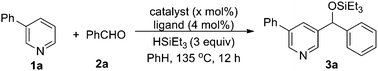DOI:
10.1039/C0SC00419G
(Edge Article)
Chem. Sci., 2011,
2, 488-493
Ir-catalyzed highly selective addition of pyridyl C–H bonds to aldehydes promoted by triethylsilane†
Received
8th August 2010
, Accepted 21st October 2010
First published on 23rd November 2010
Abstract
We report herein an iridium-catalyzed activation of pyridine C–H bond and its nucleophilic addition to benzaldehydes. The reaction was applicable to various pyridine derivatives and aryl aldehydes. In contrast to most methods that selectively functionalize the C2 pyridine C–H bond, this reaction proceeded with unusual meta-selectivity. An independently synthesized silyl iridium complex was able to catalyze the reaction, indicating that a silyl iridium intermediate might be responsible for the novel reactivity and the unusual selectivity.
Introduction
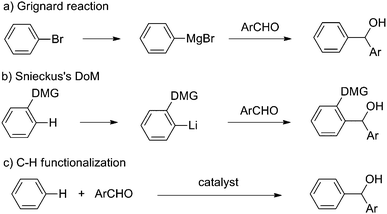
Nucleophilic addition of aryl Grignard reagents to carbonyl compounds is a fundamentally important reaction, which has been well developed and broadly utilized in alcohol synthesis.1 An inherent limitation of this traditional process is the requirement of using organohalides as the starting materials, since the preparation of organohalides commonly requires tedious procedures and causes environmental problems. As a valuable complementary method, Snieckus's directed orthometallation (DoM) employs arenes as substrates, which is advantageous from a synthetic point of view.2 However, both Grignard reaction and Snieckus's chemistry involve the manipulation of air- and moisture-sensitive main group organometallic reagents, as well as produce salt wastes.
To overcome these issues, direct utilization of aromatic C–H bonds as the surrogates of main-group organometallic reagents such as Grignard reagents would be highly appealing (Scheme 1). Despite the recent achievements made in direct C–H functionalization,3 transition-metal catalyzed direct addition of an aromatic C–H bond to a carbonyl group through C–H functionalization has rarely been reported.4–7 Murai and co-workers first reported an iridium-catalyzed coupling of imidazole with aliphatic aldehydes, in which C–H activation was not involved in the proposed mechanism.4 Later on, Takai and co-workers developed the elegant Re- and Mn-catalyzed addition of aromatic C–H bonds to aldehydes by using directing group assisted strategy.5 Recently, Shibata and co-workers described a cationic iridium-catalyzed intramolecular addition of aromatic C–H bonds to ketones, which provided an efficient method to synthesize useful benzofurans and indoles.6 Despite these significant achievements, the limited substrate scope, restrained ortho selectivity, and/or the requirement of a directing group substantially hampered the practicability of these novel reactions. Therefore, the development of an efficient catalytic system that enables diversified substrate scope and different selectivity would be of profound interest in this area.
Pyridine is an important structural unit and widely exists in natural products and medicinally relevant compounds.8 Consequently, a diverse range of catalytic methods have been developed to directly functionalize pyridine derivatives through C–H activation.9 However, most of these functionalization processes occurred regioselectively at the C2 position. Although a number of groups reported intramolecular C3 and C4 arylation of pyridine derivatives,10a to the best of our knowledge, the intermolecular C–H activation/C–C formation at C3 or C4 position of pyridine without activating or directing groups has never been achieved.10,11 Herein, we report the first example of an Ir-catalyzed nucleophilic addition of pyridine derivatives to aldehydes through C–H activation with unusual meta selectivity (Scheme 2).12 From a synthetic point of view, a meta-selective addition of pyridine to aldehydes is highly desirable since metallation of pyridine with main-group and early transition metals (Li, Mg, Mn, etc) always occurs at the ortho-position, and a meta-metallation is notoriously difficult.2e
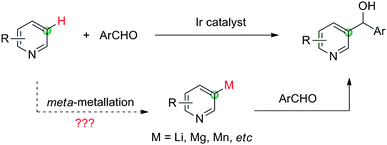 |
| | Scheme 2
Meta-selective functionalization of pyridine. | |
Results and discussion
Initial studies
3-Phenylpyridine
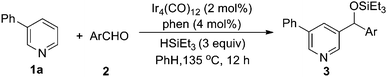
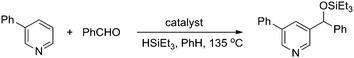
1a was first selected to explore the suitable reaction conditions (Table 1). Although several metal complexes, including Mn, Re, Ru and Rh, showed good reactivities in many direct C–H transformations,3 they completely failed after many trials (entries 1–5). To our delight, an iridium complex [Ir(COD)Cl]2 promoted the addition of the meta pyridyl C–H bond to benzaldehyde in the presence of triethylsilane in benzene (entry 6). The product was obtained as a triethylsilyl-protected benzyl alcohol. Other iridium complexes such as [Ir(COD)(OMe)]2 and [Cp*IrCl2]2, however, gave no product at all (entries 7 and 8). To our satisfaction, an iridium carbonyl complex Ir4(CO)12 remarkably improved the efficiency (entry 9). Various ligands were screened to further improve the yield. Although phosphines completely inhibited the reaction, the addition of chelating N-ligands was beneficial for this transformation (entries 10–15). When phenanthroline was used, the desired product was obtained in 73% yield as a single regioisomer (entry 12). The structure of the product 3a was confirmed by independent synthesis as well as by X-ray crystallography of the p-bromobenzoyl ester of the deprotected alcohol (Scheme 3, Fig. 1). Moreover, this reaction was very sensitive to the solvent. For example, the transformation could proceed in o-xylene and dioxane in lower yields but gave no product in more polar solvents such as DMF (entries 16–18). Different silanes also affected the yields dramatically, as the use of iPr3SiH, tBuMe2SiH, PhMe2SiH and Et2MeSiH instead of Et3SiH gave 0, 12, 29 and 61% yields, respectively.
Table 1 Optimization studya
| Entry |
Catalyst (mol%) |
Ligand |
Solvent
|
Yieldb (%) |
|
Reaction conditions: 0.25 mmol of 1a, 0.75 mmol of 2a, 0.75 mmol of HSiEt3, indicated amount of catalyst, 0.01 mmol of ligand in 1 mL solvent at 135 °C for 12 h.
Isolated yields. bpy = 2,2′-bipyridine; dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine; phen = 1,10-phenanthroline; neocuproine = 2,9-dimethyl-1,10-phenanthroline; bathophen = 4,7-diphenyl-1,10-phenanthroline; DMF = N,N-dimethylformamide.
|
| 1 |
— |
— |
Benzene
|
0 |
| 2 |
Mn2(CO)10 (4) |
— |
Benzene
|
0 |
| 3 |
Re2CO)10 (4) |
— |
Benzene
|
0 |
| 4 |
Ru3(CO)12 (3) |
— |
Benzene
|
<5 |
| 5 |
[Rh(COD)Cl]2 (4) |
— |
Benzene
|
<5 |
| 6 |
[Ir(COD)Cl]2 (4) |
— |
Benzene
|
14 |
| 7 |
[Ir(COD)(OMe)]2 (4) |
— |
Benzene
|
<5 |
| 8 |
[Cp*IrCl2]2 (4) |
— |
Benzene
|
<5 |
| 9 |
Ir4(CO)12 (2) |
— |
Benzene
|
45 |
| 10 |
Ir4(CO)12 (2) |
bpy
|
Benzene
|
65 |
| 11 |
Ir4(CO)12 (2) |
dtbpy
|
Benzene
|
70 |
|
12
|
Ir4(CO)12 (2) |
phen
|
Benzene
|
73
|
| 13 |
Ir4(CO)12 (2) |
neocuproine
|
Benzene
|
59 |
| 14 |
Ir4(CO)12 (2) |
bathophen |
Benzene
|
58 |
| 15 |
Ir4(CO)12 (2) |
PPh3 |
Benzene
|
<5 |
| 16 |
Ir4(CO)12 (2) |
phen
|
o-Xylene
|
56 |
| 17 |
Ir4(CO)12 (2) |
phen
|
Dioxane
|
57 |
| 18 |
Ir4(CO)12 (2) |
phen
|
DMF
|
<5 |
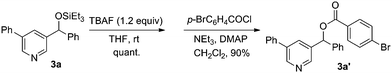 |
| | Scheme 3 Determination of the structure 3a. | |
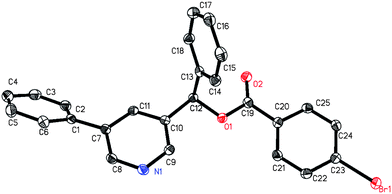 |
| | Fig. 1 ORTEP diagram of 3a′ with 50% thermal ellipsoids. | |
Synthetic scope
With the optimized condition, we next studied the scope of the aryl aldehydes (Table 2).‡ The reactions of various ortho and para substituted aryl aldehydes gave good yields (entries 2–6). Aryl halides including fluoride, chloride and even bromide were compatible with the reaction conditions, despite their tendency to undergo silylation or dehalogenation reactions (entries 7–9).13 Heteroaromatic aldehydes 2j–2l were also suitable substrates (entries 10–12). In general, electron-rich aromatic aldehydes (2f, 2j–2l) gave lower yields, presumably due to their lower electrophilicity. Aliphatic aldehyde (2m) exhibited poor reactivity since it is more electron-rich (entry 13). Conducting the reaction at 2.5 mmol scale did not affect the yield (entry 1).
Table 2 Scope of aryl aldehydesa
| Entry |
Ar |
Product |
Yieldb (%) |
|
Reaction conditions: 0.25 mmol of 1, 0.75 mmol of aldehyde, 0.75 mmol of HSiEt3, 0.005 mmol of Ir4(CO)12, 0.01 mmol of phen in 1 mL of benzene at 135 °C for 12 h.
Isolated yields.
2.5 mmol scale.
|
| 1 |
C6H5 |
2a
|
3a
|
73 (72)c |
| 2 |
2-MeC6H4 |
2b
|
3b
|
78 |
| 3 |
2,5-Me2C6H3 |
2c
|
3c
|
77 |
| 4 |
2-Naphthyl
|
2d
|
3d
|
72 |
| 5 |
4-MeC6H4 |
2e
|
3e
|
76 |
| 6 |
4-MeOC6H4 |
2f
|
3f
|
57 |
| 7 |
4-FC6H4 |
2g
|
3g
|
74 |
| 8 |
4-ClC6H4 |
2h
|
3h
|
62 |
| 9 |
4-BrC6H4 |
2i
|
3i
|
60 |
| 10 |
2-Benzofuryl
|
2j
|
3j
|
55 |
| 11 |
2-Benzothiophene
|
2k
|
3k
|
54 |
| 12 |
3-Thiophene
|
2l
|
3l
|
68 |
| 13 |
n-Hexyl
|
2m
|
3m
|
<10 |
The catalytic system was further applied to functionalize a variety of pyridine derivatives (Table 3). Various 3-arylpyridines (1a–1c) and 3-alkylpyridines (1d, 1e) reacted quite well. However, more electron-donating groups such as methoxyl and benzyloxyl substituents (1f, 1g) slightly decreased the reaction efficiency. To our interest, isoquinoline (1i) showed much better reactivity than quinoline (1h). In all cases, selective functionalization at the meta position of the nitrogen atom was exclusively obtained. When unsubstituted pyridine (1j) was tested, a mixture of mono- and di-functionalized products at meta-position was obtained, indicating that such a meta selectivity was controlled by the intrinsic feature of pyridine rather than the substituent. Although the reaction efficiency of 3,4-disubstituted pyridine (1k) was decreased by the substituent at the C4 position, this steric effect did not completely inhibit the reaction. This stood in contrast with the well known meta-selective C–H borylation and silylation reactions, in which steric bias dominated the regioselectivity.14,15 For reasons we do not fully understand, the reactions of 2-substituted pyridines such as 2-methylpyridine and 2,6-lutidine gave low yields (<5%) of products.
| Entry |
1
|
Product |
Yieldb (%) |
|
Reaction conditions: 0.25 mmol of 1, 0.75 mmol of benzaldehyde, 0.75 mmol of HSiEt3, 0.005 mmol of Ir4(CO)12, 0.01 mmol of phen in 1 mL of benzene at 135 °C for 12 h.
Isolated yields.
|
| 1 |

|

|
3a, 73 |
| 2 |
3n, 68 |
| 3 |
3o, 66 |
| 4 |
3p, 61 |
| 5 |
3q, 66 |
| 6 |
3r, 53 |
| 7 |
3s, 55 |
| |
| 8 |

|

|
3t, 27 |
| |
| 9 |

|

|
3u, 62 |
| |
| 10 |

|

|
3v, 37 |
|
3v′, 21 |
| |
| 11 |

|

|
3w, 31 |
Mechanistic studies
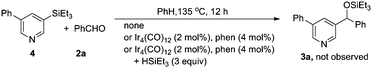
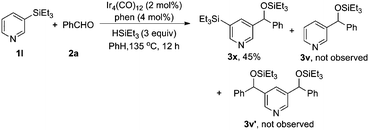
In order to gain insight into this novel reaction, mechanistic studies were carried out. We found that, (1) trialkylsilane was essential for the reaction, as no reaction occurred in its absence; (2) C–H silylation of the pyridine derivatives could not be detected at various stages of the reaction (eqn (1)). However, it could be detected in the absence of benzaldehyde, albeit in a very low yield. When norbornene or tert-butylethylene was added instead of benzaldehyde, these hydrogen acceptors did not promote the efficiency of the C–H silylation(eqn (2)).16 Thus, it is unlikely that benzaldehyde functioned as a hydrogen scavenger for the C–H silylation reaction; (3) the nucleophilic addition of the independently synthesized 3-phenyl-5-(triethylsilyl)pyridine 4 to benzaldehyde could not occur either directly or in the presence of iridium catalyst(eqn (3)); (4) when 3-triethylsilylpyridine was subjected to the standard condition, the desired coupling product was obtained. However, the product resulting from the addition of arylsilane to benzaldehyde was not detected at all (eqn (4)). This suggested that the active catalyst for the addition of a C–H bond to aldehyde was not capable to promote the addition of pyridylsilane to the aldehyde. Together, these results showed that the efficiency of the catalytic C–H silylation process was low,16 and the reaction of pyridylsilane with aldehyde was infeasible.17 Therefore, a reaction mechanism involving C–H silylation followed by the addition of the resulting silane to aldehyde is unlikely in the current catalytic system.| |  | (1) |
| | 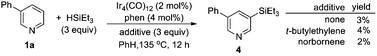 | (2) |
| |  | (3) |
| |  | (4) |
One possible explanation of the meta selectivity is the migration of the initially formed C(2)–Ir bond to the C3 position. To see whether the C–H bond at the ortho position was involved in the reaction, we synthesized the D-labeled 3-phenylpyridine and subjected it to the standard reaction condition (eqn (5)). It turned out that there was no significant loss of deuterium at the C2 position, suggesting that the ortho C–H bond was not cleaved during the reaction. Thus, the migration of the iridium from C2 to C3 is unlikely.
| |  | (5) |
It is well known that in the meta-selective aromatic C–H borylation reactions, the bulky boryl metal complex was responsible for the regioselectivity.14 Analogously, we assumed that a similar silyl iridium complex might be the key intermediate. Therefore, we synthesized a silyl iridium complex according to a recent elegant report by Tilley and co-workers (Scheme 4).18a
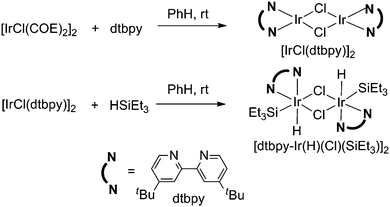 |
| | Scheme 4 Preparation of a silyl iridium complex | |
However, the direct application of this silyl iridium complex to the reaction showed no catalytic activity at all (entry 6, Table 4). This was not surprising since a combination of [Ir(COE)2Cl]2 and dtbpy did not catalyze the reaction either (entry 5). Comparing with Ir4(CO)12, [Ir(COE)2Cl]2 lacked the CO ligand and has an additional Cl− ligand. To mimic the standard reaction conditions, we carried out the silyl iridium complex catalyzed reactions either under CO atomosphere (entry 7) or in the presence of silver salt as the halide scavenger (entries 8 and 9). Although an CO atomosphere was not beneficial, the addition of silver triflate successfully promoted the silyl iridium catalyzed reaction in 35% isolated yield (entry 9). This result suggested that a silyl iridium complex might be involved in the reaction mechanism.
Table 4
Catalytic reactions using the silyl iridium complexa
| Entry |
Catalyst
|
Isolated yield (%) |
|
Reaction conditions: 0.25 mmol of 1, 0.75 mmol of benzaldehyde, 0.75 mmol of HSiEt3, indicated amount of catalyst in 1 mL of benzene at 135 °C for 12 h.
|
| 1 |
2 mol% Ir4(CO)12 |
45 |
| 2 |
2 mol% Ir4(CO)12 + 8 mol% dtbpy |
60 |
| 3 |
4 mol% [Ir(COD)Cl]2 |
14 |
| 4 |
4 mol% [Ir(COE)2Cl]2 |
5 |
| 5 |
4 mol% [Ir(COE)2Cl]2 + 8 mol% dtbpy |
0 |
| 6 |
4 mol% [(dtbpy)Ir(H)(Cl)(SiEt3)]2 |
0 |
| 7 |
4 mol% [(dtbpy)Ir(H)(Cl)(SiEt3)]2 + CO (1 atm) |
Trace |
| 8 |
4 mol% [(dtbpy)Ir(H)(Cl)(SiEt3)]2 + 8 mol% AgBF4 |
Trace |
| 9 |
4 mol% [(dtbpy)Ir(H)(Cl)(SiEt3)]2 + 8 mol% AgOTf |
35 |
Based on these results, a catalytic mechanism was proposed in Scheme 5. First, an active silyl iridium catalyst 5, which might contain CO, phenanthroline and/or silyl group as ligands, was generated from the combination of Ir precursor, ligand and silane.18 Oxidative addition of the pyridyl C–H bond to the low-valent Ir species produced the key intermediate 6. Instead of an unfavorable C–Si bond formation by reductive elimination, the intermediate was intercepted by arylaldehyde through C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O insertion into the Ir–Si bond,19 affording the pyridyl alkyl iridium species 7. Finally, C–C formation through reductive elimination led to the product 3, along with the generation of an iridium hydride species 8, which reacted with hydrosilane to regenerate the active iridium catalyst. According to the proposed mechanism, several observations could be rationalized. First, hydrosilane was necessary for the reaction since it was required for the formation of the active silyl iridium intermediate in the catalytic cycle.18 Second, the meta selectivity was induced from the oxidative addition of the pyridyl C–H bond to the iridium silyl species. Such a regioselectivity has been observed in several Ir-catalyzed C–H borylation reactions, in which iridium boryl complexes are similarly responsible for the selectivity.11 It should be pointed out that this analogy might not always hold true since borylation of pyridine at the C2 or C4 position was also observed.11b,c Finally, the silyl group in the intermediate 6 is oxophilic, thereby promoting the insertion of the benzaldehyde through the formation of a strong Si–O bond.19 Thus, the insertion of benzaldehyde to a less reactive C–Ir bond (intermediate 7′) followed by O–Si forming reductive elimination might be unfavorable.
O insertion into the Ir–Si bond,19 affording the pyridyl alkyl iridium species 7. Finally, C–C formation through reductive elimination led to the product 3, along with the generation of an iridium hydride species 8, which reacted with hydrosilane to regenerate the active iridium catalyst. According to the proposed mechanism, several observations could be rationalized. First, hydrosilane was necessary for the reaction since it was required for the formation of the active silyl iridium intermediate in the catalytic cycle.18 Second, the meta selectivity was induced from the oxidative addition of the pyridyl C–H bond to the iridium silyl species. Such a regioselectivity has been observed in several Ir-catalyzed C–H borylation reactions, in which iridium boryl complexes are similarly responsible for the selectivity.11 It should be pointed out that this analogy might not always hold true since borylation of pyridine at the C2 or C4 position was also observed.11b,c Finally, the silyl group in the intermediate 6 is oxophilic, thereby promoting the insertion of the benzaldehyde through the formation of a strong Si–O bond.19 Thus, the insertion of benzaldehyde to a less reactive C–Ir bond (intermediate 7′) followed by O–Si forming reductive elimination might be unfavorable.
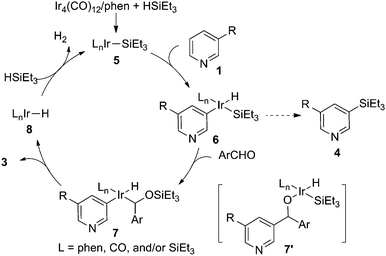 |
| | Scheme 5 Proposed mechanism. | |
Conclusions
In conclusion, we have developed an unprecedented Ir-catalyzed meta selective C–H activation of pyridine derivatives and subsequent nucleophilic addition to benzaldehyde promoted by hydrosilane. In this reaction, the unfunctionalized pyridine was used as the Grignard reagent surrogate. Mechanistic studies indicated that a silyl iridium complex might be the active catalytic intermediate. The application of chelating N-ligands provided an opportunity for asymmetric synthesis. Expansion of the substrate scope and further understanding of the reaction mechanism are currently underway in our laboratory.
Acknowledgements
Support of this work by NSFC (No. 20672006, 20821062, 20832002, 20925207, GZ419) and the “973” Project from the MOST of China (2009CB825300) is gratefully acknowledged.
Notes and references
-
(a) V. Grignard and C. R. Hebd, C. R. Seances Acad. Sci., 1900, 130, 1322 Search PubMed;
(b) H. Shinokubo and K. Oshima, Eur. J. Org. Chem., 2004, 2081 CrossRef CAS;
(c)
Grignard Reagents: New Developments, ed. H. G. Richey, Wiley, New York, 2000 Search PubMed.
-
(a) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS;
(b) M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206 CrossRef CAS;
(c) C. J. Rohbogner, G. C. Clososki and P. Knochel, Angew. Chem., Int. Ed., 2008, 47, 1503 CrossRef CAS;
(d) S. H. Wunderlich, M. Kienle and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 7256 CrossRef CAS;
(e) M. Jaric, B. A. Haag, A. Unsinn, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 5451 CrossRef CAS.
-
(a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS;
(b) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826 CrossRef CAS;
(c) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS;
(d) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS;
(e) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS;
(f) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS;
(g) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS;
(h) T. Satoh and M. Miura, Chem. Lett., 2007, 36, 200 CrossRef CAS;
(i) L. C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35;
(j) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC;
(k) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS;
(l) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013 CrossRef CAS;
(m) X. Chen, K. M. Engle, D. H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS;
(n) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS;
(o) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS;
(p) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS;
(q) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS;
(r) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS;
(s) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS;
(t) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC;
(u) R. Jazzar, H. Julien, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS.
- Y. Fukumoto, K. Sawada, M. Hagihara, N. Chatani and S. Murai, Angew. Chem., Int. Ed., 2002, 41, 2779 CrossRef.
-
(a) Y. Kuninobu, Y. Nishina, C. Nakagawa and K. Takai, J. Am. Chem. Soc., 2006, 128, 12376 CrossRef CAS;
(b) Y. Kuninobu, Y. Nishina and K. Takai, Tetrahedron, 2007, 63, 8463 CrossRef CAS;
(c) Y. Kuninobu, Y. Fujii, T. Matsuki, Y. Nishina and K. Takai, Org. Lett., 2009, 11, 2711 CrossRef CAS;
(d) Y. Kuninobu, Y. Nishina, T. Takeuchi and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 6518 CrossRef CAS.
- K. Tsuchikama, Y. Hashimoto, K. Endo and T. Shibata, Adv. Synth. Catal., 2009, 351, 2850 CrossRef CAS.
- For the addition of C–H bonds to other polar bonds:
(a) W. D. Jones, G. P. Foster and J. M. Putinas, J. Am. Chem. Soc., 1987, 109, 5047 CrossRef;
(b) C. Zhou and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 2302 CrossRef CAS;
(c) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS;
(d) Y. Kuninobu, Y. Inoue and K. Takai, Chem. Lett., 2006, 35, 1376 CrossRef CAS;
(e) Y. Kuninobu, K. Kikuchi and K. Takai, Chem. Lett., 2008, 37, 740 CrossRef CAS;
(f) B. Qian, S. Guo, J. Shao, Q. Zhu, L. Yang, C. Xia and H. Huang, J. Am. Chem. Soc., 2010, 132, 3650 CrossRef CAS;
(g) I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858 CrossRef CAS.
-
(a) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627 RSC;
(b) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC;
(c)
G. Jones, Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees, E. F. V. Scriven and A. McKillop, Pergamon, Oxford, 1996, vol. 5, p. 167 Search PubMed.
-
(a) R. F. Jordan and D. F. Taylor, J. Am. Chem. Soc., 1989, 111, 778 CrossRef CAS;
(b) E. J. Moore, W. R. Pretzer, T. J. O. Connell, J. Harris, L. LaBounty, L. Chou and S. S. Grimmer, J. Am. Chem. Soc., 1992, 114, 5888 CrossRef CAS;
(c) L. S. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS;
(d) J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332 CrossRef CAS;
(e) A. Larive, J. J. Mousseau and A. B. Charette, J. Am. Chem. Soc., 2008, 130, 52 CrossRef CAS;
(f) Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448 CrossRef CAS;
(g) S. H. Cho, S. J. Hwang and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254 CrossRef CAS;
(h) A. M. Berman, J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 14926 CrossRef CAS;
(i) L. C. Campeau, D. R. Stuart, J. P. Leclerc, M. Bertrand-Laperle, E. Villemure, H. Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef;
(j) M. Tobisu, I. Hyodo and N. Chatani, J. Am. Chem. Soc., 2009, 131, 12070 CrossRef CAS;
(k) X. J. Wu, L. Cui, G. Chen and Y. Jiang Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef CAS;
(l) J. J. Mousseau, J. A. Bull and A. B. Charette, Angew. Chem., Int. Ed., 2010, 49, 1115 CrossRef CAS;
(m) J. Barluenga, G. Lonzi, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2010, 132, 13200 CrossRef CAS.
- For intramolecular arylation of pyridine at meta position:
(a) J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20 Search PubMed, and references therein. For intermolecular arylation and alkenylation of fluoropyridine at C3 or C4 position:
(b) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754 CrossRef;
(c) M. Lafrance, D. Shore and K. Fagnou, Org. Lett., 2006, 8, 5097 CrossRef CAS;
(d) H. Q. Do and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 1128 CrossRef CAS;
(e) Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 16170 CrossRef CAS. For directed arylation of pyridine derivatives:
(f) M. Wasa, B. T. Worrell and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 1275 CrossRef CAS. After submission of this manuscript, two papers describing C4-selective C–H activation of pyridines appeared:
(g) C.-C. Tsai, W.-C. Shih, C.-.H. Fang, T.-G. Ong and G. P. A. Yap, J. Am. Chem. Soc., 2010, 132, 11887 CrossRef CAS;
(h) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666 CrossRef CAS.
- For examples of borylation of pyridine at the meta position:
(a) J. Takagi, K. Sato, J. F. Hartwig, T. Ishiyama and N. Miyaura, Tetrahedron Lett., 2002, 43, 5649 CrossRef CAS;
(b) I. A. I. Mkhalid, D. N. Coventry, D. Albesa-Jove, A. S. Batsanov, J. A. K. Howard, T. B. Marder and R. N. Perutz, Angew. Chem., Int. Ed., 2006, 45, 489 CrossRef CAS;
(c) J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS;
(d) D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926 CrossRef CAS;
(e) T. E. Hurst, T. K. Macklin, M. Becker, E. Hartmann, W. Kugel, J.-C. Parisienne-La Salle, A. S. Batsanov, T. B. Marder and V. Snieckus, Chem.–Eur. J., 2010, 16, 8155 CAS.
- For meta-selective functionalization of arenes:
(a) J.-W. Park and C.-H. Jun, ChemCatChem, 2009, 1, 69 Search PubMed;
(b) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS;
(c) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 5072 CrossRef CAS;
(d) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904 CrossRef CAS;
(e) B.-J. Li, S.-L. Tian, Z. Fang and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115 CrossRef CAS;
(f) D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072 CrossRef CAS;
(g) W. Yue, Y. Li, W. Jiang, Y. Zhen and Z. Wang, Org. Lett., 2009, 11, 5430 CrossRef CAS;
(h) S. Potavathri, K. C. Pereira, S. I. Gorelsky, A. Pike, A. P. LeBris and B. DeBoef, J. Am. Chem. Soc., 2010, 132, 14676 CrossRef CAS.
- Y. Yamanoi and H. Nishihara, J. Org. Chem., 2008, 73, 6671 CrossRef CAS.
-
(a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS;
(b) J.-Y. Cho, C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 2000, 122, 12868 CrossRef CAS;
(c) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Jr.Maleczka and M. R. Smith III, Science, 2001, 295, 305;
(d) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS;
(e) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS;
(f) T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263 CrossRef CAS.
-
(a) T. Ishiyama, K. Sato, Y. Nishio and N. Miyaura, Angew. Chem., Int. Ed., 2003, 42, 5346 CrossRef CAS;
(b) T. Saiki, Y. Nishio, T. Ishiyama and N. Miyaura, Organometallics, 2006, 25, 6068 CrossRef CAS.
- A high-yielding C–H silylation of pyridine has not been achieved yet. Instead, pyridine could be used as the directing group for C–H silylation:
(a) F. Kakiuchi, M. Matsumoto, K. Tsuchiya, K. Igi, T. Hayamizu, N. Chatani and S. Murai, J. Organomet. Chem., 2003, 686, 134 CrossRef CAS;
(b) F. Kakiuchi, K. Tsuchiya, M. Mstsumoto, E. Mizushima and N. Chatani, J. Am. Chem. Soc., 2004, 126, 12792 CrossRef CAS;
(c) M. Tobisu, Y. Ano and N. Chatani, Chem.–Asian J., 2008, 3, 1585 CrossRef CAS.
- The catalytic addition of arylsilane to aldehyde typically required activating reagent for the silicon center and basic conditions:
(a) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS;
(b) S. Oi, M. Moro and Y. Inoue, Organometallics, 2001, 20, 1036 CrossRef CAS;
(c) T. Fujii,, T. Koike, A. Mori and K. Osakada, Synlett, 2002, 298 CrossRef CAS;
(d) R. Lerebours and C. Wolf, J. Am. Chem. Soc., 2006, 128, 13052 CrossRef CAS;
(e) M. Murata, R. Shimazaki, M. Ishikura, S. Watanabe and Y. Masuda, Synthesis, 2002, 717 CrossRef CAS.
-
(a) J. L. McBee and T. D. Tilley, Organometallics, 2009, 28, 5072 CrossRef CAS. For a review, see:
(b) J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1999, 99, 175 CrossRef CAS.
-
(a) D. C. Apple, K. A. Brady, J. M. Chance, N. E. Heard and T. A. Nile, J. Mol. Catal., 1985, 29, 55 CrossRef CAS;
(b) I. Ojima, T. Kogure and M. Kumagai, J. Org. Chem., 1977, 42, 1671 CrossRef CAS, and references therein.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization of new compounds. CCDC reference number 773152. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00419g |
| ‡ General procedure for the catalytic addition of pyridine derivatives to aryl aldehydes: To an oven-dried 25 mL Schlenk tube was added Ir4(CO)12 (5.5 mg, 0.005 mmol) and phenanthroline (1.8 mg, 0.01 mmol). The tube was evacuated and refilled with nitrogen. Then 3-phenylpyridine (38.8 mg, 0.25 mmol), benzaldehyde (79.5 mg, 0.75 mmol) and triethylsilane (87.0 mg, 0.75 mmol) were successively added via syringes under a positive stream of dry nitrogen. After the addition of 1 mL of benzene, the tube was tightly sealed and heated to 135 °C in an oil-bath for 12 h. After cooling to room temperature, the resulting mixture was directly purified by flash chromatography on silica gel eluting with ethyl acetate and petroleum ether. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.