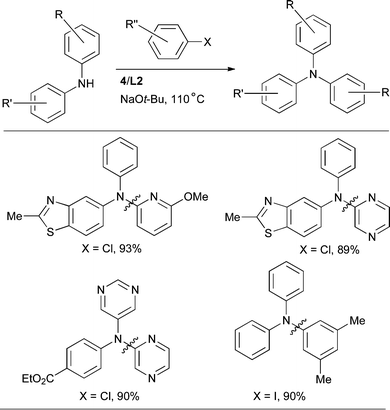 | ||
| Scheme 22 L2-based catalyst for the arylation of diarylamines. | ||
Further, utilizing a catalyst based on L9, triarylamines can be assembled in a one pot procedure from an aniline and two different aryl halides. This method provides a means for the rapid formation of the desired unsymmetrical product from readily available starting materials in one step (Scheme 23).204
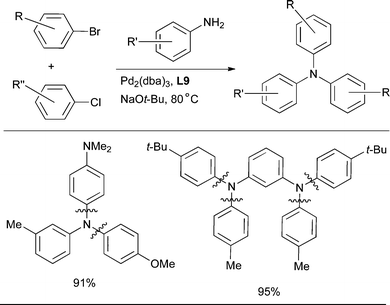 | ||
| Scheme 23 The use of an L9-based catalyst for the synthesis of triarylamines from anilines and aryl halides. | ||
3.3 Nucleophile: aliphatic amines
The arylation of primary aliphatic amines has been the subject of study with numerous classes of ligand and some highly efficient catalyst systems have been described.18,205 Dialkylbiaryl phosphines are also very successful in this context; catalysts based on L1 are the most efficacious. By exploiting this system simple aliphatic amines can be coupled with electron-neutral, -rich and -poor, as well as ortho-substituted aryl chlorides with 0.05 mol% Pd (Scheme 24). The best results are given by a base/solvent combination of NaOt-Bu/Bu2O.85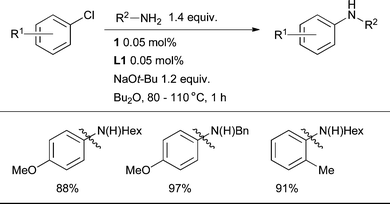 | ||
| Scheme 24 L1 permits the coupling of 1° aliphatic amines with low catalyst loadings and short reaction times. | ||
Substrates bearing protic functional groups can also be efficiently employed when LHMDS is used as base in conjunction with 1 (Scheme 25).89
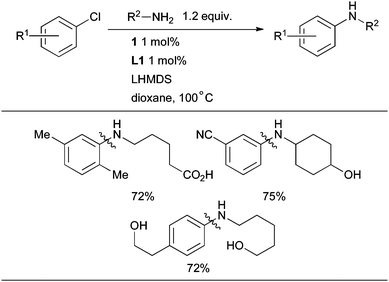
Importantly, these catalysts are much less efficient for the arylation of secondary amines which means that excellent selectivity for monoarylation of a 1° amine can be achieved. Indeed 1° amines can be arylated in the presence of 2° amines with this catalyst system (Scheme 26).85
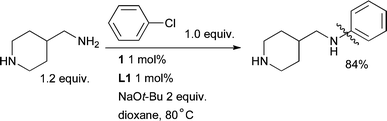 | ||
| Scheme 26 L1-based catalyst systems provide excellent selectivity for the arylation of 1° amines in the presence of 2° amines. | ||
The ability to selectively perform arylation in this way alleviates the need for protecting groups and can contribute to improving the efficiency of synthetic routes. The high selectivity exhibited by L1-based catalysts is exemplified by the ability to monoarylate methylamine, providing a convenient access to N-methyl anilines (Scheme 27). It should be noted that the selective arylation of anilines in the presence of aliphatic amines is also possible by using L3 or L7 as ligand.43,83
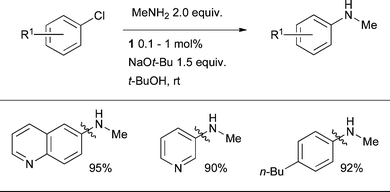 | ||
| Scheme 27 The efficient monoarylation of methylamine can be accomplished by the use of a L1-based catalyst system. | ||
The cross-coupling of secondary aliphatic amines with aryl halides was one of the earliest reaction classes to be explored in Pd-catalyzed amination.142N-Aryl cyclic amines, in particular N-aryl piperazines, are a frequent constituent of CNS-active pharmaceutical agents.13L2 is the best dialkylbiaryl phosphine for this transformation, permitting the arylation of a range of simple cyclic amines at low catalyst loadings with aryl and heteroaryl chlorides in a variety of substitution patterns (Scheme 28).89
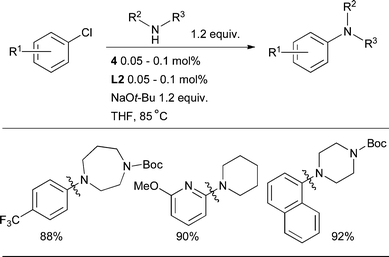 | ||
| Scheme 28 L2 can be used for the arylation of cyclic 2° aliphatic amines under a variety of conditions. | ||
The lowest catalyst loadings are generally secured by using NaOt-Bu as base; Cs2CO3 or LHMDS can alternatively be used in conjunction with more highly functionalized substrates, although somewhat higher catalyst loadings are typically required (Scheme 29).
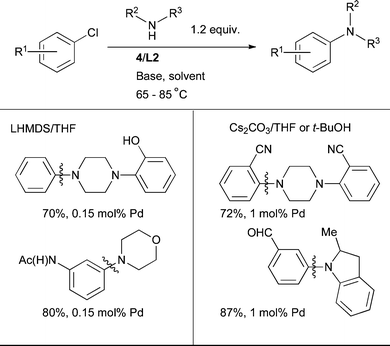 | ||
| Scheme 29 L2 can be used for the arylation of cyclic 2° aliphatic amines under a variety of conditions. | ||
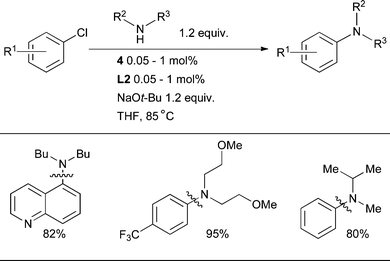
Acyclic secondary amines are often more challenging substrates, presumably as a result of the greater propensity of these substrates to undergo undesired β-hydride elimination.48L2, however, can also provide highly efficient catalysts for the arylation of this class of substrate (Scheme 30).89 At present no suitable catalyst system has been reported for the coupling of hindered secondary amines, particularly with ortho-substituted aryl halides. This is presumably due to two factors: 1) a slower rate of transmetallation (amine binding/deprotonation) and 2) a competitive rate of β-H elimination relative to reductive elimination.
The arylation of dimethylamine can present a particular difficulty, perhaps due to the further enhanced potential of the intermediate Pd amido complex to undergo undesired β-hydride elimination. This transformation can be realized, however, under mild conditions, either with L4 in conjunction with LHMDS as base at room temperature or L3 with K3PO4 at 110 °C.116 Under these conditions dimethylamine can be arylated with a range of electron-rich and electron-deficient aryl chlorides in the presence of a number of functional groups (Scheme 31).
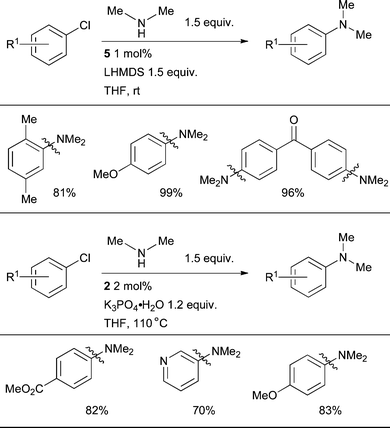 | ||
| Scheme 31 Catalysts based on L3 or L4 can effect the arylation of dimethylamine. | ||
3.4 Nucleophile: amides
The Pd-catalyzed arylation of amides provides a convenient complementary approach to the Cu-mediated Goldberg reaction.206 This process was initially demonstrated with the chelating ligand Xantphos207,208 and it was thought that a chelating ligand was necessary in order to prevent the formation of unwanted κ2 interactions of the amide with the Pd center.209 However, it was shown that L3 is able to effect the arylation of lactams, carbamates, primary amides and N-methyl formamides, including for the first time the amidation of aryl sulfonates.83,210L3 has also been established to be superior to chelating ligands for the intramolecular amidation of aryl chlorides.113Detailed studies with dialkylbiaryl phosphines have been directed towards the arylation of oxazolidinones,211sulfonamides,212 and sulfamides.213 Scientists at GlaxoSmithKline have determined that L4 is the optimal ligand for the room temperature arylation of t-Bu-carbamate.214 The use of this ligand is also fruitful in the vinylation of N-Boc hydrazine.121 Further studies revealed that ligand L5 is more effective than L3 for amidation, providing an effective catalyst system for a range of substrates including lactams, primary amides and sulfonamides and a single example of an N-methyl substituted amide.87 Subsequently the scope of aryl amidation was expanded to include ortho-substituted aryl chlorides by the introduction of L6 (Scheme 32).103 Notably, the H2O activation procedure proved to be the most effective way to generate an active catalyst (Section 2.1). In all cases amidation is best accomplished with weak bases such as K2CO3, K3PO4 or Cs2CO3.
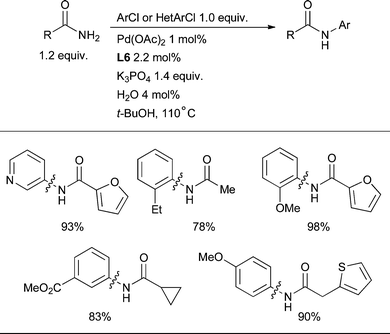 | ||
| Scheme 32 L6 is the best dialkylbiaryl phosphine for the arylation of 1° amides. | ||
Similar conditions are effective for the amidation of aryl mesylates (Scheme 13). So far L6 is the only ligand that has been reported to be effective for this transformation.104
The biaryl phosphine ligand L10 allows the intermolecular arylation of acyclic secondary amides.88 This had not previously been achieved with either Pd or Cu catalysis except for formamides and N-methyl and N-phenyl amides (Scheme 33), which explains the difficulty of the reaction and the design of the ligand. Note that the presence of molecular sieves is usually necessary in these reactions in order to prevent hydrolysis of the amide by adventitious water.
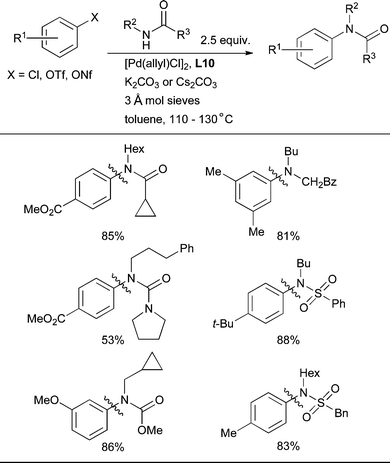 | ||
| Scheme 33 Arylation of 2° amides, ureas, carbamates and sulfonamides is possible by using L10 as ligand. | ||
3.5 Nucleophile: NH heterocycles
The Pd-catalyzed coupling of NH heterocycles with aryl halides is also an area of considerable interest. Studies with chelating ligands have suggested that reductive elimination can be challenging in these cases.215 The first application of dialkylbiaryl phosphines in this context was for the N-arylation of indoles with aryl bromides and triflates and in one example an aryl chloride.105 A range of indoles can be arylated efficiently with aryl bromides, chlorides, iodides and triflates, the preferred ligand depending both on the nature of the leaving group of the electrophile and the steric hindrance about the indole NH. For simple substrates L8 can be used. By employing a variety of other ligands including L11 and L12 it was also possible for the first time to bring about the Pd-catalyzed N-arylation of both 2- and 7-substituted indoles, as well as the N-arylation of indoles with ortho-substituted aryl bromides (Scheme 34). Previously such reactions had been plagued by competing arylation at C3 as well as the formation of bis-arylated products.216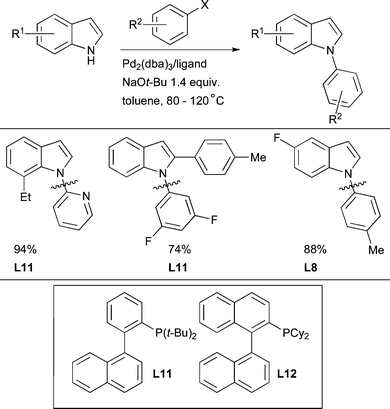
Subsequent studies by Beletskaya have shown that the use of di- or trivalent metal counterions in these reactions can result in selective arylation at the 2 or 3 position of indole (i.e., C–C rather than C–N bond formation).217 Typically, NaOt-Bu was found to be the base of choice, however, K3PO4 can be used for substrates bearing electrophilic functional groups. Unfortunately, some of the best ligands used in these studies are not trivial to access. Prior deprotonation of the heterocycle with BuLi can also be successful.178 Later it was discovered that L3 is an effective ligand for the N-arylation of indoles, even permitting the N-arylation of indoles with aryl benzenesulfonates.83
The cross-coupling of more acidic heterocycles such as indazole and pyrazole has proven to be more problematic with Pd catalysis. By the use of L4 or L5, however, it is possible to couple indazoles, pyrazoles, benzimidazole and in one case imidazole with a variety of aryl and heteroaryl halides (Scheme 35).86
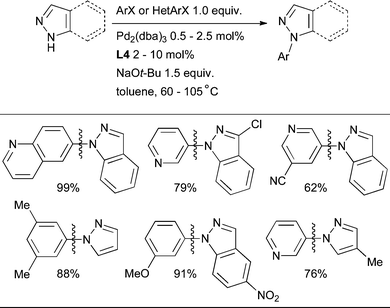
The formation of N1 or N2 arylated indazole was found to be dependent on the nature of the base and the reaction temperature. For the arylation of indazole with 3-bromoanisole, arylation occurs selectively at N1 in the presence of NaOt-Bu in toluene at 80 °C or with Cs2CO3 at 105 °C in 1,4-dioxane. If, however, NaOt-Bu is used at 100 °C mixtures of N1 and N2 arylated products are formed. A possible explanation for these observations is that kinetic binding of Pd occurs at N2 and so when deprotonation of the Pd-bound amine is rapid, arylation at N2 is observed. If, however, deprotonation of the Pd-bound amine is slow (e.g., NaOt-Bu in toluene at 80 °C) then migration of the metal to N1 can take place and arylation is seen at this position.
In order to bring about the arylation of imidazole or benzimidazole L5 is the best dialkylbiaryl phosphine (Scheme 36).86 This is, however, a challenging transformation and the reaction has not yet been extended to more complex nucleophiles of this type.
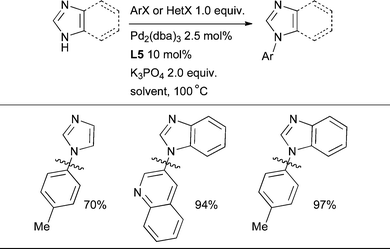 | ||
| Scheme 36 The arylation of imidazole and benzimidazole can be brought about in some cases by using L5 as ligand. | ||
3.6 Nucleophile: benzophenone imine and hydrazone
The Pd-catalyzed cross-coupling of benzophenone imine with aryl halides with subsequent hydrolysis of the resultant N-aryl imine permits the conversion of aryl halides to anilines,218 a process that has been extensively applied in organic synthesis. A number of other ammonia equivalents have been used to accomplish this goal,219–223 however, benzophenone imine remains the most widely used as the reaction can be performed in the presence of a range of functional groups, with aryl bromides, chlorides and triflates as the electrophile and with ortho-substituted electrophiles. Benzophenone imine is commercially available and the intermediate N-aryl imine can be converted to the desired aniline by hydrogenation, transamination with hydroxylamine, or by acid-catalyzed hydrolysis. The direct Pd-catalyzed coupling of free NH3 with aryl halides is also possible,108,224–226 however, the functional group tolerance remains limited unless a high pressure of NH3 is used.227 Of the dialkybiaryl phosphines, both L982 and L383,228 have been used successfully for the arylation of benzophenone imine with aryl chlorides and sulfonates. The mildest conditions reported to date are afforded by exploiting L4 (Scheme 37).157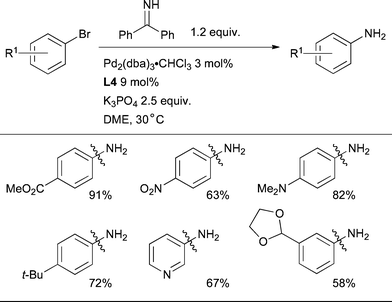
The use of the weak base K3PO4 at 30 °C provides excellent functional group tolerance, however, the reaction is limited to aryl bromides and extended reaction times are required. By using NaOt-Bu in toluene at 65 °C reactions could be completed in 0.5 h. It is important to note that it is strongly advised that commercial benzophenone imine be distilled before use as the nucleophile in any Pd-catalyzed amination reaction.229
The arylation of LiHMDS using a catalyst based on L13 has also been reported as an efficient and more atom economical method for the conversion of aryl chlorides or bromides to the corresponding anilines in high yields (Scheme 38).221 However, because LiHMDS is hindered, ortho-substituted aryl halides are poor substrates. Further, the high basicity of LiHMDS limits the functional group tolerance of this method. On this basis benzophenone imine still remains the ammonia equivalent of choice for discovery chemists.
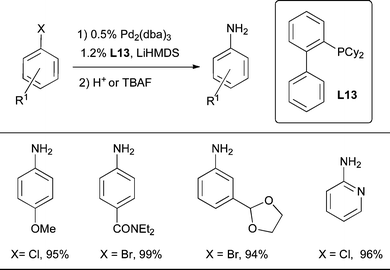 | ||
| Scheme 38 L13 is a useful ligand for the conversion of aryl halides to anilines using LiHMDS as the ammonia surrogate.221 | ||
The Pd-catalyzed arylation of benzophenone hydrazone is also of interest as the resulting N-aryl hydrazones are intermediates in the Fischer indole synthesis and as such can undergo thermal or acid-catalyzed sigmatropic rearrangement to yield indoles.230 The mildness of the conditions permits access to indoles which are challenging to make under more conventional conditions. Alternatively, reaction of the intermediate hydrazones with 1,3-diketones permits access to functionalized pyrazoles.196 As a result of the practical utility of these processes, the Pd-catalyzed arylation of benzophenone hydrazone with aryl bromides and chlorides using dialkylbiaryl phosphines has undergone thorough optimization by scientists at Rhodia (Scheme 39).98
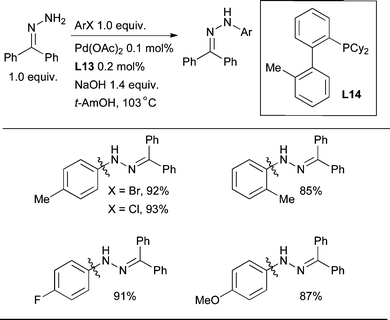 | ||
| Scheme 39 Benzophenone hydrazone can be effectively arylated with aryl chlorides and bromides. | ||
These studies revealed that L3, L8 and L14 can all constitute highly efficient catalyst systems for this reaction,83 allowing the reaction to be performed in 3–4 h with 0.1 mol% Pd. When using protic solvents such as t-AmOH, finely ground NaOH is the best base, however, in toluene NaOt-Bu is more effective. Pd(OAc)2 is a useful Pd source here despite the absence of a nucleophile capable of reducing Pd(II) to Pd(0) for entry into the catalytic cycle. In these experiments adventitious H2O-mediated reduction of Pd(II) by the phosphine must have occurred.
4 Guidelines for optimization
Fig. 6–8 provide a summary of typical reaction conditions for Pd-catalyzed cross-coupling reactions with various substrate classes. The exact choice of conditions will depend to a large extent on both the structure of the substrate as well as the goals of optimization, for example, maximization of yield, minimization of catalyst loading, minimization of reaction time or generality for a variety of substrates if analogues of a particular compound are to be synthesized. When attempting to improve a set of reaction conditions, analysis of the by-products of the reaction can be informative. For example, observation of significant amounts of reduced aryl halide (ArX → ArH) can indicate that reductive elimination is slow relative to β-hydride elimination and that a ligand which is better at promoting reductive elimination should be used (e.g., by switching from a dicyclohexylphosphino to a di-tert-butylphosphino ligand86). Sometimes formation of this by-product can also be suppressed by running reactions at a lower temperature, perhaps implying that in some instances the formation of this by-product is related to catalyst decomposition. Indeed the nature of the reductant in these reactions is not always obvious, even reactions with nucleophiles possessing no β-hydrogen atoms, such as anilines, can produce significant quantities of the reduced arene. Conversely, low conversion of starting aryl halide can be improved by raising the reaction temperature or increasing the catalyst loading. An important cause of low conversion is also inefficient formation of L1Pd, hence switching from Pd(OAc)2 or Pd2(dba)3 to precatalysts 1–5 can also be highly beneficial.93 Alternatively, if mass balance is poor this can reveal that the base is interacting adversely with a functional group in one of the substrates, suggesting the use of a weaker base, for example, Cs2CO3 in place of NaOt-Bu. Another common by-product is the phenol corresponding to the aryl halide or pseudo-halide (ArX → ArOH). This results from either desulfonylation of an aryl sulfonate substrate or from competing coupling of water with the aryl halide.231 In either case this product can be minimized by thorough drying of the reagents159 and the addition of activated molecular sieves to the reaction mixture.88 The phenol thus formed can also undergo Pd-catalyzed C–O bond formation with another molecule of aryl electrophile to generate the symmetrical diaryl ether (ArX → ArOAr).38Pd-catalyzed amination reactions can also produce symmetrical biaryls (ArX → ArAr). Various mechanistic hypotheses have been advanced to explain the formation of these by-products,232–237 however, they tend to occur most commonly in amination reactions when transmetallation is difficult. Hence switching to ligands that are less sterically encumbered or more electron-deficient at phosphorus can be helpful. Furthermore, this reaction can be more prevalent in dipolar, aprotic solvents such as DMF, DMA and DMSO,233,234 hence moving to an ethereal solvent or t-BuOH can bring improvements in the outcome of the reaction while maintaining substrate solubility (Fig. 5).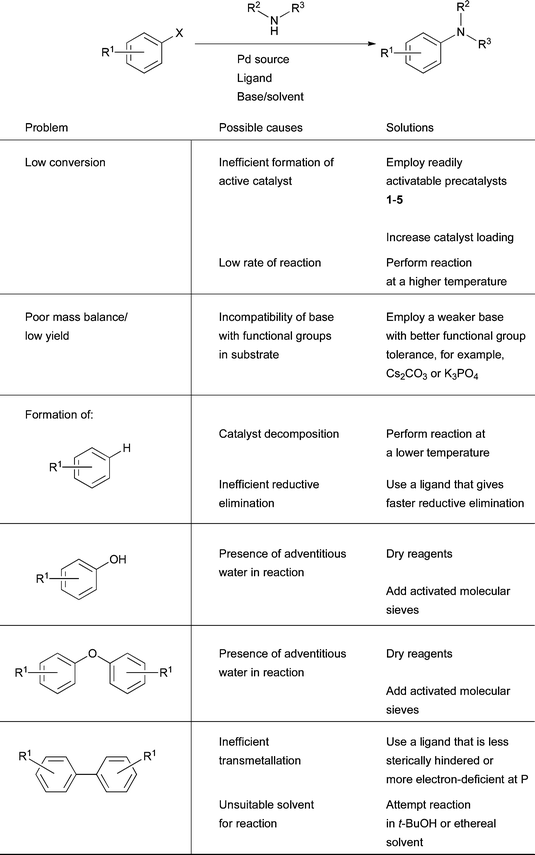 | ||
| Fig. 5 Simplistic troubleshooting guide for Pd-catalyzed amination. | ||
5 Conclusions
The Pd-catalyzed arylation of nitrogen nucleophiles using dialkylbiaryl phosphine ligands has undergone considerable development since the discovery of this ligand class. Much of this progress has been driven by innovations in ligand design, but also by attention to the optimization of reaction conditions. As a result, useful catalyst systems of this type are available for many classes of electrophile and nitrogen nucleophile. The reactions can now often be employed with complex substrates and low catalyst loading.In general, catalysts based on the ligand L1 are the most powerful for 1° amines, and those based on L2 for 2° amines. For certain classes of nucleophile such as amides and NH heterocycles, other dialkylbiaryl phosphine ligands are typically more appropriate. Individual substrate combinations may, however, necessitate a different ligand system as a result of structural peculiarities of the nucleophile and electrophile. It is also crucial to realize that the base/solvent combination is not a hard and fast selection for a given type of substrate. The reaction conditions summarized in Fig. 6–8 are given as starting points for optimization.
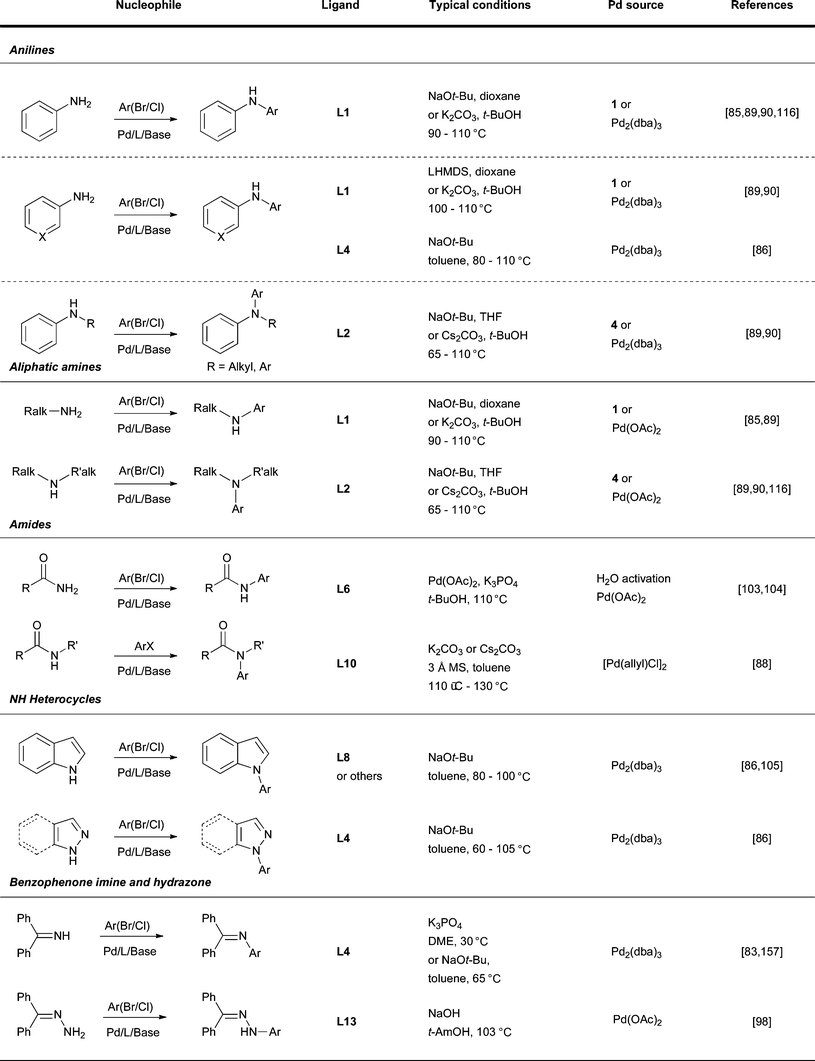 | ||
| Fig. 6 Summary of reaction conditions used for different classes of nucleophile. | ||
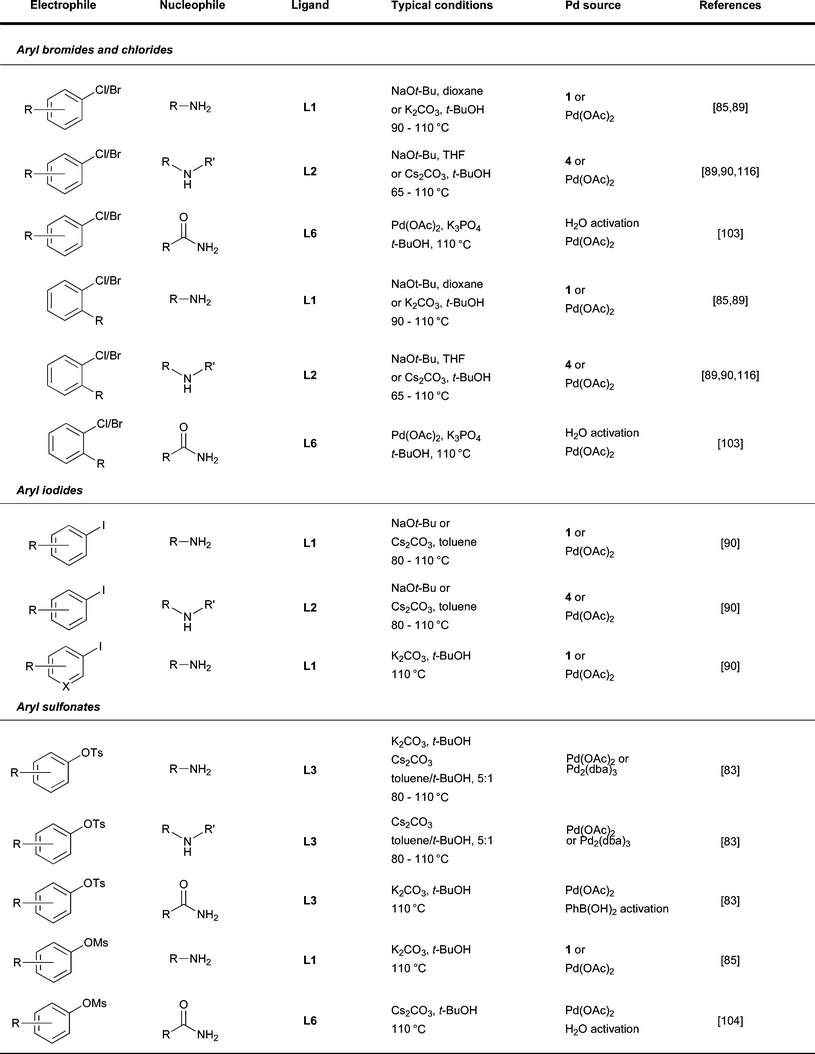 | ||
| Fig. 7 Summary of reaction conditions used for different classes of electrophile. | ||
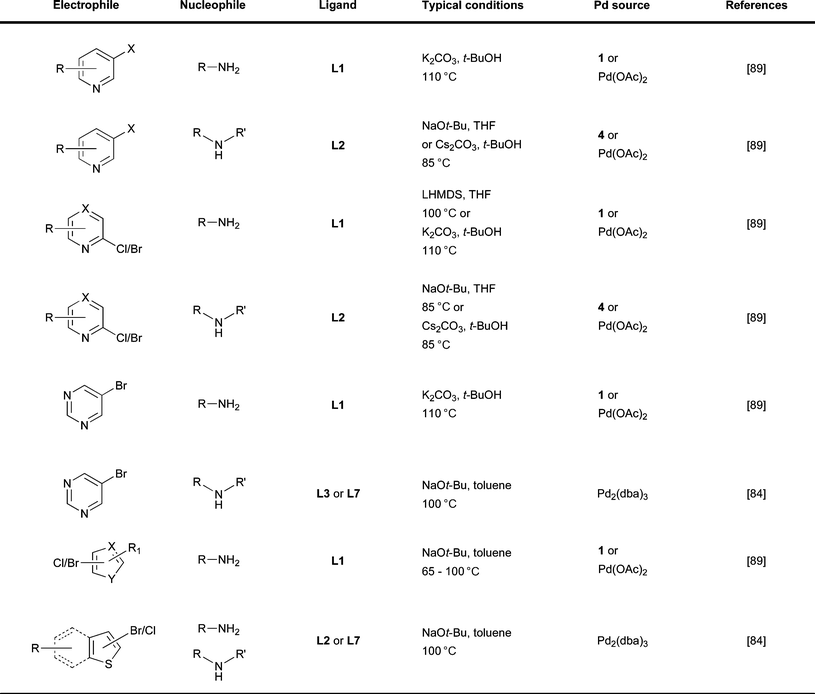 | ||
| Fig. 8 Summary of reaction conditions used for different classes of heteroaryl electrophile. | ||
It is important to re-emphasize the significance of efficient formation of catalytically active, yet kinetically stable (towards decomposition), L1Pd species under the reaction conditions, which depends on the correct choice of Pd source for the reaction. In this regard precatalysts 1–5 that activate very effectively in situ are often the most desirable.
It is hoped that this perspective has supplied some insight into the selection of reaction conditions for a given amination process and the rationale for further optimization. The reader should be aware, however, that some substrates such as certain heteroaryl halides and hindered amines are at present refractory to Pd-catalyzed cross-coupling and remain enticing challenges for the on-going further development of ever more efficient and general catalyst systems.
Acknowledgements
We thank the National Institutes of Health (NIH) for support of our work in this area (Grant GM-58160).Notes and references
- A. O. King and N. Yasuda, Top. Organomet. Chem., 2004, 6, 205–245 CAS.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS.
- A. Baeza, C. Burgos, J. Alvarez-Builla and J. J. Vaquero, Tetrahedron Lett., 2007, 48, 2597–2601 CrossRef CAS.
- R. P. Wurz, L. H. Pettus, S. M. Xu, B. Henkle, L. Sherman, M. Plant, K. Miner, H. McBride, L. M. Wong, C. J. M. Saris, M. R. Lee, S. Chmait, C. Mohr, F. Hsieh and A. S. Tasker, Bioorg. Med. Chem. Lett., 2009, 19, 4724–4728 CrossRef CAS.
- D. Bauer, D. A. Whittington, A. Coxon, J. Bready, S. P. Harriman, V. F. Patel, A. Polverino and J. C. Harmange, Bioorg. Med. Chem. Lett., 2008, 18, 4844–4848 CrossRef CAS.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS.
- J. A. Bikker, N. Brooijmans, A. Wissner and T. S. Mansour, J. Med. Chem., 2009, 52, 1493–1509 CrossRef CAS.
- A. Quintas-Cardama, H. Kantarjian and J. Cortes, Nat. Rev. Drug Discovery, 2007, 6, 834–848 CrossRef CAS.
- S. J. Brickner, D. K. Hutchinson, M. R. Barbachyn, P. R. Manninen, D. A. Ulanowicz, S. A. Garmon, K. C. Grega, S. K. Hendges, D. S. Toops, C. W. Ford and G. E. Zurenko, J. Med. Chem., 1996, 39, 673–679 CrossRef CAS.
- Fluoroquinolone Antibiotics, ed. A. R. Ronald and D. E. Low, Birkhauser, Basel, 2003 Search PubMed.
- J. W. Nilsson, F. Thorstensson, I. Kvarnstrom, T. Oprea, B. Samuelsson and I. Nilsson, J. Comb. Chem., 2001, 3, 546–553 CrossRef CAS.
- J. P. Wolfe, S. Wagaw and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 7215–7216 CrossRef CAS.
- J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 2000, 65, 1144–1157 CrossRef CAS.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 7217–7218 CrossRef CAS.
- Y. Guari, D. S. van Es, J. N. H. Reek, P. C. J. Kamer and P. van Leeuwen, Tetrahedron Lett., 1999, 40, 3789–3790 CrossRef CAS.
- Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef CAS.
- N. Marion, O. Navarro, J. G. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS.
- M. Nishiyama, T. Yamamoto and Y. Koie, Tetrahedron Lett., 1998, 39, 617–620 CrossRef CAS.
- C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS.
- D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722–9723 CrossRef CAS.
- R. A. Singer, S. Caron, R. E. McDermott, P. Arpin and N. M. Do, Synlett, 2003, 1727–1731 CAS.
- F. Rataboul, A. Zapf, R. Jackstell, S. Harkal, T. Riermeier, A. Monsees, U. Dingerdissen and M. Beller, Chem.–Eur. J., 2004, 10, 2983–2990 CrossRef CAS.
- Q. Dai, W. Z. Gao, D. Liu, L. M. Kapes and X. M. Zhang, J. Org. Chem., 2006, 71, 3928–3934 CrossRef CAS.
- R. A. Singer, M. L. Dore, J. E. Sieser and M. A. Berliner, Tetrahedron Lett., 2006, 47, 3727–3731 CrossRef CAS.
- C. A. Fleckenstein and H. Plenio, Chem.–Eur. J., 2007, 13, 2701–2716 CrossRef CAS.
- N. Schwarz, A. Tillack, K. Alex, I. A. Sayyed, R. Jackstell and M. Beller, Tetrahedron Lett., 2007, 48, 2897–2900 CrossRef CAS.
- S. Doherty, J. G. Knight, C. H. Smyth and G. A. Jorgenson, Adv. Synth. Catal., 2008, 350, 1801–1806 CrossRef CAS.
- C. M. So, Z. Zhou, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 6402–6406 CrossRef CAS.
- K. Suzuki, Y. Hori and T. Kobayashi, Adv. Synth. Catal., 2008, 350, 652–656 CrossRef CAS.
- G. J. Withbroe, R. A. Singer and J. E. Sieser, Org. Process Res. Dev., 2008, 12, 480–489 Search PubMed.
- R. Pratap, D. Parrish, P. Gunda, D. Venkataraman and M. K. Lakshman, J. Am. Chem. Soc., 2009, 131, 12240–12249 CrossRef CAS.
- J. W. Ruan, L. Shearer, J. Mo, J. Bacsa, A. Zanotti-Gerosa, F. Hancock, X. F. Wu and J. L. Xiao, Org. Biomol. Chem., 2009, 7, 3236–3242 RSC.
- K. Suzuki, Y. Hori, T. Nishikawa and T. Kobayashi, Adv. Synth. Catal., 2007, 349, 2089–2091 CrossRef CAS.
- C. H. Burgos, T. E. Barder, X. H. Huang and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 4321–4326 CrossRef CAS.
- A. V. Vorogushin, X. H. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 8146–8149 CrossRef CAS.
- W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS.
- G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48, 9240–9261 CrossRef CAS.
- M. R. Biscoe, T. E. Barder and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7232–7235 CrossRef CAS.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2047–2067.
- I. P. Beletskaya and A. D. Averin, Pure Appl. Chem., 2004, 76, 1605–1619 CrossRef CAS.
- M. Kienle, S. R. Dubbaka, K. Brade and P. Knochel, Eur. J. Org. Chem., 2007, 4166–4176 CrossRef CAS.
- D. Prim, J. M. Campagne, D. Joseph and B. Andrioletti, Tetrahedron, 2002, 58, 2041–2075 CrossRef CAS.
- J. P. Wolfe, S. Wagaw, J. F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS.
- B. H. Yang and S. L. Buchwald, J. Organomet. Chem., 1999, 576, 125–146 CrossRef CAS.
- J. F. Hartwig, in Modern Amination Methods, ed. A. Ricci, Wiley-VCH, Weinheim, 2000, p. 195 Search PubMed.
- J. F. Hartwig, in Modern Arene Chemistry, ed. P. Astruc, Wiley-VCH, Weinheim, 2002, p. 107 Search PubMed.
- J. F. Hartwig, in Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley-Interscience, New York, 2002, p. 1051 Search PubMed.
- J. F. Hartwig, in Comprehensive Coordination Chemistry II, ed. M. D. Ward, Elsevier, Amsterdam, 2003, p. 369 Search PubMed.
- J. F. Hartwig, S. Shekar, Q. Shen and F. Barrios-Landeros, in The Chemistry of Anilines, ed. Z. Rappoport, Wiley, New York, 2007, p. 455 Search PubMed.
- S. L. Buchwald and L. Jiang, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Deiderich, Wiley-VCH, Weinheim, 2004, p. 699 Search PubMed.
- J. M. Janey, in Name Reactions for Functional Group Transformations, ed. J. J. Li and E. J. Corey, Wiley, New York, 2007, p. 564 Search PubMed.
- A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131.
- J. F. Hartwig, Synlett, 1997, 329–340 CrossRef CAS.
- J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852–860 CrossRef CAS.
- J. F. Hartwig, Synlett, 2006, 1283–1294 CrossRef CAS.
- J. F. Hartwig, Inorg. Chem., 2007, 46, 1936–1947 CrossRef CAS.
- R. B. Jordan, Organometallics, 2007, 26, 4763–4770 CrossRef CAS.
- B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599–1626 CrossRef CAS.
- S. L. Buchwald, C. Mauger, G. Mignani and U. Scholz, Adv. Synth. Catal., 2006, 348, 23–39 CrossRef CAS.
- V. K. Aggarwal, A. C. Staubitz and M. Owen, Org. Process Res. Dev., 2006, 10, 64–69 Search PubMed.
- S. E. Denmark and C. R. Butler, J. Am. Chem. Soc., 2008, 130, 3690–3704 CrossRef CAS.
- H. Tomori, J. M. Fox and S. L. Buchwald, J. Org. Chem., 2000, 65, 5334–5341 CrossRef CAS.
- S. Kaye, J. M. Fox, F. A. Hicks and S. L. Buchwald, Adv. Synth. Catal., 2001, 343, 789–794 CrossRef CAS.
- T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 5096–5101 CrossRef CAS.
- J. P. Wolfe and S. L. Buchwald, Angew. Chem., Int. Ed., 1999, 38, 2413–2416 CrossRef CAS.
- D. M. Ratner, O. J. Plante and P. H. Seeberger, Eur. J. Org. Chem., 2002, 826–833 CrossRef CAS.
- S. Hosokawa, T. Ogura, H. Togashi and K. Tatsuta, Tetrahedron Lett., 2005, 46, 333–337 CrossRef CAS.
- G. Bringmann, T. Guider, M. Reichert and F. Meyer, Org. Lett., 2006, 8, 1037–1040 CrossRef CAS.
- M. D. Ganton and M. A. Kerr, J. Org. Chem., 2007, 72, 574–582 CrossRef CAS.
- W. J. Pitts, W. Vaccaro, T. Huynh, K. Leftheris, J. Y. Roberge, J. Barbosa, J. Q. Guo, B. Brown, A. Watson, K. Donaldson, G. C. Starling, P. A. Kiener, M. A. Poss, J. H. Dodd and J. C. Barrish, Bioorg. Med. Chem. Lett., 2004, 14, 2955–2958 CrossRef CAS.
- K. Senten, P. Van der Veken, I. De Meester, A. M. Lambeir, S. Scharpe, A. Haemers and K. Augustyns, J. Med. Chem., 2004, 47, 2906–2916 CrossRef CAS.
- J. Renaud, S. F. Bischoff, T. Buhl, P. Floersheim, B. Fournier, M. Geiser, C. Halleux, J. Kallen, H. Keller and P. Ramage, J. Med. Chem., 2005, 48, 364–379 CrossRef CAS.
- K. Wakabayashi, H. Miyachi, Y. Hashimoto and A. Tanatani, Bioorg. Med. Chem., 2005, 13, 2837–2846 CrossRef CAS.
- E. Isabel, R. Aspiotis, W. C. Black, J. Colucci, R. Fortin, A. Giroux, E. L. Grimm, Y. X. Han, C. Mellon, D. W. Nicholson, D. M. Rasper, J. Renaud, S. Roy, J. Tam, P. Tawa, J. P. Vaillancourt, S. Xanthoudakis and R. J. Zamboni, Bioorg. Med. Chem. Lett., 2007, 17, 1671–1674 CrossRef CAS.
- X. L. Jiang, G. T. Lee, K. Prasad and O. Repic, Org. Process Res. Dev., 2008, 12, 1137–1141 Search PubMed.
- J. T. Kuethe, K. G. Childers, G. R. Humphrey, M. Journet and Z. H. Peng, Org. Process Res. Dev., 2008, 12, 1201–1208 Search PubMed.
- J. P. Wolfe, H. Tomori, J. P. Sadighi, J. J. Yin and S. L. Buchwald, J. Org. Chem., 2000, 65, 1158–1174 CrossRef CAS.
- X. H. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 6653–6655 CrossRef CAS.
- M. D. Charles, P. Schultz and S. L. Buchwald, Org. Lett., 2005, 7, 3965–3968 CrossRef CAS.
- B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS.
- K. W. Anderson, R. E. Tundel, T. Ikawa, R. A. Altman and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 6523–6527 CrossRef CAS.
- T. Ikawa, T. E. Barder, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 13001–13007 CrossRef CAS.
- J. D. Hicks, A. M. Hyde, A. M. Cuezva and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 16720–16734 CrossRef CAS.
- D. Maiti, B. P. Fors, J. L. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2010 10.1039/c0sc00330a.
- B. P. Fors, N. R. Davis and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 5766–5768 CrossRef CAS.
- K. Suzuki, Y. Hori, T. Nishikawa and T. Kobayashi, Adv. Synth. Catal., 2007, 349, 2089–2091 CrossRef CAS.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366–374 CrossRef CAS.
- M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 6686–6687 CrossRef CAS.
- T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 12003–12010 CrossRef CAS.
- J. Muzart, J. Mol. Catal. A: Chem., 2009, 308, 15–24 CrossRef CAS.
- E. R. Strieter, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 13978–13980 CrossRef CAS.
- E. R. Strieter and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 925–928 CrossRef CAS.
- C. Mauger and G. Mignani, Adv. Synth. Catal., 2005, 347, 773–782 CrossRef CAS.
- D. Zim and S. L. Buchwald, Org. Lett., 2003, 5, 2413–2415 CrossRef CAS.
- D. B. Damon, R. W. Dugger, S. E. Hubbs, J. M. Scott and R. W. Scott, Org. Process Res. Dev., 2006, 10, 472–480 Search PubMed.
- C. Amatore, E. Carre, A. Jutand and M. A. Mbarki, Organometallics, 1995, 14, 1818–1826 CrossRef CAS.
- B. P. Fors, P. Krattiger, E. Strieter and S. L. Buchwald, Org. Lett., 2008, 10, 3505–3508 CrossRef CAS.
- B. P. Fors, K. Dooleweerdt, Q. L. Zeng and S. L. Buchwald, Tetrahedron, 2009, 65, 6576–6583 CrossRef CAS.
- K. Dooleweerdt, B. P. Fors and S. L. Buchwald, Org. Lett., 2010, 12, 2350–2353 CrossRef CAS.
- D. W. Old, M. C. Harris and S. L. Buchwald, Org. Lett., 2000, 2, 1403–1406 CrossRef CAS.
- M. C. Harris, X. H. Huang and S. L. Buchwald, Org. Lett., 2002, 4, 2885–2888 CrossRef CAS.
- K. W. Anderson, M. Mendez-Perez, J. Priego and S. L. Buchwald, J. Org. Chem., 2003, 68, 9563–9573 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 10354–10355 CrossRef CAS.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 12898–12899 CrossRef CAS.
- C. Amatore and A. Jutand, Coord. Chem. Rev., 1998, 178–180, 511–528 CrossRef CAS.
- Y. Mace, A. R. Kapdi, I. J. S. Fairlamb and A. Jutand, Organometallics, 2006, 25, 1795–1800 CrossRef CAS.
- B. J. Kotecki, D. P. Fernando, A. R. Haight and K. A. Lukin, Org. Lett., 2009, 11, 947–950 CrossRef CAS.
- E. L. Cropper, A. P. Yuen, A. Ford, A. J. P. White and K. K. Hii, Tetrahedron, 2009, 65, 525–530 CrossRef CAS.
- J. P. Stambuli, R. Kuwano and J. F. Hartwig, Angew. Chem., Int. Ed., 2002, 41, 4746–4748 CrossRef CAS.
- R. B. Bedford, C. S. J. Cazin, S. J. Coles, T. Gelbrich, P. N. Horton, M. B. Hursthouse and M. E. Light, Organometallics, 2003, 22, 987–999 CrossRef CAS.
- B. K. Lee, M. R. Biscoe and S. L. Buchwald, Tetrahedron Lett., 2009, 50, 3672–3674 CrossRef CAS.
- D. Maiti and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 17423–17429 CrossRef CAS.
- S. D. Edmondson, A. Mastracchio and E. R. Parmee, Org. Lett., 2000, 2, 1109–1112 CrossRef CAS.
- S. Tasler, J. Mies and M. Langa, Adv. Synth. Catal., 2007, 349, 2286–2300 CrossRef CAS.
- K. Ohshita, H. Ishiyama, K. Oyanagi, H. Nakata and J. Kobayashi, Bioorg. Med. Chem., 2007, 15, 3235–3240 CrossRef CAS.
- J. Barluenga, P. Moriel, F. Aznar and C. Valdes, Org. Lett., 2007, 9, 275–278 CrossRef CAS.
- K. H. Kim, A. Wissner, M. B. Floyd, H. L. Fraser, Y. D. Wang, R. G. Dushin, Y. B. Hu, A. Olland, B. Guo and K. Arndt, Bioorg. Med. Chem. Lett., 2009, 19, 5225–5228 CrossRef CAS.
- S. R. Stauffer and M. A. Steinbeiser, Tetrahedron Lett., 2005, 46, 2571–2575 CrossRef CAS.
- S. R. Stauffer and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 6977–6985 CrossRef CAS.
- H. Christensen, S. Kiil, K. Dam-Johansen, O. Nielsen and M. B. Sommer, Org. Process Res. Dev., 2006, 10, 762–769 Search PubMed.
- U. Schon, J. Messinger, M. Buchholz, U. Reinecker, H. Thole, M. K. S. Prabhu and A. Konda, Tetrahedron Lett., 2005, 46, 7111–7115 CrossRef.
- K. W. Fried, C. M. Schneider, K. W. Schramm, A. Datta, N. Chahbane, C. Corsten, D. R. Powell, D. Lenoir, A. Kettrup, P. Terranova, G. I. Georg and K. K. Rozman, ChemMedChem, 2007, 2, 890–897 CrossRef CAS.
- Aqueous-phase Organometallic Catalysis, ed. B. Cornils and W. A. Hermmann, Wiley-VCH, Weinhem, 2004 Search PubMed.
- N. E. Leadbeater, Chem. Commun., 2005, 2881–2902 RSC.
- K. H. Shaughnessy, Chem. Rev., 2009, 109, 643–710 CrossRef CAS.
- K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2005, 44, 6173–6177 CrossRef CAS.
- M. Nishimura, M. Ueda and N. Miyaura, Tetrahedron, 2002, 58, 5779–5787 CrossRef CAS.
- A. Konovets, A. Penciu, E. Framery, N. Percina, C. Goux-Henry and D. Sinou, Tetrahedron Lett., 2005, 46, 3205–3208 CrossRef CAS.
- R. R. Poondra and N. J. Turner, Org. Lett., 2005, 7, 863–866 CrossRef CAS.
- J. T. Anderson, A. E. Ting, S. Boozer, K. R. Brunden, C. Crumrine, J. Danzig, T. Dent, L. Faga, J. J. Harrington, W. F. Hodnick, S. M. Murphy, G. Pawlowski, R. Perry, A. Raber, S. E. Rundlett, A. Stricker-Krongrad, J. M. Wang and Y. L. Bennani, J. Med. Chem., 2005, 48, 7096–7098 CrossRef CAS.
- G. Van Baelen and B. U. W. Maes, Tetrahedron, 2008, 64, 5604–5619 CrossRef CAS.
- A. Ogawa and D. P. Curran, J. Org. Chem., 1997, 62, 450–451 CrossRef CAS.
- U. Schon, J. Messinger, M. Buckendahl, M. S. Prabhu and A. Konda, Tetrahedron, 2009, 65, 8125–8131 CrossRef.
- K. T. J. Loones, B. U. W. Maes, G. Rombouts, S. Hostyn and G. Diels, Tetrahedron, 2005, 61, 10338–10348 CrossRef CAS.
- S. Shekhar and J. F. Hartwig, Organometallics, 2007, 26, 340–351 CrossRef CAS.
- G. Busca, Chem. Rev., 2010, 110, 2217–2249 CrossRef CAS.
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348–1350 CrossRef CAS.
- M. Prashad, B. Hu, Y. S. Lu, R. Draper, D. Har, O. Repic and T. J. Blacklock, J. Org. Chem., 2000, 65, 2612–2614 CrossRef CAS.
- J. P. Schulte and S. R. Tweedie, Synlett, 2007, 2331–2336 CrossRef CAS.
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- G. Wullner, H. Jansch, S. Kannenberg, F. Schubert and G. Boche, Chem. Commun., 1998, 1509–1510 RSC.
- J. P. Wolfe and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6359–6362 CrossRef CAS.
- J. Ahman and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6363–6366 CrossRef CAS.
- C. Meyers, B. U. W. Maes, K. T. J. Loones, G. Bal, G. L. F. Lemiere and R. A. Dommisse, J. Org. Chem., 2004, 69, 6010–6017 CrossRef CAS.
- A. Klapars, X. H. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS.
- K. Dooleweerdt, H. Birkedal, T. Ruhland and T. Skrydstrup, J. Org. Chem., 2008, 73, 9447–9450 CrossRef CAS.
- T. A. Jensen, X. F. Liang, D. Tanner and N. Skjaerbaek, J. Org. Chem., 2004, 69, 4936–4947 CrossRef CAS.
- T. H. M. Jonckers, B. U. W. Maes, G. L. F. Lemiere and R. Dommisse, Tetrahedron, 2001, 57, 7027–7034 CrossRef CAS.
- C. T. Yang, Y. Fu, Y. B. Huang, J. Yi, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed., 2009, 48, 7398–7401 CrossRef CAS.
- A. S. Dallas and K. V. Gothelf, J. Org. Chem., 2005, 70, 3321–3323 CrossRef CAS.
- J. J. Yin, M. M. Zhao, M. A. Huffman and J. M. McNamara, Org. Lett., 2002, 4, 3481–3484 CrossRef CAS.
- S. Bhagwanth, G. M. Adjabeng and K. R. Hornberger, Tetrahedron Lett., 2009, 50, 1582–1585 CrossRef CAS.
- J. A. Cella and S. W. Bacon, J. Org. Chem., 1984, 49, 1122–1125 CrossRef CAS.
- M. Movassaghi and A. E. Ondrus, J. Org. Chem., 2005, 70, 8638–8641 CrossRef CAS.
- R. E. Tundel, K. W. Anderson and S. L. Buchwald, J. Org. Chem., 2006, 71, 430–433 CrossRef CAS.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS.
- C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254–278 CrossRef CAS.
- A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 194–202 CrossRef CAS.
- V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047–1062 CrossRef CAS.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS.
- T. Yamamoto, M. Nishiyama and Y. Koie, Tetrahedron Lett., 1998, 39, 2367–2370 CrossRef CAS.
- N. P. Reddy and M. Tanaka, Tetrahedron Lett., 1997, 38, 4807–4810 CrossRef CAS.
- M. Beller, T. H. Riermeier, C. P. Reisinger and W. A. Herrmann, Tetrahedron Lett., 1997, 38, 2073–2074 CrossRef CAS.
- J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 1996, 61, 1133–1135 CrossRef CAS.
- R. A. Widenhoefer and S. L. Buchwald, Organometallics, 1996, 15, 2755–2763 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 1997, 62, 1264–1267 CrossRef CAS.
- J. Louie, M. S. Driver, B. C. Hamann and J. F. Hartwig, J. Org. Chem., 1997, 62, 1268–1273 CrossRef CAS.
- J. Barluenga, A. Jimenez-Aquino, F. Aznar and C. Valdes, J. Am. Chem. Soc., 2009, 131, 4031–4041 CrossRef CAS.
- J. Hogermeier and H. U. Reissig, Adv. Synth. Catal., 2009, 351, 2747–2763 CrossRef.
- X. Q. Zhang and Z. Sui, Tetrahedron Lett., 2003, 44, 3071–3073 CrossRef CAS.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 7369–7370 CrossRef CAS.
- A. Y. Lebedev, V. V. Izmer, D. N. Kazyul'kin, I. P. Beletskaya and A. Z. Voskoboynikov, Org. Lett., 2002, 4, 623–626 CrossRef CAS.
- M. C. Willis and G. N. Brace, Tetrahedron Lett., 2002, 43, 9085–9088 CrossRef CAS.
- J. Barluenga, M. A. Fernandez, F. Aznar and C. Valdes, Chem. Commun., 2004, 1400–1401 RSC.
- J. Barluenga, F. Aznar, P. Moriel and C. Valdes, Adv. Synth. Catal., 2004, 346, 1697–1701 CrossRef CAS.
- T. Kino, Y. Nagase, Y. Horino and T. Yamakawa, J. Mol. Catal. A: Chem., 2008, 282, 34–51 CrossRef CAS.
- J. Barluenga, A. Jimenez-Aquino, M. A. Fernandez, F. Aznar and C. Valdes, Tetrahedron, 2008, 64, 778–786 CrossRef CAS.
- M. C. Willis, G. N. Brace, T. J. K. Findlay and I. P. Holmes, Adv. Synth. Catal., 2006, 348, 851–856 CrossRef CAS.
- J. Barluenga, M. A. Fernandez, F. Aznar and C. Valdes, Chem.–Eur. J., 2005, 11, 2276–2283 CrossRef CAS.
- J. Barluenga, A. Jimenez-Aquino, C. Valdes and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 1529–1532 CrossRef CAS.
- Y. Q. Fang and M. Lautens, Org. Lett., 2005, 7, 3549–3552 CrossRef CAS.
- M. Movassaghi and A. E. Ondrus, Org. Lett., 2005, 7, 4423–4426 CrossRef CAS.
- V. F. Slagt, A. H. M. de Vries, J. G. de Vries and R. M. Kellogg, Org. Process Res. Dev., 14, pp. 30–47 Search PubMed.
- C. Y. Legault, Y. Garcia, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 12664–12665 CrossRef CAS.
- Y. Garcia, F. Schoenebeck, C. Y. Legault, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 6632–6639 CrossRef CAS.
- F. Paul, J. Patt and J. F. Hartwig, Organometallics, 1995, 14, 3030–3039 CrossRef CAS.
- Q. L. Shen, S. Shekhar, J. P. Stambuli and J. F. Hartwig, Angew. Chem., Int. Ed., 2005, 44, 1371–1375 CrossRef CAS.
- J. W. Lim, N. Stock, R. Pracitto, J. K. Boueres, B. Munoz, A. Chaudhary, A. M. Santini, K. Orr, H. Schaffhauser, R. E. Bezverkov, J. Aiyar and S. Venkatraman, Bioorg. Med. Chem. Lett., 2004, 14, 1913–1916 CrossRef CAS.
- K. S. Gudmundsson and B. A. Johns, Org. Lett., 2003, 5, 1369–1372 CrossRef CAS.
- N. Haddad, A. Salvagno and C. Busacca, Tetrahedron Lett., 2004, 45, 5935–5937 CrossRef CAS.
- C. J. Burns, D. G. Bourke, L. Andrau, X. Y. Bu, S. A. Charman, A. C. Donohue, E. Fantino, M. Farrugia, J. T. Feutrill, M. Joffe, M. R. Kling, M. Kurek, T. L. Nero, T. Nguyen, J. T. Palmer, I. Phillips, D. M. Shackleford, H. Sikanyika, M. Styles, S. Su, H. Treutlein, J. Zeng and A. F. Wilks, Bioorg. Med. Chem. Lett., 2009, 19, 5887–5892 CrossRef CAS.
- O. Johansson, Synthesis, 2006, 2585–2589 CrossRef CAS.
- O. Johansson, L. Eriksson and R. Lomoth, Dalton Trans., 2008, 3649–3651 RSC.
- V. K. Gore, V. V. Ma, R. Tamir, N. R. Gavva, J. J. S. Treanor and M. H. Norman, Bioorg. Med. Chem. Lett., 2007, 17, 5825–5830 CrossRef CAS.
- M. W. Hooper and J. F. Hartwig, Organometallics, 2003, 22, 3394–3403 CrossRef CAS.
- M. W. Hooper, M. Utsunomiya and J. F. Hartwig, J. Org. Chem., 2003, 68, 2861–2873 CrossRef CAS.
- S. Tasler, O. Muller, T. Wieber, T. Herz, R. Krauss, F. Totzke, M. H. G. Kubbutat and C. Schachtele, Bioorg. Med. Chem. Lett., 2009, 19, 1349–1356 CrossRef CAS.
- M. C. Harris and S. L. Buchwald, J. Org. Chem., 2000, 65, 5327–5333 CrossRef.
- Q. L. Shen and J. F. Hartwig, Org. Lett., 2008, 10, 4109–4112 CrossRef CAS.
- J. Lindley, Tetrahedron, 1984, 40, 1433–1456 CrossRef CAS.
- J. J. Yin and S. L. Buchwald, Org. Lett., 2000, 2, 1101–1104 CrossRef CAS.
- J. J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 6043–6048 CrossRef CAS.
- K. Fujita, M. Yamashita, F. Puschmann, M. M. Alvarez-Falcon, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9044–9045 CrossRef CAS.
- X. N. Li and R. Vince, Bioorg. Med. Chem., 2006, 14, 5742–5755 CrossRef CAS.
- A. Ghosh, J. E. Sieser, M. Riou, W. L. Cai and L. Rivera-Ruiz, Org. Lett., 2003, 5, 2207–2210 CrossRef CAS.
- G. Burton, P. Cao, G. Li and R. Rivero, Org. Lett., 2003, 5, 4373–4376 CrossRef CAS.
- L. Alcaraz, C. Bennion, J. Morris, P. Meghani and S. M. Thom, Org. Lett., 2004, 6, 2705–2708 CrossRef CAS.
- S. Bhagwanth, A. G. Waterson, G. M. Adjabeng and K. R. Hornberger, J. Org. Chem., 2009, 74, 4634–4637 CrossRef CAS.
- G. Mann, J. F. Hartwig, M. S. Driver and C. Fernandez-Rivas, J. Am. Chem. Soc., 1998, 120, 827–828 CrossRef CAS.
- J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy and L. M. Alcazar-Roman, J. Org. Chem., 1999, 64, 5575–5580 CrossRef CAS.
- M. V. Nikulin, A. Y. Lebedev, A. Z. Voskoboinikov and I. P. Beletskaya, Dokl. Chem., 2008, 423, 326–329 CrossRef CAS.
- J. P. Wolfe, J. Ahman, J. P. Sadighi, R. A. Singer and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6367–6370 CrossRef CAS.
- S. Lee, M. Jorgensen and J. F. Hartwig, Org. Lett., 2001, 3, 2729–2732 CrossRef CAS.
- X. H. Huang and S. L. Buchwald, Org. Lett., 2001, 3, 3417–3419 CrossRef CAS.
- D. Y. Lee and J. F. Hartwig, Org. Lett., 2005, 7, 1169–1172 CrossRef CAS.
- C. W. Lim and S. Lee, Tetrahedron, 2000, 56, 5131–5136 CrossRef CAS.
- S. Jaime-Figueroa, Y. Z. Liu, J. M. Muchowski and D. G. Putman, Tetrahedron Lett., 1998, 39, 1313–1316 CrossRef CAS.
- Q. L. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 10028–10029 CrossRef CAS.
- T. Schulz, C. Torborg, S. Enthaler, B. Schaffner, A. Dumrath, A. Spannenberg, H. Neumann, A. Borner and M. Beller, Chem.–Eur. J., 2009, 15, 4528–4533 CrossRef CAS.
- R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071–4074 CAS.
- G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049–11061 CrossRef CAS.
- K. Sondergaard, J. L. Kristensen, N. Gillings and M. Begtrup, Eur. J. Org. Chem., 2005, 4428–4433 CrossRef.
- We have observed that the efficiency of these reactions is highly dependent on the quality of the benzophenone imine.
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 10251–10263 CrossRef CAS.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 CrossRef CAS.
- A. Jutand and A. Mosleh, J. Org. Chem., 1997, 62, 261–274 CrossRef CAS.
- D. D. Hennings, T. Iwama and V. H. Rawal, Org. Lett., 1999, 1, 1205–1208 CrossRef CAS.
- J. Hassan, V. Penalva, L. Lavenot, C. Gozzi and M. Lemaire, Tetrahedron, 1998, 54, 13793–13804 CrossRef CAS.
- F. Ozawa, T. Hidaka, T. Yamamoto and A. Yamamoto, J. Organomet. Chem., 1987, 330, 253–263 CrossRef CAS.
- Y. Suzaki and K. Osakada, Organometallics, 2003, 22, 2193–2195 CrossRef CAS.
- D. J. Cardenas, B. Martin-Matute and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 5033–5040 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |