DOI:
10.1039/C0SC00498G
(Edge Article)
Chem. Sci., 2011,
2, 544-551
Ru-catalyzed activation of sp3 C–O bonds: O- to N-alkyl migratory rearrangement in pyridines and related heterocycles†
Received
23rd September 2010
, Accepted 25th October 2010
First published on 10th December 2010
Abstract
We report a novel strategy for the formal activation of sp3 C–O bonds under Ru catalysis. In this reaction, an O-alkylpyridine undergoes migratory rearrangement to its corresponding N-alkylpyridone via coordination of the Lewis basic N atom. This transformation represents the first generalcatalytic approach to O- to N-alkyl migration in N-containing heterocycles. Extension of this methodology to other heterocycles, including O-alkylpyridazines and O-alkylazoles, was achieved.
Introduction
The transition metal catalyzed activation of σ bonds provides a general strategy to build molecular complexity from simple starting materials. Oxidative addition of electron-deficient bonds, such as C–X, C–N2, and C–OSO2R, is well known;1 however, direct insertion into electron-rich bonds, such as C–H, C–C, and C–OR, remains a significant challenge.2–4 Notably, ethereal C–O bonds are typically inert, unlike the C–O bonds of sulfates, esters, carbonates, and carbamates, which are active in cross-couplings5 and π-allyl chemistry.6 Ethereal variants of these reactions are quite rare.4 To this end, Kakiuchi and co-workers reported a unique strategy for sp2 C–O bond activation in ethers using a ketone directing group.7 Herein, we report a complementary method for sp3 ethereal C–O bond activation in alkoxypyridines and related heterocycles (Fig. 1).
A brief survey of the literature reveals a lack of general methods for the preparation of N-alkylated heterocycles (e.g., N-alkylpyridones),8 important structural motifs found in natural products9 and medicinal targets.10 Direct alkylation of aromatic imidates is not ideal due to competing O- and N-alkylation because of their ambivalent character.11 One attractive solution to this problem is the O- to N-alkyl migratory rearrangement reaction, a practical approach because O-alkyl heterocycles can be easily synthesized via nucleophilic aromatic substitution. Current methods for O- to N-alkyl migration use stoichiometric amounts of LiI12 or NaCl;13catalytic variants are limited to rearrangements of O-allylN-heterocycles promoted by Pd and Pt salts.14 We propose a complementary strategy involving two distinct mechanistic steps: 1) oxidative addition of an sp3 C–O bond to generate a metal imidate, and 2) reductive elimination to furnish an sp3 C–N bond.
Results and discussion
Initial studies
Our initial studies confirmed that most transition metals, including Pd, Ni, Rh, and Fe were ineffective in promoting the desired O- to N-alkyl migration reaction in 2-benzoxypyridine (1a) (Table 1, entries 1–4).‡ Surprisingly, Rucatalysts known to undergo oxidative addition into C–O bonds7 were unreactive (entries 5–6); to our delight, [Ru(p-cymene)Cl2]2 catalyzed the rearrangement efficiently (entry 7). The difference in reactivity may be attributed to the presence of the CO ligand. Substitution of the solvent by THF was tolerated, although DCE resulted in decreased reaction yields (entries 8–9), as did the use of other ligands (entries 10–13). The use of strong bases (e.g., NaOtBu, entry 15) or organic bases (e.g., NEt3) led to decreased reactivity.
Table 1 Reaction optimization
| Entrya |
Cat. [M]/ligandb |
Base
|
Solv. |
Yield (%)c |
|
Reaction conditions: Substrate, 0.1 mmol; catalyst, 10 mol% [M], ligand, 10 mol%, base, 0.11 mmol, solvent, 1 mL, 80 °C, 24 h.
Arene = p-cymene.
Conversion by 1H NMR; isolated yield in parenthesis.
Ligand, 5 mol%.
|
| 1 |
Pd(PPh3)4
|
K2CO3
|
PhMe
|
0 |
| 2 |
Ni(COD)2
|
K2CO3
|
PhMe
|
0 |
| 3 |
Fe(CO)5
|
K2CO3
|
PhMe
|
0 |
| 4 |
RhCl(PPh3)3
|
K2CO3
|
PhMe
|
0 |
| 5 |
Ru3(CO)12
|
K2CO3
|
PhMe
|
0 |
| 6 |
RuH2(CO)(PPh3)3
|
K2CO3
|
PhMe
|
0 |
| 7 |
[Ru(arene)Cl2]2/PPh3 |
K2CO3
|
PhMe
|
100 (91) |
| 8 |
[Ru(arene)Cl2]2/PPh3 |
K2CO3
|
THF
|
100 |
| 9 |
[Ru(arene)Cl2]2/PPh3 |
K2CO3
|
DCE
|
19 |
| 10 |
[Ru(arene)Cl2]2/PCy3 |
K2CO3
|
PhMe
|
1 |
| 11 |
[Ru(arene)Cl2]2/P(OPh)3 |
K2CO3
|
PhMe
|
0 |
| 12 |
[Ru(arene)Cl2]2/dpppd |
K2CO3
|
PhMe
|
4 |
| 13 |
[Ru(arene)Cl2]2/bpyd |
K2CO3
|
PhMe
|
0 |
| 14 |
[Ru(arene)Cl2]2/PPh3 |
K3PO4
|
PhMe
|
85 |
| 15 |
[Ru(arene)Cl2]2/PPh3 |
NaOtBu
|
PhMe
|
0 |
| 16 |
[Ru(arene)Cl2]2/PPh3 |
NEt3
|
PhMe
|
5 |
Synthetic scope
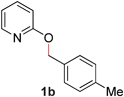
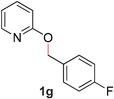
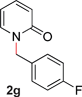
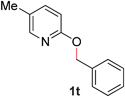
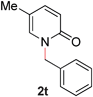
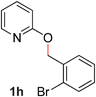
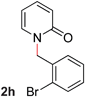
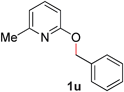
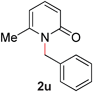
With our optimized reaction conditions, we synthesized a number of N-alkyl pyridones by catalyticO- to N-migratory rearrangement reaction. Electron-neutral, electron-deficient, and electron-rich benzyl groups were tolerated in this transformation (Table 2, entries 1–14, 2a–n, 66–99%). Halide and ester functionalities remained unchanged under our reaction conditions (entries 5–9, 11). Importantly, other ether linkages present in the molecule are preserved and the pyridine N directs selective sp3 C–O bond activation (entries 12–14). Furthermore, the presence of a bulky substituent at the ortho-position did not inhibit catalysis, although elevated temperatures were required (entry 8). Substitution on the backbone of the pyridine ring with electron-donating and electron-withdrawing groups had minimal effect on the reaction efficiency (entries 15–25, 2o–y, 85–99%). Pyridines bearing substituents at the 6-position, however, rendered the reaction unproductive—a steric effect, since both electron-donating (entry 21) and electron-withdrawing (entry 24) substituents were not tolerated. Additionally, substrates bearing secondary O-alkyl groups were completely unreactive even at higher temperatures (entry 26).
| Entrya |
Substrate |
Product |
Yield (%)b |
Entrya |
Substrate |
Product |
Yield (%)b |
|
Reaction conditions: Substrate, 0.2 mmol; [Ru(p-cymene)Cl2]2, 5 mol%; PPh3, 20 mol%; K2CO3, 0.22 mmol; PhMe, 2 mL; 24 h.
Isolated yields.
No desired product could be detected by 1H NMR spectroscopy.
|
| 1 |
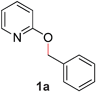
|
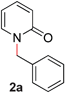
|
91 (80 °C) |
14 |
|
|
88 (90 °C) |
| 2 |
|
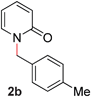
|
76 (80 °C) |
15 |
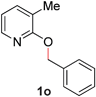
|
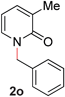
|
90 (80 °C) |
| 3 |
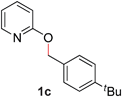
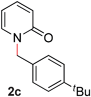
|
|
99 (90 °C) |
16 |
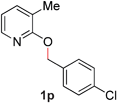
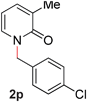
|
|
90 (80 °C) |
| 4 |
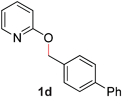
|
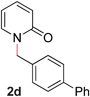
|
99 (90 °C) |
17 |
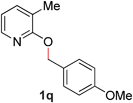
|
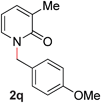
|
85 (80 °C) |
| 5 |
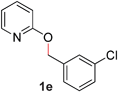
|
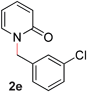
|
95 (80 °C) |
18 |
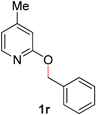
|
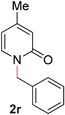
|
92 (80 °C) |
| 6 |
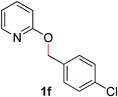
|
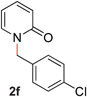
|
89 (80 °C) |
19 |
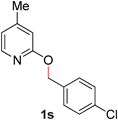
|
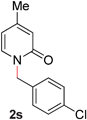
|
94 (80 °C) |
| 7 |
|
|
88 (90 °C) |
20 |
|
|
98 (100 °C) |
| 8 |
|
|
66 (120 °C) |
21 |
|
|
—c (80–110 °C) |
| 9 |
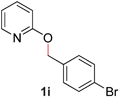
|
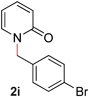
|
83 (100 °C) |
22 |
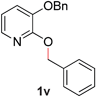
|
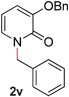
|
99 (90 °C) |
| 10 |
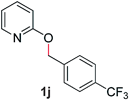
|
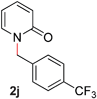
|
96 (100 °C) |
23 |
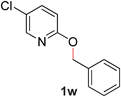
|
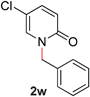
|
87 (110 °C) |
| 11 |
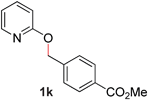
|
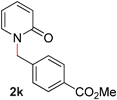
|
99 (90 °C) |
24 |
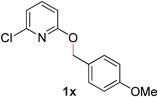
|
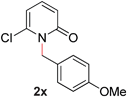
|
—c (80–110 °C) |
| 12 |
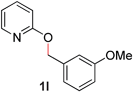
|
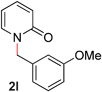
|
78 (80 °C) |
25 |
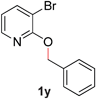
|
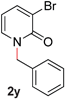
|
99 (100 °C) |
| 13 |
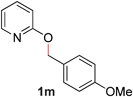
|
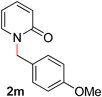
|
99 (90 °C) |
26 |
|
|
—c (80–120 °C) |
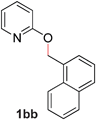
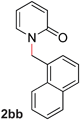
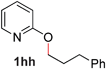
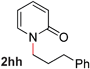
Pyridines bearing larger aryl- and heteroaryloxy substituents, such as naphthyl, benzodioxolyl, furyl and benzofuryl at the 2-position underwent the desired O- to N-alkyl migration efficiently (Table 3, entries 1–6, 2aa–ff, 81–99%). Simple alkoxy chains could also undergo the migratory rearrangement, although higher temperatures were required (entries 7–9). A silyl ether was tolerated in our catalytic reaction (entry 9), although other ether and thioether functionalities led to loss of reactivity (entries 10–11), presumably because chelation renders the catalyst inactive.
| Entrya |
Substrate |
Product |
Yield (%)b |
Entrya |
Substrate |
Product |
Yield (%)b |
|
Reaction conditions: Substrate, 0.2 mmol; [Ru(p-cymene)Cl2]2, 5 mol%; PPh3, 20 mol%; K2CO3, 0.22 mmol; PhMe, 2 mL; 24 h.
Isolated yields.
No desired product could be detected by 1H NMR spectroscopy.
|
| 1 |
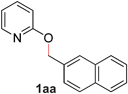
|
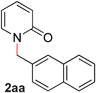
|
99 (80 °C) |
7 |
|
|
64 (110 °C) |
| 2 |
|
|
92 (90 °C) |
8 |
|
|
92 (110 °C) |
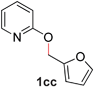
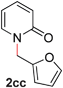
| 3 |
|
|
99 (90 °C) |
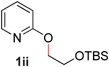
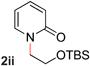
9 |
|
|
65 (120 °C) |
| 4 |
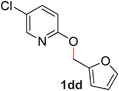
|
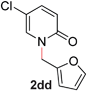
|
95 (90 °C) |
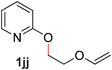
10 |
|
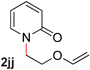
|
—c (80–120 °C) |
| 5 |
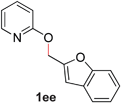
|
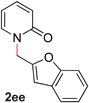
|
81 (90 °C) |
11 |
|
|
—c (120 °C) |
| 6 |
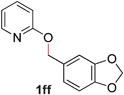
|
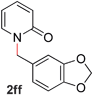
|
96 (90 °C) |
|
|
|
|
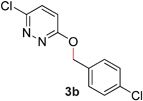
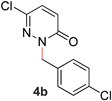
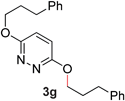
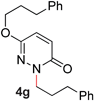
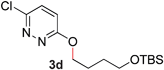
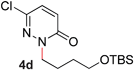
During the course of our studies, we found that pyridazines could direct the desired sp3 C–O bond activation (Table 4, entries 1–7, 4a–g, 60–94%). A number of 3-alkoxypyridazines were successfully transformed to the corresponding 2-alkylpyridazones. Electron-neutral and electron-deficient benzyloxy substituents migrated efficiently (entries 1 and 2), although migratory rearrangement of an electron-rich benzyl group and an aliphatic silyl ether required elevated temperatures (entries 3 and 4). With pyridazines bearing two O-alkyl groups (entries 5–7), only one alkyl migration occurred. Presumably, either the electronic nature of the resulting 2-alkylpyridazone is not sufficient to direct the Rucatalyst to activate the second C–O bond or the increased steric bulk on the 2-position hinders the approach of the Rucatalyst.
| Entrya |
Substrate |
Product |
Yield (%)b |
Entrya |
Substrate |
Product |
Yield (%)b |
|
Reaction conditions: Substrate, 0.2 mmol; [Ru(p-cymene)Cl2]2, 5 mol%; PPh3, 20 mol%; K2CO3, 0.22 mmol; PhMe, 2 mL; 24 h.
Isolated yields.
|
| 1 |
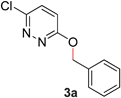
|
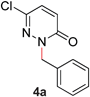
|
70 (90 °C) |
5 |
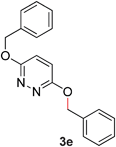
|
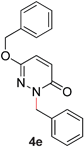
|
94 (80 °C) |
| 2 |
|
|
92 (90 °C) |
6 |
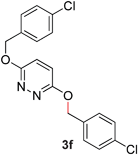
|
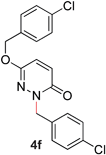
|
94 (80 °C) |
| 3 |
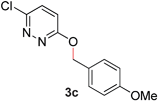
|
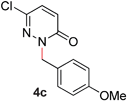
|
64 (120 °C) |
7 |
|
|
64 (110 °C) |
| 4 |
|
|
60 (120 °C) |
|
|
|
|
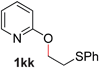
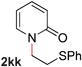
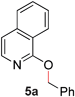
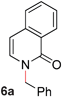
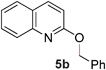
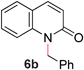
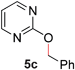
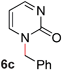
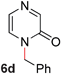
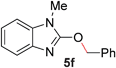
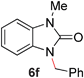
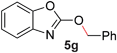
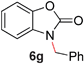
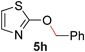
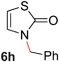
To demonstrate the generality of our strategy for sp3 C–O bond activation, several other O-alkyl N-containing heterocycles were examined (Table 5). The synthesis of 2-benzylisoquinolinone (6a) could be achieved in good yield at 80 °C (96%), while the isomeric 2-benzylquinolinone (6b) was not detected via O- to N-alkyl migration, in agreement with the unreactive 6-substituted O-alkoxylpyridines (1u,x). With the successful conversion of alkoxypyridazines (see Table 4), pyrimidine and pyrazine directing groups were also examined. 2-Benzyloxypyrimidine (5c) failed to undergo the desired O- to N-alkyl migration, but 2-benzyloxypyrazine (5d) underwent successful migration, although 120 °C was required (33%). An analogous S- to N-alkyl migratory rearrangement was also examined. However, we found that 2-thiopyridine5e was unreactive under our optimized conditions, in stark contrast to the 2-alkoxy analogue (cf.Table 3, entry 3) where essentially quantitative yield of the substituted pyridine (2cc) was obtained. Migratory rearrangement using 5-membered N-containing heterocycles as directing groups had varying results. Gratifyingly, both benzo[d]imidazole5f and benzo[d]oxazole5g were efficiently rearranged to their respective products (6f, 6g); in comparison, 2-alkoxypyridine substrates bearing substituents at the 6-position (cf.1u,x and 5b) were completely unreactive. Interestingly, benzyloxythiazole5h generated thiocarbamate6h, albeit in a lower yield (55%), contrasting the lack of reactivity of 2-(2-(phenylthio)ethoxy)pyridine (1kk) and 2-((furan-2-ylmethyl)thio)pyridine (5e). In this case, chelation of the substrate to the Rucatalyst is not possible.
| Entrya |
Substrate |
Product |
Yield (%)b |
|
Reaction conditions: Substrate, 0.2 mmol; [Ru(p-cymene)Cl2]2, 5 mol%; PPh3, 20 mol%; K2CO3, 0.22 mmol; PhMe, 2 mL; 24 h.
Isolated yields.
No desired product could be detected by 1H NMR spectroscopy.
|
| 1 |
|
|
96 (80 °C) |
| 2 |
|
|
—c (80–120 °C) |
| 3 |
|
|
—c (80–120 °C) |
| 4 |
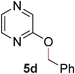
|
|
33 (120 °C) |
| 5 |
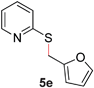
|
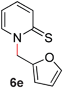
|
—c (80–120 °C) |
| 6 |
|
|
95 (100 °C) |
| 7 |
|
|
70 (100 °C) |
| 8 |
|
|
55 (110 °C) |
Mechanistic discussion
In our reaction design, we hypothesized that oxidative addition of an sp3 C–O bond of 2-alkoxypyridines (and related heterocycles) could generate a metal imidate, similar in structure to known Pd π-allyl complexes.14a–c This Ru-imidate could undergo subsequent reductive elimination to furnish the new sp3 C–N bond.
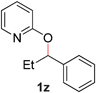
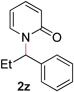
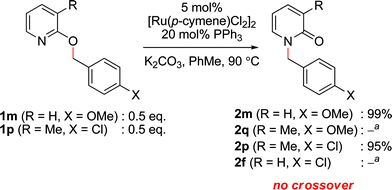
To study the reaction mechanism, we first conducted a competition study as depicted in Fig. 2. Combining 2-alkoxypyridines 1m and 1p in equimolar amounts and subjecting the mixture to the optimized catalytic conditions resulted in production of N-alkylpyridones 2m and 2p, respectively. No products arising from intermolecular exchange processes (i.e., 2q, and 2f) were observed. This result suggests that Ru is promoting a selective intramolecular alkyl transfer process. If a metal imidate is formed during the course of the reaction, no exchange between two Ru complexes occurs.
Next, we synthesized deuterated 3-(benzyloxy)-6-chloropyridazine (3a-d2) and subjected this substrate to the reaction conditions (Fig. 3). Since Lewis basic pyridine rings are known to facilitate sp3 C–H bond functionalization,15 this experiment should probe the possibility of C–H bond activation at the α protons. We observed loss of the D label in both recovered starting material and isolated product. This observation suggests that reversible sp3 C–H bond activation is taking place.16
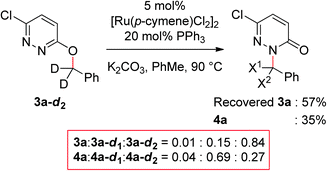 |
| | Fig. 3 Competition experiment. Ratios were determined by ESI-MS. For details, see Electronic Supplementary Information.† | |
Hence, we propose a mechanism involving α-C–H bond metalation to generate a Ru-hydride, α-C–O bond elimination to yield a Ru-methylidene, and finally a 1,2-hydride migration to produce the formal product of an sp3 C–O bond activation (Fig. 4a). This mechanism is based on Krogh-Jespersen and Goldberg's work on stoichiometric studies involving activation of the ethereal C–O bond of anisole with Ir pincer complexes.17 However, our studies do not rule out a non-productive C–H bond activation event. Indeed, both Kakiuchi's direct oxidative addition of ethereal C–O bonds (Fig. 4b)7 and concerted processes such as concerted suprafacial [1,4]-sigmatropic rearrangements (Fig. 4c)18,19 are possible. Further mechanistic studies are underway.
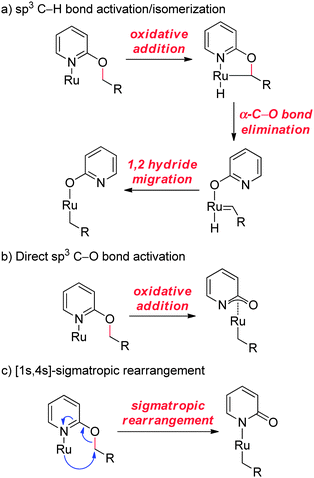 |
| | Fig. 4 Proposed mechanisms for sp3 C–O bond activation (see text for discussion). | |
Conclusions
A Ru-catalyzed O- to N-alkyl migratory rearrangement reaction has been developed. This is a mechanistically novel and distinct strategy for sp3 ethereal C–O bond activation towards the preparation of N-alkylated-2-pyridones and related heterocycles. Directing groups of differing electronics had little effect on the desired migration; steric bulk, however, hinders catalysis. Numerous 2-benzoxypyridines and a diverse array of other N-containing heterocycles are efficiently transformed.
Acknowledgements
Funding is provided by the University of Toronto, Canada Foundation for Innovation, Ontario Research Fund, Boehringer Ingelheim Ltd. Canada, and the Natural Sciences and Engineering Research Council (NSERC) of Canada. VMD is grateful for an Alfred P. Sloan Fellowship and CSY for an NSERC André Hamer Award and a Canada Graduate Scholarship.
Notes and references
- For reviews, see:
(a) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS;
(b) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS.
- For reviews on C–H bond activation, see:
(a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS;
(b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS;
(c) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS;
(d) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1770 CrossRef CAS.
- For reviews on C–C bond activation, see:
(a) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC;
(b) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef.
- For reviews on C–O bond activation, see:
(a) Y.-S. Lin and A. Yamamoto, Topics in Organometallic Chemistry, 1999, 3, 161–192 Search PubMed;
(b) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010 DOI:10.1021/ar100082d. For selected examples of C–OR bond activation involving π-allyl chemistry, see:
(c) T. Nishikata and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 12103–12105 CrossRef CAS;
(d) R. Moser, T. Nishikata and B. H. Lipshutz, Org. Lett., 2010, 12, 28–31 CrossRef CAS;
(e) M. Fisher and R. C. D. Brown, Tetrahedron Lett., 2001, 42, 8227–8230 CrossRef CAS;
(f) H. Murakami, T. Minami and F. Ozawa, J. Org. Chem., 2004, 69, 4482–4486 CrossRef CAS;
(g) G. Mora, O. Piechaczyk, X. F. L. Goff and P. le Floch, Organometallics, 2008, 27, 2565–2569 CrossRef CAS;
(h) H. Yasui, K. Mizutani and H. Yorimitsu, Tetrahedron, 2006, 62, 1410–1415 CrossRef CAS;
(i) Y. Masayuma and M. Marukawa, Tetrahedron Lett., 2007, 48, 5963–5965 CrossRef CAS. For selected examples of C–OR bond activation involving Ni-catalyzed Kumada couplings, see:
(j) J. W. Dankwardt, Angew. Chem., Int. Ed., 2004, 43, 2428–2432 CrossRef CAS;
(k) B.-T. Guan, S.-K. Xiang, B.-Q. Wang, Z.-P. Sun, Y. Wang, K.-Q. Zhao and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 3268–3269 CrossRef CAS.
- For selected examples, see:
(a) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661–1664 CrossRef CAS;
(b) K. W. Quasdorf, M. Riener, K. V. Petrova and N. K. Garg, J. Am. Chem. Soc., 2009, 131, 17748–17749 CrossRef CAS;
(c) A. Antoft-Finch, T. Blackburn and V. Snieckus, J. Am. Chem. Soc., 2009, 131, 17750–17752 CrossRef CAS;
(d) B.-T. Guan, Y. Wang, B.-J. Li, D.-G. Yu and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 14468–14470 CrossRef CAS;
(e) L. Zhang, T. Meng and J. Wu, J. Org. Chem., 2007, 72, 9346–9349 CrossRef CAS;
(f) Z.-Y. Tang and Q.-S. Hu, J. Am. Chem. Soc., 2004, 126, 3058–3059 CrossRef CAS;
(g) V. Percec, G. M. Golding, J. Smidrkal and O. Weichold, J. Org. Chem., 2004, 69, 3447–3452 CrossRef CAS;
(h) D. Zim, V. R. Lando, J. Dupont and A. L. Monteiro, Org. Lett., 2001, 3, 3049–3051 CrossRef CAS.
-
Handbook of Organopalladium Chemistry for Organic Synthesis; John Wiley & Sons, Inc.: New York, 2002; Vol. 1, p. 1661–2117 Search PubMed.
-
(a) F. Kakiuchi, S. Kan, K. Igi, N. Chatani and S. Murai, J. Am. Chem. Soc., 2003, 125, 1698–1699 CrossRef CAS;
(b) F. Kakiuchi, M. Usui, S. Ueno, N. Chatani and S. Murai, J. Am. Chem. Soc., 2004, 126, 2706–2707 CrossRef CAS.
- For reviews, see :
(a) M. D. Hill and M. Movassaghi, Chem.–Eur. J., 2008, 14, 6836–6844 CrossRef CAS;
(b) M. Torres, S. Gil and M. Parra, Curr. Org. Chem., 2005, 9, 1757–1779 CrossRef CAS.
- For selected examples of natural products with N-alkylpyridone rings, see :
(a) D. Gray and T. Gallagher, Angew. Chem., Int. Ed., 2006, 45, 2419–2423 CrossRef CAS;
(b) D. Stead, P. O'Brien and A. J. Sanderson, Org. Lett., 2005, 7, 4459–4462 CrossRef CAS;
(c) I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1–50 CrossRef CAS;
(d) B. Rutkowski, E. Slominska, M. Szolkiewicz, R. T. Smolenski, C. Striley, P. Rutkowski and J. Swierczynski, Kidney Int., 2003, 63(s84), 19–S21 CrossRef;
(e) N. A. Adibatti, P. Thirugnanasambantham, C. Kulothungan, S. Viswanathan, L. Kameswaran, K. Balakrishna and E. Sukumar, Phytochemistry, 1991, 30, 2449–2450 CrossRef CAS;
(f) A. H. Hunt, J. S. Mynderse, S. K. Samlaska, D. S. Fukuda, G. M. Maciak, H. A. Kirst, J. L. Occolowitz, J. K. Swartzendruber and N. D. Jones, J. Antibiot., 1988, 41, 771–779 CAS;
(g) C. M. Richards and A. Hofmann, Aust. J. Chem., 1978, 31, 187–191 CrossRef CAS;
(h) T. R. Govindachari, K. R. Ravindranath and N. Viswanathan, J. Chem. Soc., Perkin Trans. 1, 1974, 1215–1217 RSC;
(i) G. E. Lindenblad, M. Kaihara and J. M. Price, J. Biol. Chem., 1956, 219, 893–901 CAS.
- For selected examples of medicinal targets with N-alkylpyridone rings, see:
(a) J. A. Pfefferkorn, J. Lou, M. L. Minich, K. J. Filipski, M. He, R. Zhou, S. Ahmed, J. Benbow, A.-G. Perez, M. Tu, J. Litchfield, R. Sharma, K. Metzler, F. Bourbonais, C. Huang, D. A. Beebe and P. J. Oates, Bioorg. Med. Chem. Lett., 2009, 19, 3247–3252 CrossRef CAS;
(b) C. A. Martinez, D. R. Yazbeck and J. Tao, Tetrahedron, 2004, 60, 759–764 CrossRef CAS;
(c) J. J. Parlow and M. S. South, Tetrahedron, 2003, 59, 7695–7701 CrossRef CAS;
(d) J. W. Huffman, J. Lu, G. Hynd, J. L. Wiley and B. R. Martin, Bioorg. Med. Chem., 2001, 9, 2863–2870 CrossRef CAS.
-
(a) M. Breugst and H. Mayr, J. Am. Chem. Soc., 2010, 132, 15380–15389 CrossRef CAS;
(b) D. Conreaux, E. Bossharth, N. Monteiro, P. Desbordes and G. Balme, Tetrahedron Lett., 2005, 46, 7917–7920 CrossRef CAS;
(c) M. Sugahara, Y. Moritani, T. Kuroda, K. Kondo, H. Shimadzu and T. Ukita, Chem. Pharm. Bull., 2000, 48, 589–591 CAS;
(d) H. Liu, S.-B. Ko, H. Josien and D. P. Curran, Tetrahedron Lett., 1995, 36, 8917–8920 CrossRef CAS.
- E. L. Lanni, M. Bosscher, B. D. Ooms, C. A. Chandro, B. A. Ellsworth and C. E. Anderson, J. Org. Chem., 2008, 73, 6425–6428 CrossRef CAS.
- M. Ikehara and S. Tanaka, Tetrahedron Lett., 1974, 15, 497–500 CrossRef.
-
(a) A. Rodrigues, E. E. Lee and R. A. Batey, Org. Lett., 2010, 12, 260–263 CrossRef CAS;
(b) K. Itami, D. Yamazaki and J.-i. Yoshida, Org. Lett., 2003, 5, 2161–2164 CrossRef CAS;
(c) A. C. S. Reddy, B. Narsaiah and R. V. Venkataratnam, Tetrahedron Lett., 1996, 37, 2829–2832 CrossRef CAS;
(d) G. Balavoine and F. Guibe, Tetrahedron Lett., 1979, 20, 3949–3952 CrossRef;
(e) H. F. Stewart and R. P. Seibert, J. Org. Chem., 1968, 33, 4560–4561 CrossRef CAS.
- For selected examples of Ru-catalyzed transformations, see:
(a) C.-H. Jun, D.-C. Hwang and S.-J. Na, Chem. Commun., 1998, 1405–1406 RSC;
(b) F. Kakiuchi, K. Tsuchiya, M. Matsumoto, E. Mizushima and N. Chatani, J. Am. Chem. Soc., 2004, 126, 12792–12793 CrossRef CAS;
(c) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935–10941 CrossRef CAS. For selected examples of Rh-catalyzed transformations, see:
(d) N. Chatani, T. Asaumi, T. Ikeda, S. Yorimitsu, Y. Ishii, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2000, 122, 12882–12883 CrossRef CAS;
(e) Y. Ishii, N. Chatani, F. Kakiuchi and S. Murai, Organometallics, 1997, 16, 3615–3622 CrossRef CAS. For selected examples of Pd-catalyzed transformations, see:
(f) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS;
(g) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904–11905 CrossRef CAS;
(h) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924–1935 CrossRef CAS;
(i) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134–7135 CrossRef CAS;
(j) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141–1144 CrossRef CAS;
(k) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483–11498 CrossRef CAS;
(l) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330–7331 CrossRef CAS;
(m) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300–2301 CrossRef CAS;
(n) X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634–12635 CrossRef CAS;
(o) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS;
(p) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS;
(q) D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657–3659 CrossRef CAS;
(r) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef CAS;
(s) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS;
(t) J. Zhang, E. Khaskin, N. P. Anderson, P. Y. Zavalij and A. N. Vedernikov, Chem. Commun., 2008, 3625–3627 RSC.
- Presumably, the H originates either from the PhMesolvent or p-cymene or adventitious water. Similar results were observed when PhMe was substituted with PhH. When substrate 3a was subjected to stoichiometric amounts of [Ru(p-cymene)Cl2]2 and PPh3, product 4a was formed with complete conversion. No Ru-hydrides could be observed by 1H NMR spectroscopy. Instead, Ru(3a) and Ru(4a) complexes were detected, suggesting that these species are catalyst resting states. For details, see Electronic Supplementary Information†.
- J. Choi, Y. Choliy, X. Zhang, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2009, 131, 15627–15629 CrossRef CAS.
- U. Schöllkpf and I. Hoppe, Tetrahedron Lett., 1970, 11, 4527–4530 CrossRef.
- A Fries-type rearrangement involving formation of distinct Ru-imidate and benzyltriphenylphosphonium cations viadealkylation is an alternative mechanism. Alkylation of the N atom of alkoxypyridines is known using activated methylating agents:
(a) K. W. C. Poon and G. B. Dudley, J. Org. Chem., 2006, 71, 3923–3927 CrossRef CAS;
(b) W. R. Bowman and C. F. Bridge, Synth. Commun., 1999, 29, 4051–4059 CAS. Based on our crossover studies (Fig. 2), we favor the mechanisms highlighted in Fig. 4.
Footnotes |
| † Electronic Supplementary Information (ESI) available: General procedures for migratory rearrangement, and spectroscopic data. See DOI: 10.1039/c0sc00498g/ |
| ‡ General procedure for Ru-catalyzed O- to N-alkyl migration: In a N2-filled glovebox, in a one-dram vial equipped with a Teflon cap, [Ru(p-cymene)Cl2]2 (6.2 mg, 0.01 mmol, 5 mol%) and PPh3 (10.5 mg, 0.04 mmol, 20 mol%) were dissolved in PhMe (2 mL). The resulting solution was added to the O-alkyl N-containing heterocycle. Subsequently, K2CO3 (30.4 mg, 0.22 mmol) was added. The vial was stirred on a heating block at the appropriate temperature for 24 h. The resulting mixture was passed through a pad of Celite, concentrated in vacuo and the resulting residue was purified by preparative thin-layer chromatography (eluent: hexanes/EtOAc or CH2Cl2/MeOH) to afford the pure N-alkylheterocyclic migratory rearrangement products. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 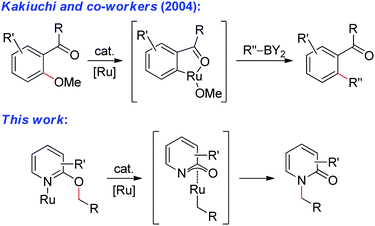





![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) No desired product could be detected by 1H NMR spectroscopy.
No desired product could be detected by 1H NMR spectroscopy.