Green synthesis of metal nanoparticles: Biodegradable polymers and enzymes in stabilization and surface functionalization
Jurate
Virkutyte†
* and
Rajender S.
Varma
*
Sustainable Technology Division, National Risk Management Research Laboratory, US Environmental Protection Agency, MS 443, 26 West M. L. K. Drive, Cincinnati, Ohio 45268, USA. E-mail: Virkutyte.Jurate@epa.gov; Varma.Rajender@epa.gov; Fax: +1 513 569-7677; Tel: +1 513 487-2701
First published on 23rd December 2010
Abstract
Current breakthroughs in green nanotechnology are capable of transforming many of the existing processes and products that enhance environmental quality, reduce pollution, and conserve natural and non-renewable resources. Successful use of metal nanoparticles and nanocomposites in various catalytic applications, electronics, biology and biomedical applications, material science, physics, environmental remediation and interdisciplinary fields as well as their toxicity essentially depends on the structural features such as size, shape, composition and the surface chemistry of nanomaterials. Moreover, to prolong the life span of metal nanoparticles and avoid undesired effects such as aggregation in aqueous solutions and organic solvents, to prevent contamination of the environment as well as to reuse and recycle nanoparticles, it is vital to select stabilizing agents and functionalization pathways that are environmentally friendly, non toxic and easy to implement. In recent years, stabilization and surface functionalization of metal nanoparticles became ‘greener’ to the extent that biocompatible stabilizing agents, e.g. biodegradable polymers and enzymes among others were introduced. These agents were able to produce a great variety of extremely stable spherical-, rod- or flower-shaped metal nanoparticles that opened up vast opportunities for their utilization and potential mass production. This review summarizes the state-of-the-art in the use of biocompatible and biodegradable homo- and copolymers as well as enzymes for the production of stable, environmentally benign, selective and active metal nanoparticles for desired applications.
 Jurate Virkutyte | Dr. Jurate Virkutyte received her BSc (1996) from Vilnius Pedagogical University (Lithuania) in Chemistry and her MSc (1999) from the Royal Institute of Technology (Sweden) in Environmental Engineering. She then moved to Finland where she received her PhD (2005) in Environmental Science from Kuopio University. She worked on the development of electrokinetic methods and advanced catalytic oxidation processes in the Netherlands, Portugal and Australia. In 2009 she was awarded with NRC Research Associateship to work with Prof. R. Varma at US-EPA, Cincinnati OH, synthesizing “green” nanocatalysts for environmental remediation. Currently, she is employed with Pegasus Technical Services Inc. Cincinnati, OH (USA). |
 Rajender S. Varma | Prof. Rajender S. Varma, Ph.D. (1976, Delhi University) did his postdoctoral research at Robert Robinson Laboratories and was a faculty member at Baylor College of Medicine, Senior Scientist at Houston Advanced Research Center and Research Professor at Sam Houston State University prior to joining as a senior scientist in the Sustainable Technology Division at US Environmental Protection Agency in 1999. He has over 35 years of experience in multi-disciplinary technical programs ranging from natural products chemistry to nanomaterials and development of environmentally friendlier alternatives for synthetic methods using microwaves, ultrasound, etc. He has published over 300 papers and has 7 US Patents. |
Introduction
Nanotechnology is mainly associated with the synthesis of nanoparticles of variable sizes, shapes, chemical compositions and controlled dispersity and their potential use for human benefits.1 Over the last few years, nanotechnology has become a topic of intense interest that has attracted significant attention due to the importance of environmental issues. The effective development of green nanotechnology has a potential to transform many currently available processes and products that enhance environmental quality, reduce pollution and conserve natural and non-renewable resources. Recently, in her report on Green Nanotechnology, Schmidt2 stated that the combination of nanotechnology and the principles and practices of green chemistry is the key to building an environmentally sustainable society in the 21st century. Moreover, nanotechnology offers a possibility to manufacture products using greener, safer and more environmentally-friendly pathways, as well as to control, monitor and truly make processes ‘green’ from the ‘cradle to grave.’According to Anastas and Warner,3 green chemistry is a set of principles or rather a chemical philosophy that encourages the design of products and processes that reduce or eliminate the use and generation of hazardous substances. All 12 principles together with a corresponding green nanotechnology approach are summarized in Table 1.
| Green Chemistry Principles | Green Nanotechnology approach |
|---|---|
| 1. Prevent waste | Develop new ‘greener’ chemical syntheses of nanomaterials that minimize or completely eliminate waste streams and products by designing pollution prevention at the source. |
| 2. Manufacture safer chemicals and products | Fabricate nanomaterials that posses the desired physicochemical properties and which have little or no toxicity. |
| 3. Design less hazardous chemical syntheses | Discover and develop syntheses that utilize benign reagents and solvents and generate substances with little or no toxicity to the environment and humans. |
| 4. Use renewable feedstocks | Use raw materials and feedstock that are renewable and benign. |
| 5. Utilize catalysts | Minimize waste by utilizing nanocatalysts synthesized in a green manner. Develop catalytic methods that can greatly enhance reaction selectivity and overall process efficiency. |
| 6. Avoid chemical derivatives | Avoid using blocking or protecting groups or any temporary modifications if possible. Eliminate solvent-intensive syntheses by utilizing selective nanosyntheses. |
| 7. Maximize atom economy | Design and develop syntheses so that the final product contains the maximum proportion of the starting materials. |
| 8. Use safer solvents and reaction conditions | Avoid using hazardous solvents, separation agents, or other auxiliary chemicals. Utilize bottom-up approach to enhance materials efficiency and eliminate steps. |
| 9. Increase energy efficiency | Develop synthesis protocols so that they are carried out at ambient temperature and pressure whenever possible. |
| 10. Design chemicals and products to degrade after use | Assess and quantify the nanomaterial’s behavior in the environment, study their fate and major chemical interactions with other particles or organisms. Design nanomaterials to degrade to innocuous substances after use so that they do not accumulate and harm the environment. |
| 11. Real-time monitoring and process control to prevent pollution | Utilize real-time monitoring to optimize reaction chemistry, minimize energy costs and to minimize or eliminate the formation of by-products. Develop Life Cycle Assessment (LCA) of nanoscience, which is an essential tool for managing risks in research, development and commercialization of this field. |
| 12. Minimize the potential for accidents | Produce chemicals and their forms (solid, liquid, or gas) to minimize the potential for chemical accidents including explosions, fires, and releases into the environment. |
The 12 green chemistry principles and their application in green nanotechnology include the design and development of syntheses using renewable high energy efficient materials, benign reaction media and non hazardous as well as non toxic solvents, utilizing catalysts, determining the interactions of nanomaterials in the environment, their fate and transport as well as the development of real-time monitoring and process control to avoid or minimize accidents.4
Application of green chemistry principles has already reduced the use of hazardous solvents or other toxic ingredients, optimized reaction conditions, improved materials and energy efficiency of chemical processes, increased the utilization of renewable resources and enhanced the design of products for end of life.4 Furthermore, the primary goals of green nanotechnology are processes and products.5 Processes include the production of nanomaterials without harming the environment or human health and products involve the design and development of nanomaterials that provide sustainable solutions to environmental problems. Therefore it is obvious that the main purpose of both, green chemistry and nanotechnology is similar, that is to make processes function more like ecosystems or cells, in which benign materials are used at ambient temperatures, chemical reactions are conducted in water rather than hazardous and toxic solvents, waste streams are recycled and energy is used efficiently.2
Although all green chemistry principles are important, there are several that are directly related to the main goals of green nanotechnology and therefore will be emphasized in this work. Principles 3 and 8 are directly linked to the utilization of environmentally benign solvents and the eco-friendly reaction conditions during less hazardous chemical syntheses of target nanoparticles and principles 2 and 10 are associated with the manufacturing of safer metal nanoparticles that degrade after use (Table 1).
Successful use of nanomaterials in various catalytic applications, electronics, biology and biomedical applications, material science, physics, environmental remediation and interdisciplinary fields as well as their toxicity critically depends on the structural features such as size, shape, composition and the surface chemistry of the nanoparticles.4,6 Thus, in order to extend the array of properties, it is necessary to develop various environmentally-friendly routes leading to nanoparticles of various shapes and sizes relevant to a desired application.7
This work will summarize the ‘state of the art’ in the exploitation of biodegradable homo and block copolymers and enzymes to synthesize various metal nanoparticles for a great variety of purposes.
Functionalization and stabilization of metal nanoparticles
The high chemical activity of engineered nanoparticles with a developed surface is usually the main reason for undesirable strong and often irreversible processes such as aggregation.8 Aggregation reduces the specific surface area and the interfacial free energy, thereby diminishing particle reactivity (Scheme 1).9 Thus, it is extremely important to increase the stability of the developed nanoparticle during storage, transportation and its overall life cycle. As emphasized by Stubbs and Gilman,10 the majority of the stabilization methods involve dispersant molecules such as surfactants or polyelectrolytes which not only alter the chemistry and physics of the nanoparticle surface, but also produce a tremendous waste stream because they occupy a significant (more than 50%) mass fraction of a nanoparticle system. Therefore, to avoid contamination of and subsequent adverse effects on the environment, there is a need to search for environmentally-benign stabilization and functionalization pathways as well as biocompatible, i.e. non-immunogenic, non-toxic and hydrophilic stabilizing agents.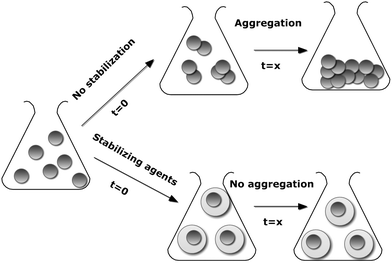 | ||
| Scheme 1 Schematized aggregation of nanoparticles in the presence and absence of stabilizing agents | ||
Various stabilizing agents are available to prevent the nanoparticles from aggregating as well as to functionalize the particles for the desired applications at the same time.11–18 However, usually severe reaction conditions and toxic chemicals may not be suitable for biological and biochemical applications.19 Currently, there are several ‘green’ stabilizing agents such as polyphenols, enzymes, citric acid, vitamins (B, C, D, K), biodegradable polymers and silica, to name a few available, that are able to stabilize and functionalize metal nanoparticles without the undesirable consequences on the environment and biosystems. Nonetheless, due to the tremendous amount of information available, this review will be selectively restricted to the use of biodegradable polymers and enzymes in the stabilization of metal nanoparticles.
Recoverability and reusability of metal nanoparticles
It is of paramount importance to functionalize and stabilize metal nanoparticles for diverse applications, however, easy and relatively inexpensive recoverability and therefore reusability of nanoparticles is currently gaining an increased attention amongst the scientific community. For a number of years, magnetic nanoparticles have been extensively used in the fields of metal ions and dye recovery, drug delivery, enzyme immobilization, protein and cell separation because magnetic separation offers unique advantages of high efficiency, cost effectiveness and rapidity in comparison to other nanoparticles.20–22 Nowadays, magnetic nanoparticles are consistently emerging as heterogeneous supports (so called magnetic nano-cores) in various catalytic transformations providing added benefits of easy recoverability with a simple magnet, thereby eliminating the necessity of solvent swelling before or catalyst filtration after the reaction.23–26Biodegradable polymers
Biodegradable polymers may be derived from various renewable sources (corn, wood cellulose, polylactides, thermoplastic starch, plant oils, gelatin, chitosan), petroleum sources (aliphatic polyesters or aliphatic-aromatic copolyesters), can be synthesized by bacteria from small molecules or may be obtained from mixtures of biomass and petroleum.28 Rozenberg and Tenne29 argued that nanoparticles stabilized by surface-active polymers, which adsorb strongly onto the particle surface, due to van del Waals attractive forces between the surface and monomer units of the polymer chain, are not prone to aggregation because of a large decrease of their surface energy in comparison with native particles. Block polymers are even more powerful nanoparticle stabilizers than homopolymers because they demonstrate phase separation and exhibit unique properties such as surface reactivity, elasticity, selectivity and resistance.30Stabilization of metal nanoparticles can be achieved by a simple entrapment of metals in a polymer gel, by a free radical polymerization with a radical initiator,31 by thiol supported adsorption of polymer27 or by in situ formation of metal nanoparticles during polymerization.27,32,33
Polylactides (PLA) and block copolymers
Polylactide (PLA) is a biodegradable, thermoplastic, aliphatic polyester derived from renewable resources, such as corn starch or sugarcane. Due to its biocompatibility and biodegradability, PLA is well suited to drug delivery, tissue engineering and stabilization of nanoparticles (Scheme 2).27 Consequently, PLA- (homopolymer or block copolymers) stabilized metal (magnetic in particular) nanoparticles may be potentially used to deliver drugs, for magnetic cell separations and magnetic hyperthermia therapy for treatment of tumors.31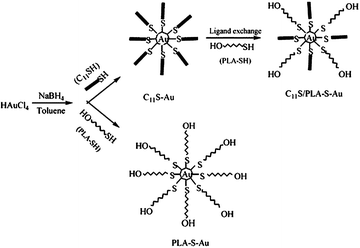 | ||
| Scheme 2 The route to fabricate and stabilize gold nanoparticles.27 Reproduced with permission from American Chemical Society, copyright 2004 | ||
Representative preparation of magnetic nanoparticles stabilized by diblock poly(D,L-lactide-co-glycolide) (PLGA) and triblock (poly(lactide-b-siloxane-b-lactide) (PLSL) copolymers (M ≈ 11 540) involves dissolution of polymers in an organic solvent (e.g. toluene) and placing into an oil bath at 65 and 100 °C or an ice bath in the presence of ultrasound irradiation.34 It is important to note the effect of pH on the degradation of copolymers. To avoid hydrolysis of the polylactide blocks, pH must be adjusted to 7. It is observed that at circum-neutral pH, magnetite has both cationic and anionic charges (the magnetite isoelectric point = pH 6.8) and therefore acid groups in the copolymer are in the anionic salt form.31
Freshly prepared copolymer is re-dissolved in an organic solvent, magnetic nanoparticles are added and the solution is thoroughly mixed for 18 h to ensure a complete adsorption of the copolymer onto the magnetic nanoparticle.31 Magnetic nanoparticles stabilized with PLA block copolymers (Fig. 1) showed minor or no aggregation over a significant period of time and retained spherical nanostructures.34
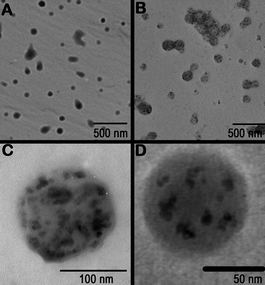 | ||
| Fig. 1 Representative TEMs of (A) PLGA (B) PLGA particles with incorporated magnetite, (C) PLGA particles with magnetite made from a 5 mg M−1 solution and (D) PLGA particles with magnetite made from a 1 mg M−1 solution.34 Reproduced with permission from Elsevier, copyright 2007. | ||
Gold nanoparticles (AuNPs) have been extensively used in biological or medical applications because of their biocompatibility, size, ease of characterization and rich history of surface chemistry.35,36 Thus, to expand the array of applications, AuNPs can be stabilized using a phase separation approach. Qiu and colleagues27 developed a simple metal nanoparticle preparation and a subsequent stabilization procedure which involved dissolution of a magnetic nanoparticle precursor in water and a subsequent transfer to the organic phase (N(C8H17)4Br in toluene) under a vigorous mixing. Then, thiol end-capped PLA block copolymer (PLA-SH, M ≈ 2400) was added and the whole mixture was stirred for about 4 h. This way, predominantly spherical gold nanoparticles stable in non-polar and weakly polar solvents such as heptanes, toluene and chloroform were formed, whereas stability of gold nanoparticles in DMSO increased with the amount of PLA-SH attached to the surface.27
Polysaccharides
Polysaccharides are linear or branched polymeric carbohydrate structures, joined together by the glycosidic bonds. Polysaccharide surfaces provide the potential for significant surface modification using well-established carbohydrate chemistry, which allows tailoring the surface functionality of metal nanoparticles.37Carboxymethyl cellulose (CMC)
Cellulose is one of the most abundant polysaccharides, a natural and renewable biopolymer, which can be easily modified to sodium carboxymethyl cellulose (CMC) by replacing the native CH2OH group in the glucose unit with a carboxymethyl group.9,38 CMC-stabilized nanomaterials are constantly gaining importance in various technological38,39 (e.g. land and water remediation), biological and biomedical applications (e.g. antibacterial and antifungal coatings, food packaging, biolabeling, biomedical devices)40 due to their unique properties, e.g. low toxicity and high sensitivity. Unfortunately, CMC is not able to produce Au, Pt and Pd nanoparticles because noble metals do not chelate with carboxyl groups at room temperature,40 whereas Fe nanoparticles can be stabilized with CMC in aqueous solutions regardless of the temperature.9To produce homogeneous CMC-stabilized (M ≈ 90 000) noble metal salt nanoparticles with smaller sizes, Nadagouda and Varma40 developed a protocol where microwave irradiation (100 °C, 5 min) was employed and CMC acted both, as a capping and reducing agent (Fig. 2). Aqueous solutions of noble metal salts with appropriate amounts of CMC were subjected to the microwave irradiation until homogeneous colored nanocomposites of various morphologies were formed.
 | ||
| Fig. 2 Selected TEM images of CMC nanocomposites with (a) Cu, (b) In, (c) Fe, and (d) Ag.40 Reproduced with permission from American Chemical Society, copyright 2007. | ||
Interestingly, the Cu-CMC composite showed needle-like nanostructures in the form of bushes, In and Fe demonstrated spherical nanoparticles uniformly embedded into the CMC matrix and Ag had a spherical morphology with a majority of cube-shaped nanoparticles.40 Stable (9 days fully dispersed in water) Fe nanoparticles prepared in aqueous solution under vacuum (reaction time approx. 30 min) in the presence of a reducing agent sodium borohydride,9 were also spherical and similar in size to Fe nanoparticles formed under the microwave irradiation. On the other hand, CMC-stabilized and ascorbic acid reduced Pd nanoparticles, synthesized in aqueous solution at 22, 50, 80 and 95 °C (reaction time 5 min and ageing for 24 h) showed spherical nanostructures and were slightly (up to 10%) larger than nanoparticles obtained during more conventional reduction and stabilization processes.38,41 Regardless of the size, CMC-Pd particles offered comparable reaction rates and greater surface catalytic reactivity to degrade trichlorethene (TCE) in aqueous solutions under otherwise identical conditions.38 In addition, CMC-stabilized (remarkably stable for more than 9 months in aqueous solutions) Pt nanoparticles in the presence of a reducing agent sodium borohydride,42 were spherical and faceted (Fig. 3).
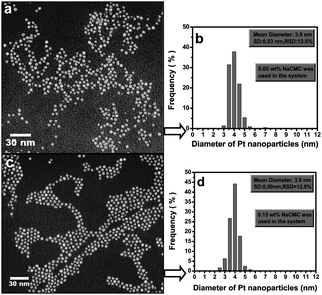 | ||
| Fig. 3 Selected TEM images of Pt nanoparticles stabilized with (a) 0.05 wt % and (c) 0.15 wt % CMC and their corresponding particle size distribution histograms (b and d).42 Reproduced with permission from American Chemical Society, copyright 2007. | ||
Thus, it is possible to conclude that when CMC was added as a stabilizing agent (regardless of the presence or the absence of the reducing agent), spherical noble and magnetic metal nanoparticles with an average diameter of less than 4 nm (with the exception of CMC-Fe nanoparticles with diameters <17 nm)9,43 were predominantly formed.
The stability and size of metal nanoparticles highly depended on the CMC to metal molar ratio and initial metal concentration and medium pH as well as the concentration of macroelements (Na+ and Ca2+) in water.38,39 Interestingly, concentration of CMC had no significant effect on the size of Pt particle42 and an increase in temperature resulted in a significant decrease in both, the size and size distribution of the CMC-stabilized Pd nanoparticles.38,41
Chitosan
Chitosan (2-amino-2-deoxy-b-β-glucan) is basic, non-toxic, hydrophilic, biocompatible, and biodegradable deacylated derivative of chitin44 that is widely used in drug delivery, biosensors, food, cosmetics and pharmaceutical industry, wastewater treatment45,46 and material sciences.47,48 The interest of a scientific community to use chitosan and its derivatives in the stabilization of nanoparticles is consistently growing (Fig. 4).49,50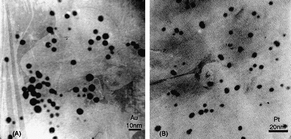 | ||
| Fig. 4 TEM images of gold (A) and platinum (B) nanoparticles in Au- and Pt-chitosan nanocomposites.49 Reproduced with permission from Elsevier, copyright 2004. | ||
Wang and Cui48 noted that chitosan was able to stabilize and/or reduce Au nanoparticles by virtue of its abundant amino and hydroxyl groups in the presence of luminol. Moreover, gold nanoparticles with average diameters of 18 ± 5 nm (spherical) and 10–20 nm (nanoflowers) were formed as a result of aqueous synthesis of precursors in acetic acid (2%) solution. Remarkably, when solely chitosan or luminol were employed, spherical Au nanoparticles were formed (Fig. 5a–c), whereas the use of both, stabilizing (chitosan) and reducing (luminol) agents resulted in the formation of well-defined nanoflowers (Fig. 5d–f).48
 | ||
| Fig. 5 High resolution (HRTEM) images of Au nanoparticles.48 Reproduced with permission from American Chemical Society, copyright 2008. | ||
Similar synthesis of chitosan-stabilized Ag nanoparticles resulted in the formation of stable, selective and active catalysts that can be efficiently (yields 70–98%) utilized in C–C coupling reactions.51
Also, Wei and colleagues45 reported the successful preparation of single-crystal Au nanosheets in the presence of chitosan, which can be potentially translated into the mass production. They found that the formation of a nanosheet highly depended on the precursor (Au salt) concentration, e.g. at high (8 to 12 mM) salt concentrations, micro-sized Au nanosheets with regular shape were formed, whereas smaller salt concentrations yielded a large number of Au particles (50 nm in size) along with a few nanosheets.
To increase the effectiveness and to avoid the necessity of employing a reducing agent, biocompatible bio-conjugates that simultaneously stabilize and reduce metal nanoparticles may be exploited. For instance, a novel chitosan ninhydrin (CHIT-NH) bioconjugate to stabilize Au nanoparticles at physiological (37 °C) temperature was proposed.52 The preparative procedure involved aqueous synthesis in acetic acid (2%) solution and resulted in the formation of highly branched spherical assemblies with an average diameter of 18 nm stable over a considerably long time periods.
Other polysaccharides
Dextran is a polysaccharide polymer predominantly composed of α-D-glucopyranosyl units with varying degrees of chain length and branching.47 It is extremely biocompatible, non-toxic and hydrophilic, therefore may be used to coat various metal and polymer nanoparticles.53 Jiang et al.53 prepared magnetic nanoparticles (average diameter of 25.3 nm) in aqueous solutions (pH 10–11) in the presence of ammonium hydroxide (NH4OH), urea and dextran. They argued that the addition of urea CO(NH2)2 resulted in the more narrow size distribution of nanoparticles in comparison to particles obtained through a conventional co-precipitation procedure and could be controlled with the varying amount of decomposed urea.Philip54 pionereed the stabilization of Au nanoparticles in water at room temperature (reaction time within 30 min) with honey, which is a known source of fructose, glucose and aminoacids (Fig. 6).
 | ||
| Fig. 6 Selected TEM images of colloids with: (a) 20 mL and (b) 25 mL of honey; (c) representative high-resolution image and (d) SAED pattern.54 Reproduced with permission from Elsevier, copyright 2009. | ||
Hypothetically, the main agents responsible for the reduction and simultaneous nanoparticle stabilization are monosaccharide fructose, vitamin C, gluconic acid, glucose, sucrose and enzymes with fructose being the most preferred reducing and protein (free amine groups) stabilizing agent. However, the exact mechanism of Au salt reduction and a stabilization of nanoparticles, highly warrant further research. The lower concentrations of honey resulted in unstable triangular, whereas the higher concentrations resulted in stable spherical (average size 15 nm) Au nanoparticles.
Moreover, one-dimensional (1D) carbon nanomaterials wrapped by spherical and well dispersed silver nanoparticles were fabricated by Niu and colleagues55via a facile and environmentally benign route with the assistance of supercritical carbon dioxide and glucose as a reducing agent. In addition, a recent and very comprehensive review on the green synthesis of Ag nanoparticles utilizing polysaccharides among other reducing agents was published pointing out and discussing in detail various methods of nanoparticle preparation.56 Importantly, both works emphasized the applicability of these nanostructures for antibacterial activities. Raveendran and co-workers57,58 used β-D-glucose as a reducing agent and starch as a capping agent to synthesize silver, gold and Ag–Au alloys nanoparticles in a sustainable manner. Synthesized nanoparticles were extremely stable (no aggregation observed for several months), comparable in size (from 1 to 8 nm, mean particle diameter of 5.3 nm and 6–10 nm for Ag and Au, respectively) and polydispersity to those produced using more conventional methods.
Polyethylene glycol (PEG) and block copolymers
The utilization of polyethylene glycol (PEG) of molecular weight from 200 to tens of thousands and block copolymers has attracted much attention because they are inexpensive, do not exhibit any toxicity, are hydrophilic and their properties can be tuned by changing the molecular weight.29,59,60 PEG-based (PEGylated) metal nanoparticles can be used for a great array of biological, chemical and biomedical applications, e.g. to deliver drugs, genes and proteins (tumor necrosis factor) to solid tumors or infections,61 as diagnostic contrast agents for computed tomography62 as well as in colloidal biosensors, semiconductors63,64 and catalysis.59,65,66 To stabilize metal nanoparticles in water or in organic solvents, PEG homopolymer or block copolymers are often deployed.The synthesis and simultaneous stabilization of metal (Ag, Pt, Fe, etc.) nanoparticles with PEG in aqueous solutions was recently developed by Nadagouda and Varma.67 They used a simple and straightforward one-pot procedure of mixing appropriate aqueous metal salt solution with PEG (M ≈ 300) at room temperature and then irradiated using microwave synthesis system at 100 °C for 1 h with a maximum pressure of 280 psi (Scheme 3). Such a method did not require any surfactants or reducing agents, did not produce hazardous wastes and, thus, could be applied for a wide variety of applications.
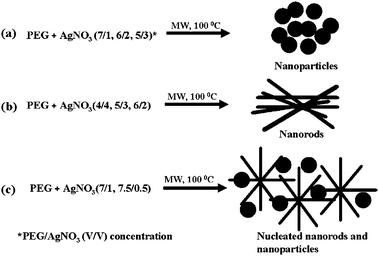 | ||
| Scheme 3 Schematic presentation of Ag a) nanoparticles, b) nanorods and c) nucleated nanorods and nanoparticles formation mechanisms.67 Reproduced with permission from American Chemical Society, copyright 2008 | ||
It was found that the concentration of PEG was the main factor in controlling the morphology and the size of stable metal nanoparticles and a limiting value was reached when the surface of the nanoparticle was saturated with PEG.68 For instance, at higher PEG concentrations nanoparticles were predominantly formed whereas at low concentrations, nanorods nucleation with nanoparticles was observed. Metal nanoparticles prepared in aqueous solutions with PEG homopolymer were stable in the medium and no precipitation was observed for several months.68
It is also possible to stabilize metal nanoparticles with PEG homopolymer in the presence of organic solvents.66 Thus, appropriate metal salt is dissolved in the mixture of solvents (e.g. toluene, n-heptane, etc.) and the solution is transferred to a stainless steel autoclave, where various concentrations of PEG are subsequently added. The autoclave is then flushed and pressurized with H2 for a desired period of time. Such a procedure, can provide an extremely stable metal nanocatalyst that can be recycled 6 times without the significant loss in activity and selectivity.66
In addition, not only PEG homopolymer, but also di- and tri-block as well as branch copolymers α-acetal-poly-(ethylene glycol)-b-poly(D,L-lactide) (α-acetal-PEG-PLA), polyethyleneoxide-polyethyleneimine (PEO)n-b-PEI), PEG containing both, mercapto and acetal terminal groups (α-acetal-ω-mercapto-PEG, acetal-PEG-SH), amphiphilic polystyrene-poly(ethylene glycol) (PS-PEG) and poly(ethylene glycol)-b-poly(4-vinylpyridine)-b-poly(N-isopropylacrylamide) (PEG-b-PVP-b-PNIPAM) were extremely efficient in preventing a non-specific aggregation of metal (Au, Rh, Pd and Pt) nanoparticles and thus increasing their stability under physiological conditions.64,65,68,69 Sidorov and colleagues70 discovered that the addition of strong acids (low pH) decreased whereas the addition of NaOH (pH 11.7) significantly increased the stability of metal nanoparticles regardless the species of the initial metal salt used. Also, Nakao and co-workers65 reported that e.g. PS-PEG stabilized Pd nanoparticles exhibited an excellent stability and the oxidation, as well as the reduction, of selected organic compounds (yields up to 99%) took place under mild, aqueous and aerobic conditions.
Therefore, the stability of the engineered metal nanoparticles was due to the steric stabilization that resulted in thermodynamically stable system that PEG provided71,72 and highly depended on the block copolymer composition (block, branch), polymer to metal ratio, polymer density and the type of the reducing agent (H2, NaBH4).47,69,70
Importantly, PEG block copolymers can act as efficient stabilizers of metal nanoparticles only in non-polar organic solvents (toluene, heptanes, etc.) at a higher temperature66 because then they become miscible and because appropriate polymers are not able to simultaneously form micelles in water and bind metals in the core.64 Such miscibility at a higher temperature and a phase split at a lower temperature is particularly important in the case of homogeneous or heterogeneous catalysis, which provides easier separation of PEGylated nanoparticles from the solution.66 On the other hand, stabilization of metal nanoparticles is only possible in water, when the branched copolymer (PEO)n-b-PEI), (PEG)n-b-PEI or similar type is used where the whole copolymer is soluble in water and only one part of the copolymer is responsible for the stabilization of metal nanoparticles.70
Enzymes
Over the last few decades, enzymes have become very attractive for a wide range of industrial processes including biocatalysis, bioremediation, biomedical applications and biosensors73 with the expressed objective of reducing energy and raw material consumption and amounts of waste and toxic by-products formation.74 Moreover, enzymes may also be used to functionalize as well as to stabilize the formation of various nanoparticles. According to Scott et al.,73 efficient stabilization of metal nanoparticles with enzymes would provide a defined number of initiation/reduction sites, enable the formation of metal nanoparticles with controlled size and would expand our understanding of biomimetic nanoparticle syntheses methods.For instance, Au nanoparticles functionalized with a redox enzyme can act as an electron relay between the biocatalyst and the electrode12 and therefore provide a hybrid electrically active biomaterial that can be used in various sensors applications.75 They can also increase the routing of electrons from the redox enzymes to the electrodes and facilitate the synthesis of the nanoparticles by electron-relay-mediated biocatalysis.76
It was recently discovered that E. coli glutathione reductase, an active redox enzyme of a known structure was efficiently used to stabilize metal nanoparticles via interactions with an active site cysteine (Cys42).73 In fact, this discovery presented a breakthrough in the field of biomimetic nanoparticle synthesis proving that the direct reduction of metal ions leading to the formation of metal nanoparticles that are tightly adsorbed onto the active site, is viable. Moreover, the same research group has confirmed that enzymes can function as nano-reactors, allowing the defined nucleation and growth of metal nanoparticles that are protected by sequestration in the enzyme active sites.73
According to Xiao and co-workers,75 a metallic nanoparticle (Au nanoparticle functionalized with a single N-hydroxysuccinimide functionality and reacted with N6-(2-aminoethyl)-flavin adenine dinucleotide to yield Au nanoparticle-FAD) reconstituted apo–glucose oxidase (apo-GOx) with appropriate dimensions (surface coverage of the FAD-functionalized Au nanoparticle of 1.1 × 10−11 mol cm−2 and surface-reconstituted GOx enzyme of 0.96 × 10−12 mol cm−2) could act as a current nano-collector and as an electron relay to a macroelectrode. Thus, nanoparticles can function as nano-electrodes for transferring electrons between the active site and the electrode, activating the bioelectrocatalytic functions of the enzymes.76 Indeed, the reconstituted GOx exceeded the electron transfer features of the native enzyme, i.e. an electron transfer rate constant of 5 × 103 s−1 was achieved in comparison to 700 s−1 of a native GOx with O2 as an electron acceptor. Bioelectrocatalytic properties were insensitive to increasing O2 concentrations or the addition of oxidative agents such as ascorbic acid.75 Obviously, functionalization of nanoparticle with redox enzyme greatly increased its maximum turnover rate.
In addition, different oxidases may also be used to stimulate synthesis and growth of stable metal nanoparticles. Willner and colleagues76 reported the enzyme-mediated, acetylcholine esterase (AChE), production of Au nanowires. In brief, AChE stimulated hydrolysis of acetylthiocholine to thiocholine and subsequently the reduction of AuCl4 in the presence of glucose. The formation and growth (from 2–3 nm to 10–30 nm) of the Au nanoparticles was significantly faster (growth time interval of 10 min) in the presence of electron relay than when hydrogen peroxide H2O2 was utilized as the reducing agent.76 Karam and co-workers77 used glucose oxidase (GOx), a homodimer glycoprotein (M ≈ 160 000 and 16% carbohydrate content) to stabilize Pt nanoparticles in the presence of H2 as the reducing agent, that can be exploited in the fabrication of biosensors. The GOx-stabilized (pH 7) Pt nanoparticles (average dimensions of 4 nm) retained their electrocatalytic activity for several processes including H2O2 and glucose oxidation.
It is important to note that enzymes can be efficiently utilized as both, the reductive agent and the stabilizer for metal nanoparticles utilizing covalent or usually non-specific routes of attachment.19,78
Rangnekar et al.19 reported the synthesis (at 37 °C for 48 h, pH 7) and simultaneous surface functionalization of spherical Au nanoparticles using a pure enzyme such as α-amylase with the preserved enzymatic activity over a considerably long period. They found that the hydrolysis was nearly complete in 400 min for these particular reaction conditions and the rate of starch digestion by both, the functionalized nanoparticle as well as the free enzyme was similar indicating that the enzyme was stable and biologically active for more than 6 h. Similar retention of enzymatic activity (in fact, comparable to that of the free enzyme in the solution) was observed by Brennan and colleagues78 who researched lipase (Thermomyces lanuginosus) (Scheme 4) and by Gole and colleagues79 who assessed the behavior of aspartic protease (Aspergillus satoi) stabilized Au nanoparticles.
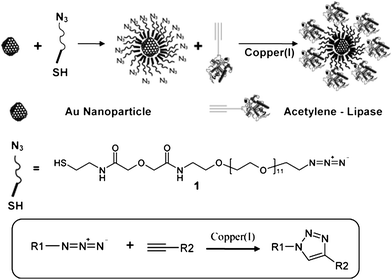 | ||
| Scheme 4 Functionalization of gold nanoparticles using click chemistry.78 Reproduced with permission from American Chemical Society, copyright 2006 | ||
Hen egg white lysozyme is a small highly cationic and amphiphilic protein that is a part of the innate immune system in higher organisms and primarily acts against bacteria through enzymatic hydrolysis of the peptidoglycian layer that surrounds the cell membrane.80 Also, due to its properties (affinity to adsorb to ionic and hydrophobic surfaces), lysozyme can also be used to engineer hydrophilic and biocompatible, extremely stable (e.g. 10 months at ca. 4 °C without precipitation) spherical Au nanoparticle in the presence or absence of reducing agents (e.g. NaBH4), Fig. 7, that can be efficiently used in biology or medicine.12,80
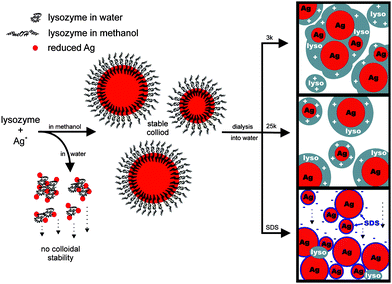 | ||
| Fig. 7 Proposed Ag–lyso nanoparticle formation mechanism.80 Reproduced with permission from American Chemical Society, copyright 2009. | ||
It was discovered that lysozyme monolayer-protected Au nanoparticles exhibited such an excellent stability that the high speed centrifugation (20 000 rpm for 30 min at room temperature) or boiling of nanoparticles for 10 min did not result in their precipitation.
Conclusive remarks
Although extensive scientific efforts are directed towards the ‘greening’ of nanotechnology, there is still room for innovations that can result in significant improvements within this particular scientific area. The use of biocompatible and biodegradable homo- and copolymers and enzymes as reducing and/or stabilizing agents have resulted in extremely efficient (stable up to 10 months in aqueous or organic solvents) stabilization of metal nanoparticles and functionalization of their surfaces. Moreover, the vast majority of metal nanoparticles studied were spherical (with an exception of some flower and rod-shaped nanoparticles when polymers and\or enzymes were utilized as stabilizing and NaBH4, H2, etc. as reducing agents), with an average diameter of 4 nm regardless of the synthesis pathway or stabilizing agents used. However, morphology and stability of metal nanoparticles using ‘greener’ pathways is still in its infancy and better understanding of reduction and stabilization mechanisms is needed. For instance, NaBH4 is hazardous and toxic, however, it is an effective, highly selective and relatively inexpensive conventional and “historically safe” accepted reducing agent, which was used for many years to successfully synthesize various organic molecules and compounds. Thus, the overall “greening” of the nanoparticle synthesis must be gradually but not drastically accomplished by introducing other much safer, sustainable and reliable reducing as well as capping agents and finally avoiding the use of hazardous chemicals such as NaBH4 and hydrazine. Preparation and a subsequent study of engineered ‘green’ nanoparticles offer the abundance of potential and already defined applications in biomedicine, biology, material science, electronics, biosensors, pharmaceutical, food and cosmetic industry and environmental remediation field.Acknowledgements
This work was accomplished while J.V. held a National Research Council Research Associateship Award at US EPA.References
- V. Kumar and S. K. Yadav, J. Chem. Technol. Biotechnol., 2009, 84, 151–157 CrossRef CAS.
- K. Schmidt, in American Chemical Society Meeting, Woodrow Wilson International Center for Scholars and The Pew Charitable Trusts, Washington DC, 2006 Search PubMed.
- P. Anastas and J. Warner, Green chemistry: Theory and practice, Oxford University Press, USA, 1998 Search PubMed.
- J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228–2269 CrossRef CAS.
- B. Karn, in NanoECOMonte Verità, Switzerland 2008 Search PubMed.
- W. E. Buhro and V. L. Colvin, Nat. Mater., 2003, 2, 138–139 CrossRef CAS.
- P. Mohanpuria, N. Rana and S. Yadav, J. Nanopart. Res., 2008, 10, 507–517 CrossRef CAS.
- A. D. Pomogailo and V. N. Kestelman, ed., Metallopolymer Nanocomposites, Springer Berlin HeidelbergNew York, 2005 Search PubMed.
- F. He, D. Zhao, J. Liu and C. B. Roberts, Ind. Eng. Chem. Res., 2007, 46, 29–34 CrossRef CAS.
- D. Stubbs and P. Gilman, Oak Ridge Center for Advanced Studies, 2007.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2008, 10, 859–862 RSC.
- T. Yang, Z. Li, L. Wang, C. Guo and Y. Sun, Langmuir, 2007, 23, 10533–10538 CrossRef CAS.
- M. N. Nadagouda, V. Polshettiwar and R. S. Varma, J. Mater. Chem., 2009, 19, 2026–2031 RSC.
- B. Baruwati and Rajender S. Varma, ChemSusChem, 2009, 2, 1041–1044 CrossRef CAS.
- M. N. Nadagouda, G. Hoag, J. Collins and R. S. Varma, Cryst. Growth Des., 2009, 9, 4979–4983 CrossRef CAS.
- M. N. Nadagouda, A. B. Castle, R. C. Murdock, S. M. Hussain and R. S. Varma, Green Chem., 2010, 12, 114–122 RSC.
- M. C. Moulton, L. K. Braydich-Stolle, M. N. Nadagouda, S. Kunzelman, S. M. Hussain and R. S. Varma, Nanoscale, 2010, 2, 763–770 RSC.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC.
- A. Rangnekar, T. K. Sarma, A. K. Singh, J. Deka, A. Ramesh and A. Chattopadhyay, Langmuir, 2007, 23, 5700–5706 CrossRef CAS.
- C. Chang, D. B. Shieh, C. H. Chang and D. H. Chen, J. Biomed. Nanotechnol., 2005, 1, 196–201 Search PubMed.
- Y.-C. Chang and D.-H. Chen, J. Colloid Interface Sci., 2005, 283, 446–451 CrossRef CAS.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS.
- R. Abu-Reziq, H. Alper, D. Wang and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279–5282 CrossRef.
- B. Baruwati, V. Polshettiwar and R. S. Varma, Tetrahedron Lett., 2009, 50, 1215–1218 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Org. Biomol. Chem., 2009, 7, 37–40 RSC.
- V. Polshettiwar and Rajender S. Varma, Chem.–Eur. J., 2009, 15, 1582–1586 CrossRef CAS.
- H. Qiu, J. Rieger, B. Gilbert, R. Jerome and C. Jerome, Chem. Mater., 2004, 16, 850–856 CrossRef CAS.
- S. Sinha Ray and M. Bousmina, Prog. Mater. Sci., 2005, 50, 962–1079 CrossRef.
- B. A. Rozenberg and R. Tenne, Prog. Polym. Sci., 2008, 33, 40–112 CrossRef CAS.
- K. Christodoulakis, D. Palioura, S. Anastasiadis and M. Vamvakaki, Top. Catal., 2009, 52, 394–411 CrossRef CAS.
- R. T. Ragheb and J. S. Riffle, Polymer, 2008, 49, 5397–5404 CrossRef CAS.
- J. R. Morones and W. Frey, Langmuir, 2007, 23, 8180–8186 CrossRef CAS.
- J. Garcia-Serrano, U. Pal, A. M. Herrera, P. Salas and C. Angeles-Chavez, Chem. Mater., 2008, 20, 5146–5153 CrossRef CAS.
- R. A. Wassel, B. Grady, R. D. Kopke and K. J. Dormer, Colloids Surf., A, 2007, 292, 125–130 CrossRef CAS.
- J. M. Tam, J. O. Tam, A. Murthy, D. R. Ingram, L. L. Ma, K. Travis, K. P. Johnston and K. V. Sokolov, ACS Nano, 2010, 4, 2178–2184 CrossRef CAS.
- L. Hosta-Rigau, I. Olmedo, J. Arbiol, L. J. Cruz, M. J. Kogan and F. Albericio, Bioconj. Chem., 2010 Search PubMed.
- Y. Habibi and A. Dufresne, Biomacromolecules, 2008, 9, 1974–1980 CrossRef CAS.
- F. He, J. Liu, C. B. Roberts and D. Zhao, Ind. Eng. Chem. Res., 2009, 48, 6550–6557 CrossRef CAS.
- F. He and D. Zhao, Environ. Sci. Technol., 2007, 41, 6216–6221 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Biomacromolecules, 2007, 8, 2762–2767 CrossRef CAS.
- J. Liu, F. He, T. M. Gunn, D. Zhao and C. B. Roberts, Langmuir, 2009, 25, 7116–7128 CrossRef CAS.
- J. Liu, J. Sutton and C. B. Roberts, J. Phys. Chem. C, 2007, 111, 11566–11576 CrossRef CAS.
- G. Naja, A. Halasz, S. Thiboutot, G. Ampleman and J. Hawari, Environ. Sci. Technol., 2008, 42, 4364–4370 CrossRef CAS.
- A. Zhu, L. Yuan, W. Jin, S. Dai, Q. Wang, Z. Xue and A. Qin, Acta Biomater., 2009, 5, 1489–1498 CrossRef CAS.
- D. Wei, W. Qian, Y. Shi, S. Ding and Y. Xia, Carbohydr. Res., 2007, 342, 2494–2499 CrossRef CAS.
- X. Liu, Q. Hu, Z. Fang, X. Zhang and B. Zhang, Langmuir, 2009, 25, 3–8 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- W. Wang and H. Cui, J. Phys. Chem. C, 2008, 112, 10759–10766 CrossRef CAS.
- H. Huang, Q. Yuan and X. Yang, Colloids Surf., B, 2004, 39, 31–37 CrossRef CAS.
- E. Kharlampieva, J. M. Slocik, T. Tsukruk, R. R. Naik and V. V. Tsukruk, Chem. Mater., 2008, 20, 5822–5831 CrossRef CAS.
- A. Murugadoss, P. Goswami, A. Paul and A. Chattopadhyay, J. Mol. Catal. A: Chem., 2009, 304, 153–158 CrossRef CAS.
- Y. Wang, Y. F. Li and C. Z. Huang, J. Phys. Chem. C, 2009, 113, 4315–4320 CrossRef CAS.
- W. Jiang, H. C. Yang, S. Y. Yang, H. E. Horng, J. C. Hung, Y. C. Chen and C.-Y. Hong, J. Magn. Magn. Mater., 2004, 283, 210–214 CrossRef CAS.
- D. Philip, Spectrochim. Acta, Part A, 2009, 73, 650–653 CrossRef.
- A. Niu, Y. Han, J. Wu, N. Yu and Q. Xu, J. Phys. Chem. C, 2010, 114, 12728–12735 CrossRef CAS.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS.
- P. Raveendran, J. Fu and S. L. Wallen, J. Am. Chem. Soc., 2003, 125, 13940–13941 CrossRef CAS.
- P. Raveendran, J. Fu and S. L. Wallen, Green Chem., 2006, 8, 34–38 RSC.
- X. Ma, T. Jiang, B. Han, J. Zhang, S. Miao, K. Ding, G. An, Y. Xie, Y. Zhou and A. Zhu, Catal. Commun., 2008, 9, 70–74 CrossRef CAS.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64–82 RSC.
- T. K. Jain, M. A. Morales, S. K. Sahoo, D. L. Leslie-Pelecky and V. Labhasetwar, Mol. Pharmaceutics, 2005, 2, 194–205 CrossRef CAS.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef CAS.
- R. Nakao, H. Rhee and Y. Uozumi, Org. Lett., 2005, 7, 163–165 CrossRef CAS.
- T. S. Huang, Y. H. Wang, J. Y. Jiang and Z. L. Jin, Chin. Chem. Lett., 2008, 19, 102–104 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Cryst. Growth Des., 2008, 8, 291–295 CrossRef CAS.
- S. Sawoo, D. Srimani, P. Dutta, R. Lahiri and A. Sarkar, Tetrahedron, 2009, 65, 4367–4374 CrossRef CAS.
- D. Li, Q. He and J. Li, Adv. Colloid Interface Sci., 2009, 149, 28–38 CrossRef CAS.
- S. N. Sidorov, L. M. Bronstein, P. M. Valetsky, J. Hartmann, H. Cölfen, H. Schnablegger and M. Antonietti, J. Colloid Interface Sci., 1999, 212, 197–211 CrossRef CAS.
- K. Babu and R. Dhamodharan, Nanoscale Res. Lett., 2009, 4, 1090–1102 CrossRef CAS.
- L. Rosenthal-Toib, K. Zohar, M. Alagem and Y. Tsur, Chem. Eng. J., 2008, 136, 425–429 CrossRef CAS.
- D. Scott, M. Toney and M. Muzikar, J. Am. Chem. Soc., 2008, 130, 865–874 CrossRef CAS.
- E. V. Rokhina, P. Lens and J. Virkutyte, Trends Biotechnol., 2009, 27, 298–306 CrossRef CAS.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS.
- I. Willner, R. Baron and B. Willner, Adv. Mater., 2006, 18, 1109–1120 CrossRef CAS.
- P. Karam, Y. Xin, S. Jaber and L. I. Halaoui, J. Phys. Chem. C, 2008, 112, 13846–13850 CrossRef CAS.
- J. L. Brennan, N. S. Hatzakis, T. R. Tshikhudo, V. Razumas, S. Patkar, J. Vind, A. Svendsen, R. J. M. Nolte, A. E. Rowan and M. Brust, Bioconjugate Chem., 2006, 17, 1373–1375 CrossRef CAS.
- A. Gole, C. Dash, C. Soman, S. R. Sainkar, M. Rao and M. Sastry, Bioconjugate Chem., 2001, 12, 684–690 CrossRef CAS.
- D. M. Eby, N. M. Schaeublin, K. E. Farrington, S. M. Hussain and G. R. Johnson, ACS Nano, 2009, 3, 984–994 CrossRef CAS.
Footnote |
| † NRC Research Associate. Present address: Pegasus Technical Services Inc., 46 East Hollister Street, Cincinnati, OH 45219-1704, USA. E-mail: E-mail: Jurate.Virkutyte@ptsied.com; Tel.: +513 365-4766 |
| This journal is © The Royal Society of Chemistry 2011 |