Mechanisms of chemical protein 19F-labeling and NMR-based biosensor construction in vitro and in cells using self-assembling ligand-directed tosylate compounds†
Yousuke
Takaoka
a,
Yedi
Sun
a,
Shinya
Tsukiji‡
ab and
Itaru
Hamachi
*ab
aDepartment of Synthetic Chemistry and Biological Chemistry, Kyoto University, Katsura, Nishikyo-Ku, Kyoto, 615-8510, Japan. E-mail: ihamachi@sbchem.kyoto-u.ac.jp
bJapan Science and Technology Agency (JST), CREST, 5 Sanbancho, Chiyoda-ku, Tokyo 102-0075, Japan
First published on 1st December 2010
Abstract
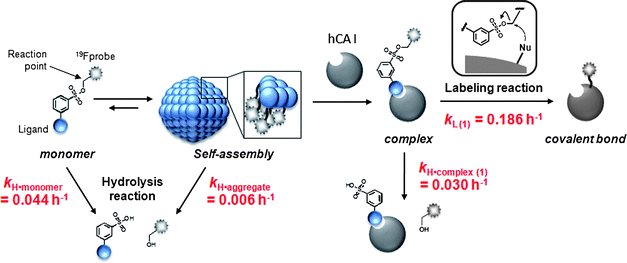
Chemical labeling methods that convert a specific endogenous protein into a semisynthetic biosensor offer numerous new opportunities for biological research and drug discovery. We recently developed a novel protein labeling scheme, termed ligand-directed tosyl (LDT) chemistry, which can site-specifically introduce a synthetic probe to a protein with the concomitant release of the affinity ligand. In previous work, we demonstrated that LDT reagent 1 can be used to modify carbonic anhydrase I (CAI) with a 19F probe, converting it into a 19F NMR-based biosensor for CAI inhibitors either in vitro or in red blood cells (RBCs). We herein report the chemical properties of 1, and the mechanisms controlling biosensor construction. It was revealed that the LDT reagent forms self-assembled aggregates in the absence of the target protein. In the aggregated state, nonproductive hydrolysis of the reagent was significantly suppressed, which suggests the potential utility of self-assembly in the design of labeling reagents that have increased stability. In the presence of the target protein, the aggregates were disrupted to form a noncovalent protein–reagent complex, and protein 19F-labeling proceeded to generate 19F-labeled CAI. The ligand-binding pocket of the labeled CAI retained the cleaved ligand fragment in vitro, whereas the pocket was vacant in RBC. Further biochemical studies suggested that an anion transporter might play a role in eliminating the cleaved ligand from the interior to the exterior of the cells. The findings provide a fundamental basis for the rational design of reagents applicable to selective protein labeling and biosensor construction in biological contexts.
Introduction
Chemical protein labeling (or bioconjugation) is a very attractive strategy for the construction of protein-based biosensors. This semisynthetic approach permits the use of a variety of non-genetically encoded molecules, including organic fluorescent dyes and other biophysical probes such as a NMR-active 19F nuclide, as reporter units that transduce an analyte-binding event into a detectable signal.1 The small size of synthetic probes compared to reporter proteins, such as green fluorescent protein and luciferase, is also thought to be advantageous in minimizing loss of protein function. In past decades, much progress has been made in the preparation of semisynthetic biosensors that can specifically detect various biologically relevant species.2,3 However, the application of these biosensors in intracellular environments has been challenging since semisynthetic biosensors are typically constructed in vitro (in test tubes) first, then transferred into cells, which is technically demanding. Consequently, in recent years, researchers in the chemical biology community have begun developing an alternate (more appealing) approach: the in situ construction of semisynthetic biosensors.4 In this approach, a target protein (scaffold) in living cells is specifically modified by a detectable synthetic probe, yielding a labeled protein that allows the monitoring of specific analytes and/or biological events in an in situ manner. The ability to prepare semisynthetic biosensors directly within living systems presents numerous new opportunities in basic biological research, drug discovery and medical diagnosis.5Toward this goal, we recently developed a method, termed ligand-directed tosyl (LDT) chemistry, whereby a target (endogenous) protein can be selectively labeled with a desired probe within its native environment (Fig. 1a).6,7 Because LDT chemistry is based on the principle of affinity labeling, the labeling site is specific for an amino acid(s) located near the active site of the protein. In addition, in contrast to existing affinity labeling reactions, LDT-based labeling is traceless because the ligand moiety is cleaved off concomitantly with the SN2-type labeling reaction.8 In previous work,6 we designed compound 1 as a tool for introducing a 19F NMR probe into human carbonic anhydrase I (CAI)9,10 (Fig. 1a). Compound 1 contains a 3,5-bis(trifluoromethyl)benzene derivative (FB) carrying six magnetically equivalent 19F nuclei connected through a phenylsulfonate (tosyl) ester bond to a protein ligand, benzenesulfonamide (SA),11 which is specific for CA. We demonstrated that CAI could be converted to a 19F NMR-based biosensor for CA inhibitors not only in a purified form (in vitro), but also in intact red blood cells (RBCs) using 1. However, many details regarding the basic properties of 1, and the mechanisms controlling biosensor construction, remained unexplored.
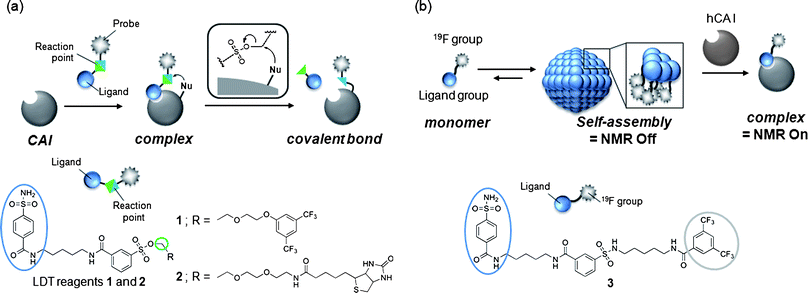 | ||
| Fig. 1 (a) Schematic of ligand-directed tosyl (LDT) chemistry for protein labeling and molecular structures of LDT reagents 1 and 2. (b) Schematic of the self-assembling 19F NMR probe for protein imaging and molecular structure of 19F-carrying protein imaging probe 3. | ||
The aim of this work was to investigate the chemical properties of 1 and related LDT compound 2 (Fig. 1a), and the mechanisms behind the processes involved in converting native CAI to a 19F NMR-based biosensor in vitro and in cells. Based on results from our other work (Fig. 1b),12 it was speculated that 1 would form aggregates in aqueous solution in the absence of CAI. Our earlier study also suggested that the tosyl linkage undergoes both a protein labeling reaction and hydrolysis.7a We thus first investigated and compared the aggregation properties, labeling efficiencies and hydrolysis rates of the two LDT compounds, and found that 1 self-assembles in aqueous solution and that the formation of aggregates effectively suppresses nonproductive hydrolysis of the tosyl moiety. Next, we investigated the mechanisms controlling in vitro and in cell biosensor construction. The high environmental sensitivity of 19F NMR chemical shifts allowed the labeling processes to be monitored in real time and the different forms of FB-labeled CAI in its free and complexed state to be characterized. In the in vitro system, CAI labeling with 1 produced FB-modified CAI that remained noncovalently complexed with the cleaved ligand-containing fragment. Removal of the cleaved ligand required an extra purification step, resulting in a CAI-based biosensor with a vacant ligand-binding pocket. In contrast, the in-cell labeling directly generated FB-labeled CAI with an empty ligand-binding pocket, indicating that the ligand-containing fragment is spontaneously removed from labeled CAI in RBCs. Further biochemical and pharmacological studies suggested that an anion transporter in RBCs might play a role in this process by mediating the export of the byproduct. Understanding these aspects will allow us to further modulate the properties and efficacy of LDT reagents and generalize the application of this method to other proteins and other cell types.
Results and discussion
Self-assembly and affinity labeling properties of LDT reagent 1
We previously developed probe 3 for 19F NMR-based protein detection and imaging (Fig. 1b).12a Compound 3, designed to be a self-assembling molecule, is comprised of the FB group connected to the CA ligand, benzenesulfonamide, via a stable (unreactive) phenylsulfonamide bond. This probe is NMR-silent in aqueous solution in the absence of CAI due to its self-assembly, but gives a distinct 19F signal in response to the target protein due to recognition-mediated disassembly of the probe aggregate. Given the high structural similarity between 1 and 3, it was expected that 1 might also form aggregates in aqueous solution.We first investigated the self-assembly properties of LDT reagent 1 using several spectroscopic techniques. Atomic force microscopy (AFM) showed spherical or oval aggregates of 1 ranging in diameter from 200 to 500 nm (Fig. 2a). Dynamic light scattering (DLS) measurements confirmed the size of the aggregates in a buffer solution (Fig. S1a, ESI†, mean diameter ca. 250 nm). Furthermore, optical density measurements at 600 nm revealed that a solution containing only 1 is turbid, and that turbidity decreases (by 10-fold) upon the addition of CAI (Fig. S1b, ESI†), indicating that the aggregates efficiently collapse through recognition of and binding by CAI. All these properties of 1 were very similar to those of 3 reported previously.12a
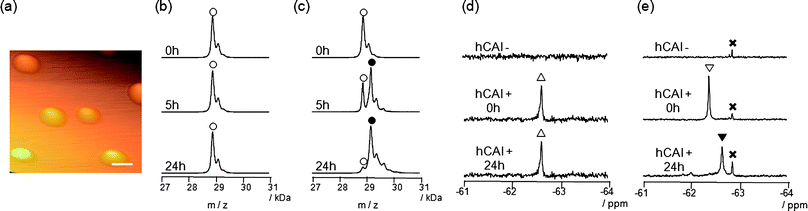 | ||
Fig. 2
Self-assembly and protein labeling properties of reagents 1 and 3. (a) AFM image of self-assembled reagent 1 (25 μM) (scale bar, 500 nm). (b, c) MALDI-TOF mass spectra of a reaction mixture containing CAI (150 μM) and 3 (300 μM) (b) or 1 (300 μM) (c). (○) Native CAI (Mw = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 781); (●) FB-CAI (Mw = 29082). (d, e) 19F NMR spectra of a reaction mixture containing CAI (150 μM) and 3 (300 μM) (d) or 1 (300 μM) (e). (△) Complex of CAI and 3; (▽) complex of CAI and 1; (▼) FB-CAISA; (×) FBOHOH. 781); (●) FB-CAI (Mw = 29082). (d, e) 19F NMR spectra of a reaction mixture containing CAI (150 μM) and 3 (300 μM) (d) or 1 (300 μM) (e). (△) Complex of CAI and 3; (▽) complex of CAI and 1; (▼) FB-CAISA; (×) FBOHOH. | ||
The most obvious difference between 1 and 3 is in protein labeling: the former is reactive, but the latter is unreactive. After mixing CAI (150 μM) with 1 or 3 (300 μM) in a buffer solution, the labeling processes were monitored by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). The labeling reaction was essentially complete within 24 h in the case of 1 (Fig. 2c), whereas no reaction occurred in the case of 3, as expected (Fig. 2b). CAI labeling by 1 was completely abolished in the presence of ethoxzolamide (EZA),11 a strong inhibitor of CA, indicating that the labeling occurs via an affinity-driven proximity effect.6 Both the self-assembly/disassembly and labeling processes were next monitored by 19F NMR spectroscopy. First, when 3 was dissolved in a buffer solution containing trifluoroacetic acid (TFA; internal standard at −75.6 ppm), no 19F signal was observed because of the formation of aggregates (Fig. 2d).12a However, a sharp signal appeared at −62.6 ppm (△) upon the addition of CAI, apparently due to disassembly of the aggregates and the formation of a protein–probe complex. This signal remained unchanged over 24 h, consistent with 3 being unreactive. Similarly, a solution containing only 1 showed no signal, but gave a sharp signal at −62.3 ppm (▽) in the presence of CAI (Fig. 2e).13 In contrast, the signal corresponding to the complex of CAI and 1 disappeared and a new signal appeared at −62.6 ppm (▼) over a 24 h period as the labeling reaction proceeded (Fig. 2e). A new minor signal also appeared at −62.9 ppm (×) during the incubation, due to a FB-containing fragment (FBOHOH) generated by the hydrolysis of 1 (Scheme 1).14 Collectively, these results indicate that (1) LDT reagent 1 self-assembles and thus is NMR-silent in aqueous solution in the absence of CAI, (2) the 19F signal appears upon recognition-mediated disassembly of the aggregates and (3) after complexation with CAI, 1 reacts with an amino acid residue on the surface of CAI, which further induces a change in the 19F NMR chemical shift. It was also shown that 1 undergoes an intrinsic hydrolysis process.
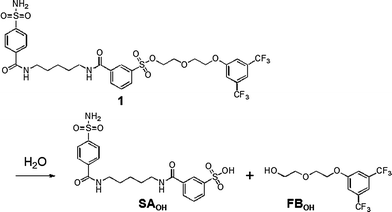 | ||
| Scheme 1 Hydrolytic reaction scheme of reagent 1. | ||
Implications of the self-assembly properties of LDT reagents for protein labeling and hydrolysis efficiencies
The self-assembly properties of labeling reagents may influence on protein labeling processes in LDT chemistry. We thus investigated in detail the labeling efficiency of LDT reagents 1 (FB-type) and 2 (biotin-type) (Fig. 1a). MALDI-TOF MS analysis indicated that the yield of CAI labeling after 24 h incubation varied among the reagents: 90 and 32% for 1 and 2, respectively (in the case of 1:1 mixture of CAI (50 μM) with 1 or 2 (50 μM)). In these structures, the distance between the ligand and the reactive site (tosylate) is identical. However, it appears that their hydrophobicities greatly differ. In fact, turbidity measurements of the reagents in buffer solution gave significantly different optical densities at 600 nm: 0.074 and 0.001 for 1 and 2, respectively (Fig. 3a). DLS measurements also showed that 1 form aggregates with a mean diameter of 250 nm in buffer solution (as above described), whereas negligible DLS intensity was apparent in the case of 2. These results indicate that 1 is self-assembling LDT reagents, whereas 2 is homogeneously soluble in aqueous solution.
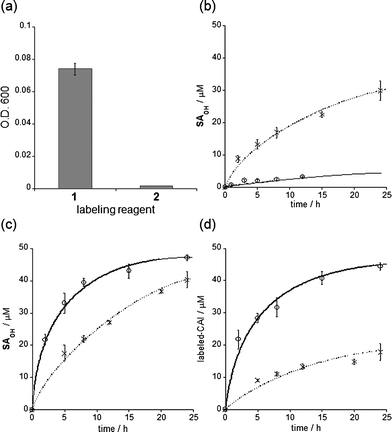 | ||
| Fig. 3 Self-assembly properties, hydrolysis rates and labeling efficiencies of LDT reagents 1 and 2. (a) Optical density (O.D.) at 600 nm of an aqueous solution containing each reagent (25 μM). Experiments were performed in triplicate to obtain mean and standard deviation values (shown as error bars). (b) Time profiles of hydrolysis of 1 (○) and 2 (◆) in buffer solution at 37 °C. The hydrolysis product SAOHOH was detected using HPLC. Fitting curves generated using eqn (1) (see Experimental section) are shown as lines. (c) Time profiles of the generation of the SAOHOH fragment during labeling experiments. The labeling reaction was carried out as shown in (d), and the SAOHOH fragment was detected using HPLC. Fitting curves generated using eqn (2), given in the Experimental section, are shown as lines. (d) Time profiles of CAI labeling (50 μM) with 1 (○) and 2 (◆) (50 μM) in buffer solution at 37 °C. The labeling reaction was monitored by MALDI-TOF MS analysis. Fitting curves generated using eqn (3), described in the Experimental section, are shown as lines. All experiments in (b–d) were performed in triplicate to obtain mean and standard deviation values (shown as error bars). | ||
LDT reagents intrinsically undergo nonproductive hydrolysis at the phenylsulfonate ester bond in aqueous medium, along with the SN2 reaction for protein labeling.7a Indeed, as mentioned above, the hydrolysis product (FBOHOH) was detected by 19F NMR in a solution containing CAI and 1 (Fig. 2e). Interestingly, we found that the amount of cleaved fragment SAOHOH, which is produced by both hydrolysis and protein labeling, was larger than the amount of labeled protein formed, in the case of 2. This observation also suggests that the hydrolysis reaction concomitantly occurs with the protein labeling reaction. We next conducted a kinetic study to evaluate the labeling rates and hydrolysis rates of the LDT reagents in the presence and absence of CAI. The hydrolysis rates of the reagents (kH) in buffer solution without CAI were determined by monitoring the time-dependent increase in SAOHOH by HPLC analysis, giving 0.006 and 0.044 h−1, for 1 and 2, respectively (Fig. 3b and Fig. S3, ESI†). Based on the difference in the aggregation property, it is reasonably considered that the value for 1 corresponds to the hydrolysis rate from aggregates (kH·aggregate), and the value for 2 corresponds to the hydrolysis rate from monomeric state of the LDT reagent (kH·monomer). Under the labeling conditions (i.e., with CAI), we determined the apparent rates of the formation of the cleaved SAOHOH fragment, which correspond to the sum of the rates of the labeling and hydrolysis reactions from the noncovalent CAI-reagent complex, i.e., (kL + kH·complex) (Fig. 3c). We also evaluated the time profiles of CAI labeling by MALDI-TOF MS analysis (Fig. 3d). Using nonlinear least-squares curve-fitting, the labeling rates (kL) were determined to be 0.186 and 0.031 h−1, and the rates of the hydrolysis reaction from the complex (kH·complex) were determined to be 0.030 and 0.038 h−1, for 1 and 2, respectively (Table 1).15
| 1 | 2 | |
|---|---|---|
| a k H·monomer: rate of hydrolysis in the monomeric state in the case of 2; kH·aggregate: rate of hydrolysis in the aggregated state in the case of 1 (in the absence of CAI); kL: rate of labeling; kH·complex: rate of hydrolysis in the complexed state (in the presence of CAI). b Not determined because 1 aggregated in aqueous solution. c Not determined because 2 did not aggregate in aqueous solution. | ||
| k H·monomer/h−1 | —b | 0.044 ± 0.005 |
| k H·aggregate/h−1 | 0.006 ± 0.0008 | —c |
| k L/h−1 | 0.186 ± 0.009 | 0.031 ± 0.002 |
| k H·complex/h−1 | 0.030 ± 0.003 | 0.038 ± 0.002 |
Scheme 2 summarizes the mechanism and reaction rates of the processes involved in CAI labeling by 1. As mentioned above, we show the rate of LDT reagent hydrolysis in the monomer state using 2. The most striking finding is that simple hydrolysis of 1 in the absence of the target protein was markedly suppressed by self-assembly: the hydrolysis rate (kH·aggregate) was more than 7-fold slower than when the reagent was in the monomeric state (kH·monomer, the value of 2). This is most likely due to sequestering of the reactive tosylate moiety within the hydrophobic interior of the self-assembled aggregates. Generally, in protein labeling using electrophilic reagents, the nucleophilic attack by water is inevitable, leading to decomposition of the reagent. Self-assembly might be a promising strategy for suppressing such undesired side reactions during chemical protein labeling. Upon binding with CAI, self-assembled 1 rapidly disassembled to form a noncovalent protein-reagent (1:1) complex. At this stage, the hydrolysis of 1 on the complex occurred at a faster rate that approached the hydrolysis rate of monomeric reagent 2, because the tosylate group was now exposed to the aqueous phase. However, the SN2 reaction of the reactive tosylate functionality by the nucleophilic amino acid present on the CAI surface (His in this case, see below) was 6-fold faster than the competing hydrolysis reaction, resulting in efficient labeling with greater than 90% yield. The hydrolysis rates from the protein–reagent complex were very similar between the two LDT reagents, whereas the labeling rate was larger for the hydrophobic probe 1, compared to 2. Therefore, the labeling efficiency of 1 was greater than 2.
 | ||
| Scheme 2 Overall scheme of the self-assembly, hydrolysis and labeling processes in 1-mediated CAI labeling. | ||
Functions of 19F-labeled CAI as a 19F NMR-based biosensor in vitro and in RBCs
As mentioned above, the 19F NMR spectrum of the post-labeling solution containing CAI and 1 (after 24 h incubation) showed a signal at −62.6 ppm (▼, Fig. 2e and Fig. 4a). The labeled CAI was then purified by gel filtration to remove small molecules including FBOHOH and SAOHOH (and unreacted 1, if any). The original signal at −62.6 ppm disappeared and a distinct new signal appeared at −62.0 ppm (□, Fig. 4a). Based on these data, the 19F signals observed at −62.6 and −62.0 ppm could be assigned to a noncovalent complex of FB-labeled CAI and SAOHOH (FB-CAISA) and the ligand-free, labeled CAI (FB-CAI), respectively. We previously confirmed that a single FB group attaches specifically at His67 of CAI, which is located in the proximity of the active site.6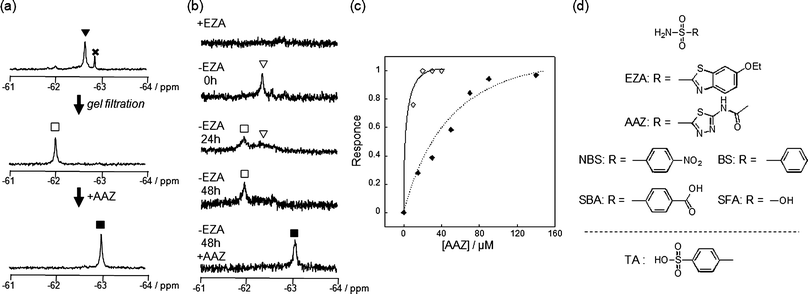 | ||
| Fig. 4 Conversion of CAI and endogenously expressed CAI (eCAI) to a NMR-based semisynthetic biosensor. (a) 19F NMR spectra of the post-in vitro-labeling solution containing CAI (150 μM) and 1 (300 μM) before (top) and after (middle) purification by gel filtration. To the purified FB-CAI solution was added AAZ (bottom). (b) 19F NMR spectra of a RBC suspension containing 1 with (top spectrum) or without (the middle three spectra) EZA. After 48 h incubation, AAZ was added to the post-in-cell-labeling suspension (bottom spectrum). (c) Quantitative NMR detection of AAZ by FB-(e)CAI in vitro (◇) and in RBCs (◆). The response shown on the y axis is defined as the relative area of peak ■ (FB-CAIAAZ) divided by the sum of the relative areas of peak □ (FB-CAI) and ■ and was plotted against the concentration of AAZ. (d) Molecular structures of CAI inhibitors used in this study. The abbreviations are as follows: EZA, 6-ethoxy-2-benzothiazolesulfonamide; AAZ, acetazolamide; NBS, 4-nitrobenzenesulfonamide; SBA, 4-sulfamoylbenozic acid; BS, benezenesulfonamide; SFA, sulfamic acid; TA, 4-toluenesufonic acid. | ||
We next evaluated the function of FB-CAI as a 19F NMR-based biosensor. When a strong inhibitor, acetazolamide (AAZ, Fig. 4d),11 was added to the FB-CAI solution, the peak shifted from −62.0 to −63.0 ppm due to the complexation of FB-CAI with AAZ (FB-CAIAAZ, Fig. 4a). This result clearly indicates that FB-CAI can sensitively readout the presence or absence of a CAI ligand by a chemical shift change in 19F NMR spectroscopy. Titration with AAZ resulted in a decrease in the 19F signal of FB-CAI (−62.0 ppm, □) which was synchronized with the increase in the 19F signal of FB-CAIAAZ (−63.0 ppm, ■) (Fig. S4a, ESI†). In the case of EZA, a 19F signal from the complex of FB-CAI and EZA (FB-CAIEZA) was observed at −62.5 ppm,6 whereas no change was observed upon the addition of a non-aromatic-ligand sulfamic acid (SFA, Fig. S4c, ESI†).11 These data strongly indicate that the observed changes in chemical shift were caused by a change in the microenvironment of the labeled FB group, which varies depending on the chemical structure of the inhibitor. The titration curve of the chemical shift change with AAZ showed saturation behavior (Fig. 4c). Similar titration curves were obtained by the addition of other sulfonamide class inhibitors (Fig. S4f, ESI†). Table 2 summarizes the chemical shifts and the association constants of FB-CAI determined from those titration curves for various benzenesulfonamide derivatives. The relative order of the affinity of the tested compounds for FB-CAI was very similar to that for native CAI.16
| Inhibitor | Chemical shift/ppm | K app/M−1 | |||||
|---|---|---|---|---|---|---|---|
| in vitro | In RBCs | in vitro a | In RBCsa | Native-CAIb | |||
| Before | After | Before | After | ||||
| a Determined by nonlinear curve-fitting analysis. b Reported values determined by in vitro assays.11 c Not determined because no signal change was observed. d Not performed. e No literature value is available. | |||||||
| EZA | −62.0 | −62.5 | −62.0 | −62.5 | >106 | >106 | 1.3 × 108 |
| AAZ | −62.0 | −63.0 | −62.0 | −63.0 | >106 | 5.9 × 104 | 4.0 × 107 |
| NBS | −62.0 | −63.2 | −62.0 | −63.2 | >106 | 1.2 × 104 | 1.6 × 107 |
| SBA | −62.0 | −62.3 | −62.0 | −62.3 | 6.4 × 105 | 1.2 × 103 | 2.9 × 106 |
| BS | −62.0 | −62.6 | −62.0 | −62.6 | 5.5 × 104 | 1.0 × 104 | 3.0 × 106 |
| SFA | −62.0 | —c | −62.0 | —d | —c | —d | 4.7 × 103 |
| TA | −62.0 | —c | −62.0 | —d | —c | —d | —e |
In previous work we demonstrated that LDT reagent 1 is cell-permeable and thus applicable to the specific modification of endogenously expressed CAI (eCAI) in human RBCs (note that CAI is a cytosolic protein).6,17 eCAI labeling in RBCs was monitored by in-cell19F NMR. At first, when 1 (200 μM) and EZA (1 mM) were added to a suspension of RBCs, no 19F signal was observed (Fig. 4b). In a separate experiment, we confirmed that 1 did indeed enter the RBCs (data not shown). Since no hemolysis occurred in any of the RBC-based experiments, these results indicate that 1 is NMR-silent even inside RBCs when it is not recognized by eCAI.18 This phenomenon can be explained by the self-assembly properties of 1 discussed above, and is similar to the result previously obtained using 3.12a Without EZA, a 19F signal was clearly observed at −62.3 ppm (▽) in a suspension containing 1 and RBCs, which is identical to the chemical shift obtained in a simple mixture of (purified) CAI and 1 (also see Fig. 2e). This signal, corresponding to the complex of 1 and eCAI, gradually disappeared, and a new signal appeared at −62.0 ppm over a period of 48 h. Interestingly, the chemical shift observed in the cell-based post-labeling solution was different from that obtained in the aforementioned in vitro post-labeling solution, i.e., −62.6 ppm (FB-CAISA). Instead, it was consistent with the chemical shift of FB-CAI (−62.0 ppm, □ in Fig. 4a) in the ligand-free state, indicating that RBC-based labeling directly led to the formation of 19F-labeled eCAI with a free active site pocket inside the cells (more details regarding this phenomenon are discussed in the next section). After intracellular labeling, the cells were washed and incubated with several CA inhibitors. By increasing the extracellular AAZ concentration, a new peak appeared at −63.0 ppm (■, FB-eCAIAAZ) in a saturating manner (Fig. 4c). Table 2 summarizes the chemical shifts and the association constants of FB-eCAI determined from titration curves generated using various benzenesulfonamide derivatives. Comparison of the in vitro data and cell-based data indicates that the chemical shifts of the inhibitor-bound FB-CAI and FB-eCAI are essentially the same in both settings. On the other hand, the affinity values in the cell-based system were lower than compared with those of native CAI determined by in vitro experiments. This can be explained by lower effective concentrations of the inhibitors inside RBCs, which is due to the limited cell permeability of the molecules as well as their potential nonspecific interactions with other components in the cells such as cell membranes or non-target proteins.
Mechanisms of 19F NMR biosensor construction in red blood cells
The in vitrolabeling of CAI with 1 generated 19F-labeled CAI which remained complexed with the cleaved SAOHOH, i.e., FB-CAISA, so that subsequent purification was required for the removal of the SAOHOH fragment to afford FB-CAI with a free active pocket. These observations initially led us to consider that a purification step is essential for the construction of a NMR biosensor in cells, as is the case in vitro. However, in the RBC experiments, it was found that simple incubation of a RBC suspension with 1 gave rise to direct (in situ) formation of FB-eCAI whose ligand-binding pocket is vacant. Consequently, we were able to convert endogenous CAI (eCAI) into the FB-eCAI biosensor inside the cells without special purification. However, the molecular mechanism of this intriguing phenomenon remained unclear. Thus, we first focused our attention on the relative intracellular concentrations of 1 and eCAI. If eCAI is present in excess compared to 1 inside the cells under the labeling conditions, the SAOHOH ligand would be transferred to non-labeled, native eCAI via equilibrium, allowing the ligand-binding pocket of the labeled FB-eCAI to be reopened. We investigated this scenario by determining the quantities of eCA and 1 present inside the cells in the labeling suspension (at 0 h). Using densitometric analysis following SDS-PAGE (CBB staining), the intracellular concentration of eCA was determined to be approximately 520 μM in 1 mL bed volume of RBCs (Fig. S5, ESI†). With this, the mole number of total eCA in the labeling sample (1 mL bed volume of RBCs, total 2 mL suspension volume) was calculated to be ca. 520 nmol. On the other hand, it was estimated that ca. 190 ± 14 nmol of 1 was present (uptaken) inside the total RBCs used, as determined by HPLC analysis of the supernatant (after centrifugation) of the labeling suspension (Fig. 5a).19 Thus, approximately 48% of total reagent 1 used for the labeling experiment (400 nmol) was incorporated into the cells. Overall, comparison of the intracellular mole numbers of eCA (520 nmol) and 1 (190 nmol) revealed that eCA was present in 2.7-fold excess over 1 in the RBC labeling setting. This moderate excess ratio is probably one factor responsible for the observation that the ligand-binding pocket of FB-eCAI reopens in RBC-based labeling.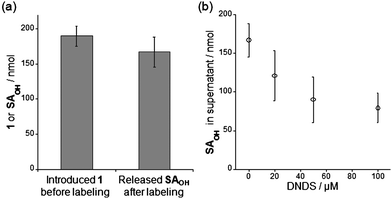 | ||
| Fig. 5 Investigation of the involvement of an anion transporter in the RBC-based labeling system. (a) Quantification of the amount of reagent 1 incorporated in RBCs before incubation (left) and of the SAOHOH fragment released into the supernatant of the post-labeling solution after incubation (right). (b) Dependence of the extracellular concentration of the SAOHOH ligand on DNDS concentration. All experiments in this figure were performed in triplicate to obtain mean and standard deviation values (shown as error bars). | ||
Human red blood cells possess a membrane anion transporter protein, termed anion exchanger 1 (AE1) or band 3.20AE1 catalyzes transport of various anionic substrates such as halogen ions, carbonate, phosphate and sulfate. Interestingly, even larger synthetic molecules, including NBD-taurine, a fluorescent sulfonate derivative, can act as a substrate for AE1-mediated anion excretion.20a Thus we hypothesized that AE1 or other transporters might be involved in the release of the anionic SAOHOH fragment from the interior to the extracellular region of the RBCs during the labeling process. To explore this possibility, we investigated whether the SAOHOH fragment could be detected in the supernatant of the post-labeling suspension after 48 h incubation.21 Surprisingly, approximately 167 ± 21 nmol of SAOHOH was present in the supernatant, which corresponds to ca. 88% of the total amount of 1 observed inside the cells before incubation (Fig. 5a). This is in agreement with the observation that the formation of ca. 160 ± 20 nmol of FB-eCAI was detected by 19F NMR measurements of the post-labeling suspension (Fig. 4b). Interestingly, the addition of 4,4′-dinitro-2,2′-stillbenedisulfonic acid (DNDS), a specific inhibitor of AE1-mediated anion transport,20a to the labeling suspension considerably lowered the extracellular concentration of SAOHOH in a DNDS-concentration-dependent manner (Fig. 5b). These results strongly suggest that the SAOHOH fragment was efficiently eliminated from the RBCs with the assistance of a natural anion transporter, AE1.
Conclusions
It is now known that many organic molecules form (colloid-like) aggregates at micromolar concentrations in aqueous media.22 The relevance and application of such aggregation to biological systems has recently begun to be addressed in the areas of drug discovery22 and drug delivery.23 In this work, we demonstrated that the LDT-based 19F-labeling reagent 1 forms self-assembled aggregates in the absence of the target protein in buffer solution. Through a comprehensive study involving the microscopic observation of aggregation morphology, light scattering measurements, product analysis and 19F NMR spectroscopy, it was shown that 1 has the following characteristics: (1) the reagent alone is NMR-silent because of its aggregation, (2) the aggregates are disrupted in the presence of the target protein (CAI) to form a protein–reagent complex that displays a clear 19F signal in a turn-on manner, and (3) after the complexation with the target protein, the reagent reacts with an amino acid residue (His67) on the protein surface, resulting in 19F-labeling (Scheme 2). Additionally, detailed kinetic investigations revealed that the self-assembly properties of the LDT reagent effectively suppresses its nonproductive hydrolytic decomposition. As well as the CAI LDT reagent, we found the suppression of hydrolysis by self-assembly in the case of LDT reagents for FKBP12 labeling (data not shown).6 Utilization of the self-assembly properties of reagents might serve as a new promising chemical strategy for selective protein labeling under biological contexts, including cells, tissues and whole organisms, using chemical electrophiles. Since the reactivity can be significantly lowered (masked) in the aggregated state until the reagent encounters the target protein, i.e., its reactivity is restored only when the reagent is recognized by the target protein, both hydrolysis and nonspecific (intermolecular) labeling of non-target proteins would be effectively suppressed. Aggregation (self-assembly) is therefore an important but largely neglected factor for determining the efficiency and selectivity of chemical labeling reagents.Detailed investigation of the processes involved in the construction of the biosensor indicated that the ligand-binding pocket of the 19F-labeled endogenous CAI (FB-eCAI) is empty, even without any special treatment after labeling in RBCs. This is in sharp contrast to in vitrolabeling, which generates FB-CAI whose active site is occupied by the cleaved ligand-containing fragment (SAOHOH). Based on the results from 19F NMR measurements, product analysis and biochemical inhibitor-based assays, this unexpected removal of the cleaved SAOHOH is likely due to: (1) re-distribution of the cleaved ligand from the FB-eCAI pocket to the native, unmodified eCAI (because the latter is present in excess) via equilibrium, and (2) the effective elimination of the anionic ligand from the interior to the exterior of the RBCs with the help of a cellular anion transporter system. Although not established, cellular transporter systems could be useful in removing labeling byproducts (in this case, a ligand-tethered phenylsulfonic acid derivative), from the cells, which might otherwise interfere with subsequent applications. Consequently, we demonstrated that it is feasible to convert an endogenous natural protein to a semisynthetic biosensor within its native habitat (in cells) in a ship-in-a-bottle manner by exogenously adding a chemical labeling reagent. We believe that these new findings reported in this work will provide a fundamental basis for the design of more general and versatile chemical methods for selective protein labeling and biosensor generation in crude biological environments.
Experimental procedures
General materials and methods
All proteins and chemicals were obtained from commercial suppliers (Sigma, Aldrich, Tokyo Chemical Industry (TCI), or Wako Pure Chemical Industries) and used without further purification. UV-visible spectra were recorded on a Shimadzu UV-2550 spectrometer. AFM measurements were performed with a SEIKO SPA-400 microscope in tapping mode. DLS measurements were performed using a NICOMP 380zls. The scattering angle was 108° and the wavelength of the laser was 520 nm. MALDI-TOF MS spectra were recorded on a Bruker Autoflex III using sinapinic acid as the matrix. 19F NMR spectra were recorded on a JEOL ECX-400P (376.5 MHz) spectrometer and calibrated with TFA (−75.6 ppm). Reversed-phase HPLC (RP-HPLC) was carried out on a Hitachi LaChrom L-7100 system equipped with a LaChrom L-7400 UV detector. UV-visible detection was performed at 254 nm, with a flow rate of 1.0 mL min−1. All runs used a mixture of acetonitrile containing 0.1% TFA (solvent A) and 0.1% aqueous TFA (solvent B).Dynamic light scattering and optical density measurements
The optical density was measured at 25 °C in 50 mM HEPES buffer (pH 7.2, 0.2 mM TFA) using a quartz cell (1 cm). A DMSO stock solution of each reagent (1 or 2) was slowly added to the buffer solution to give a final concentration of 25 μM (0.25% DMSO (v/v)). The DLS measurements were performed under the same conditions with a tubular-type cell. All measurements were carried out in triplicate.Evaluation of in vitro CAI labeling using 1 and 3
The concentration of human CAI (Sigma) was determined by measuring the absorbance at 280 nm using a molar absorption coefficient of 49000 M−1 cm−1.24CAI (150 μM) was incubated with 1 or 3 (300 μM) in 50 mM HEPES buffer (pH 7.2, 0.2 mM TFA, 10% D2O (v/v), 1.2% DMSO (v/v)) at 37 °C. Control reactions with EZA (750 μM) were also conducted. The reactions were monitored by MALDI-TOF MS and 19F NMR spectroscopy.
Kinetic studies of hydrolysis and labeling reactions
To determine the hydrolysis rates of the LDT reagents, each reagent (1 or 2: 50 μM) was dissolved in 50 mM HEPES buffer (pH 7.2, 0.2 mM TFA, 0.5% DMSO (v/v), glass tube) and incubated at 37 °C. Aliquots were taken at various times, mixed with EZA (100 μM, as an internal standard), and subjected to RP-HPLC equipped with a YMC-Pack PROTEIN column (5 μm, 250 × 4.6 mm). The elution conditions (solvent A/solvent B (v/v)) were as follows: from 0 to 10 min, 5/95; from 10 to 50 min, a linear gradient from 5/95 to 85/15. In the case of 1, the aliquots were diluted with acetonitrile (to 20%) in order to homogenize the aggregates prior to HPLC analysis. The concentrations of the hydrolysis product, SAOHOH, were estimated by determining the relative peak area of SAOHOH to the internal standard. The first-order rate constants of the hydrolysis reaction (kH·monomer or kH·aggregate) were obtained by fitting a plot of the SAOH concentration ([SAOH]) as a function of incubation time (t) to eqn (1) using KaleidaGraph (Synergy Software).| [SAOHOH] = [reagent]0(1 − exp(−kHt)) | (1) |
To determine the reaction rates of protein labeling and hydrolysis from the protein-reagent complex (kL and kH·complex, respectively), each reagent (1 or 2: 50 μM) and CAI (50 μM) was dissolved in 50 mM HEPES buffer (pH 7.2, 0.2 mM TFA, 0.5% DMSO (v/v)), and incubated at 37 °C. Aliquots were taken at various times, mixed with EZA (100 μM), and subjected to RP-HPLC as described above. First, the apparent first-order rate constants of SAOHOH formation, which correspond to (kH·complex + kL), were obtained by fitting a plot of the SAOHOH concentration ([SAOHOH]) as a function of incubation time (t) to eqn (2).
| [SAOHOH] = [reagent]0(1 − exp(−(kH·complex + kL)t)) | (2) |
In separate experiments based on MALDI-TOF MS analysis, the concentrations of labeled CAI were estimated by determining the relative peak area of labeled CAI to parental CAI. Finally, the first-order rate constants of protein labeling (kL) and hydrolysis from the protein-reagent complex (kH·complex) were obtained by fitting a plot of the concentration of labeled CAI ([labeled CAI]) as a function of incubation time to eqn (3).
| [labeled CAI] = (kL/(kH·complex + kL))[reagent]0(1 − exp(−(kH·complex + kL)t)) | (3) |
Titration of 19F NMR biosensor in vitro
19F-labeling of CAI was performed as described in the Evaluation of in vitroCAI Labeling using 1 and 3 section using CAI (150 μM) and 1 (300 μM). After 24 h incubation, the nearly quantitative labeling was confirmed by MALDI-TOF MS. The 19F-labeled CAI (FB-CAISA) was purified by size-exclusion chromatography using a TOYOPEARL HW-40F column (Tosoh Corp.). The purified FB-CAI was dissolved in 50 mM HEPES buffer (pH 7.2, 0.2 mM TFA, 10% D2O (v/v)). The in vitrotitration experiments with various sulfonamide derivatives were carried out using a solution containing FB-CAI (20 μM) in 50 mM HEPES buffer (pH 7.2, 0.2 mM TFA, 10% D2O (v/v)) in a NMR tube (550 μL) at 25 °C. The titration curves were analyzed with the nonlinear least-squares curve-fitting method to evaluate the apparent binding constants (Kapp, M−1).eCAI labeling and titration in RBCs
Blood was taken from one of the authors by Dr Eishi Ashihara (Department of Molecular Cell Physiology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine). After anti-coagulation treatment with heparin, RBCs were separated from plasma by centrifugation. The separated RBC suspensions were washed three times with HEPES-buffered saline (20 mM HEPES, 107 mM NaCl, 6 mM KCl, 1.2 mM MgSO4, 2 mM CaCl2, 11.5 mM glucose, pH 7.4, HBS). A 1 mL solution of reagent 1 (133 μM) in HBS was mixed with a 1-mL (bed volume) suspension of RBCs, incubated at room temperature for a few minutes, and centrifuged (1500 rpm, 10 min). After removing the supernatant, the same procedure was repeated two more times to treat the cells with a total of 400 nmol of 1. The 1-treated RBCs were resuspended in HBS containing 20% D2O (v/v) and 200 μM TFA (HBSDT, 1 mL, yielding a final TFA concentration of 100 μM), and subjected at several time points to 19F NMR measurements. After 48 h of incubation at 25 °C, the labeled RBCs were collected by centrifugation, and resuspended in fresh HBSDT. The in-cell titration experiments with various sulfonamide derivatives were carried out using a portion of the RBC suspension (125 μL bed volume) in a NMR tube (500 μL) at 25 °C. The titration curves were analyzed with the nonlinear least-squares curve-fitting method to evaluate the apparent binding constants (Kapp, M−1).Quantification of reagent 1 and SAOHOH following RBC-based labeling
RBC labeling with 1 was performed as described in the previous section. To determine the mole number of 1 uptaken into the cells at 0 h, the supernatants obtained during the administration of 1 were combined, mixed with EZA (100 μM, as an internal standard), and subjected to RP-HPLC on a YMC-Pack PROTEIN column (5 μm, 250 × 4.6 mm). The mole number of reagent 1 that was not uptaken into the cells was first estimated by determining the ratio between the sum of the peak areas of 1 and SAOHOH (which was produced by hydrolysis during the experiment) and the area of the internal standard. By subtracting the obtained value from 400 nmol, the mole number of 1 present in the cells was determined. After 48 h of incubation, the supernatant was collected from the labeled RBC suspension. The supernatant was mixed with EZA (100 μM) and subjected to RP-HPLC. The mole number of SAOHOH released from the cells was estimated by determining the relative peak area of SAOH to the internal standard. In the AE1 inhibition experiments, following the treatment of RBCs with 1, RBCs were resuspended in HBSDT containing DNDS (0, 20, 50 or 100 μM). After 48 h of incubation, the supernatant was obtained and analyzed as described above.Acknowledgements
We thank Drs Masahiro Shirakawa and Hidehito Tochio (Kyoto University) for assistance with 19F NMR measurements during the early stages of this project. We thank Dr Eishi Ashihara (Kyoto Prefectural University of Medicine) for his help in taking blood samples. Y. T. and Y. S. acknowledge the JSPS Research Fellowships for Young Scientists.Notes and references
- Selected reviews of semisynthetic biosensors: (a) K. A. Giuliano, P. L. Post, K. M. Hahn and D. L. Taylor, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 405–434 CrossRef CAS; (b) H. W. Hellinga and J. S. Marvin, Trends Biotechnol., 1998, 16, 183–189 CrossRef CAS; (c) R. Jelinek and S. Kolusheva, Chem. Rev., 2004, 104, 5987–6015 CrossRef CAS; (d) H. Wang, E. Nakata and I. Hamachi, ChemBioChem, 2009, 10, 2560–2577 CrossRef CAS.
- (a) M. Brune, J. L. Hunter, J. E. T. Corrie and M. R. Webb, Biochemistry, 1994, 33, 8262–8271 CrossRef CAS; (b) G. Gilardi, L. Q. Zhou, L. Hibbert and A. E. Cass, Anal. Chem., 1994, 66, 3840–3847 CrossRef CAS; (c) J. S. Marvin, E. E. Corcoran, N. A. Hattangadi, J. V. Zhang, S. A. Gere and H. W. Hellinga, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4366–4371 CrossRef CAS; (d) J. S. Marvin and H. W. Hellinga, J. Am. Chem. Soc., 1998, 120, 7–11 CrossRef CAS; (e) K. Hirose, H. Takeshima and M. Iino, Anal. Commun., 1999, 36, 175–177 RSC; (f) M. Renard, L. Belkadi, N. Hugo, P. England, D. Altschuh and H. Bedouelle, J. Mol. Biol., 2002, 318, 429–442 CrossRef CAS; (g) R. M. de Lorimier, J. J. Smith, M. A. Dwyer, L. L. Looger, K. M. Sali, C. D. Paavola, S. S. Rizk, S. Sadigov, D. W. Conrad, L. Loew and H. W. Hellinga, Protein Sci., 2009, 11, 2655–2675 CrossRef; (h) T. Morii, K. Sugimoto, K. Makino, M. Otsuka, K. Imoto and Y. Mori, J. Am. Chem. Soc., 2002, 124, 1138–1139 CrossRef CAS; (i) A. Toutchkine, V. Kraynov and K. M. Hahn, J. Am. Chem. Soc., 2003, 125, 4132–4145 CrossRef CAS; (j) P. H. Chan, H. B. Liu, Y. W. Chen, K. C. Chan, C. W. Tsang, Y. C. Leung and K. Y. Wong, J. Am. Chem. Soc., 2004, 126, 4074–4075 CrossRef CAS; (k) P. Nalbant, L. Hodgson, V. Kraynov, A. Toutchkine and K. M. Hahn, Science, 2004, 305, 1615–1619 CrossRef CAS; (l) B. E. Cohen, A. Pralle, X. Yao, G. Swaminath, C. S. Gandhi, Y. N. Jan, B. K. Kobilka, E. Y. Isacoff and L. Y. Jan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 965–970 CrossRef CAS; (m) S. Namiki, H. Sakamoto, S. Iinuma, M. Iino and K. Hirose, Eur. J. Neurosci., 2007, 25, 2249–2259 CrossRef; (n) J. Zhu and D. Pei, ACS Chem. Biol., 2008, 3, 110–119 CrossRef CAS.
- (a) I. Hamachi, T. Nagase and S. Shinkai, J. Am. Chem. Soc., 2000, 122, 12065–12066 CrossRef CAS; (b) T. Nagase, S. Shinkai and I. Hamachi, Chem. Commun., 2001, 229–230 RSC; (c) T. Nagase, E. Nakata, S. Shinkai and I. Hamachi, Chem.–Eur. J., 2003, 9, 3660–3669 CrossRef CAS; (d) E. Nakata, T. Nagase, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 490–495 CrossRef CAS; (e) Y. Koshi, E. Nakata and I. Hamachi, ChemBioChem, 2005, 6, 1349–1352 CrossRef CAS; (f) E. Nakata, Y. Koshi, E. Koga, Y. Katayama and I. Hamachi, J. Am. Chem. Soc., 2005, 127, 13253–13261 CrossRef CAS; (g) Y. Takaoka, H. Tsutsumi, N. Kasagi, E. Nakata and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 3273–3280 CrossRef CAS; (h) Y. Koshi, E. Nakata, H. Yamane and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10413–10422 CrossRef CAS; (i) T. Anai, E. Nakata, Y. Koshi, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2007, 129, 6232–6239 CrossRef CAS; (j) E. Nakata, H. Wang and I. Hamachi, ChemBioChem, 2008, 9, 25–28 CrossRef CAS; (k) Y. Koshi, E. Nakata, M. Miyagawa, S. Tsukiji, T. Ogawa and I. Hamachi, J. Am. Chem. Soc., 2008, 130, 245–251 CrossRef CAS.
- (a) C. Hoffmann, G. Gaietta, M. Bünemann, S. R. Adams, S. Oberdorff-Maass, B. Behr, J.-P. Vilardaga, R. Y. Tsien and M. H. Lohse, Nat. Methods, 2005, 2, 171–176 CrossRef CAS; (b) D. Maurel, L. Comps-Agrar, C. Brock, M.-L. Rives, E. Bourrier, M. A. Ayoub, H. Bazin, N. Tinel, T. Durroux, L. Prézeau, E. Trinquet and J.-P. Pin, Nat. Methods, 2008, 5, 561–567 CrossRef CAS; (c) C. A. Jost, G. Reither, C. Hoffman and C. Schultz, ChemBioChem, 2008, 9, 1379–1384 CrossRef CAS; (d) A recent example in which the basic strategy is likely to be applicable to cellular systems: M. A. Brun, K.-T. Tan, E. Nakata, M. J. Hinner and K. Johnsson, J. Am. Chem. Soc., 2009, 131, 5873–5884 Search PubMed.
- (a) J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS; (b) K. Johnsson, Nat. Chem. Biol., 2009, 5, 63–65 CrossRef CAS.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341–343 CrossRef CAS.
- (a) S. Tsukiji, H. Wang, M. Miyagawa, T. Tamura, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 9046–9054 CrossRef CAS; (b) S. Uchinomiya, H. Nonaka, S. Fujishima, S. Tsukiji, A. Ojida and I. Hamachi, Chem. Commun., 2009, 5880–5882 RSC.
- Other recent examples of traceless-type affinity-based labeling: (a) Y. Hwang, P. R. Thompson, L. Wang, L. Jiang, N. L. Kelleher and P. A. Cole, Angew. Chem., Int. Ed., 2007, 46, 7621–7624 CrossRef CAS; (b) C. C. Hughes, Y.-L. Yang, Y.-T. Liu, P. C. Dorrestein, J. J. La Clair and W. Fenical, J. Am. Chem. Soc., 2009, 131, 12094–12096 CrossRef CAS; (c) H. Nonaka, S. Fujishima, S. Uchinomiya, A. Ojida and I. Hamachi, Bioorg. Med. Chem. Lett., 2009, 19, 6696–6699 CrossRef CAS.
- (a) C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS; (b) V. M. Krishnamurthy, G. M. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946–1051 CrossRef CAS.
- Other examples of 19F NMR probe-incorporated proteins: (a) M. A. Danielson and J. J. Falke, Annu. Rev. Biophys. Biomol. Struct., 1996, 25, 163–195 CrossRef CAS; (b) J. C. Jackson, J. T. Hammill and R. A. Mehl, J. Am. Chem. Soc., 2007, 129, 1160–1166 CrossRef CAS; (c) C. Li, G.-F. Wang, Y. Wang, R. C. Allen, E. A. Lutz, H. Scronce, K. M. Slade, R. A. S. Ruf, R. A. Mehl and G. J. Pielak, J. Am. Chem. Soc., 2010, 132, 321–327 CrossRef CAS.
- (a) P. W. Taylor, R. W. King and A. S. V. Burgen, Biochemistry, 1970, 9, 2638–2645 CrossRef CAS; (b) A. Innocenti, S. Zimmerman, J. G. Ferry, A. Scozzafavaa and C. T. Supuran, Bioorg. Med. Chem. Lett., 2004, 14, 4563–4567 CrossRef CAS; (c) J.-Y. Winum, J.-M. Dogné, A. Casini, X. de Leval, J.-L. Montero, A. Scozzafava, D. Vullo, A. Innocenti and C. T. Supuran, J. Med. Chem., 2005, 48, 2121–2125 CrossRef CAS.
- (a) Y. Takaoka, T. Sakamoto, S. Tsukiji, M. Narazaki, T. Matsuda, T. Tochio, M. Shirakawa and I. Hamachi, Nat. Chem., 2009, 1, 557–561 CrossRef CAS; (b) K. Mizusawa, Y. Ishida, Y. Takaoka, M. Miyagawa, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 7291 CrossRef CAS.
- The 19F NMR signal of 1 was silent with CAI and EZA over 24 h in vitro.
- Chemically synthesized FBOHOH in buffer solution also showed a signal at −62.9 ppm (Fig. S2, ESI†).
- Since we set the experimental conditions where all of the labeling reagents are complexed with CAI, the determined values of kL and kH·complex are considered as the rates from the complexed state between CAI and the reagents.
- It should be noted that because of the need for a relatively high concentration of FB-CAI (>20 μM) for 19F NMR measurements, we were unable to determine the precise affinity values of FB-CAI towards the strong inhibitors, EZA, AAZ and NBS.
- The labeling site of FB-eCAI was shown to be at His67, which is identical to the site identified for FB-CAI prepared in vitro.
- Under the conditions using less than 400 μM of 1, we did not observe any toxicity for RBCs over the time period of the assay (over 48 h).
- The validity of this mole number was also supported by in-cell19F NMR measurements, in which the intracellular concentration of 1 can be determined from an integral of the signal at −62.3 ppm corresponding to the 1-eCAI complex (Fig. 4b). The mole number estimated in this manner was 194 ± 2 nmol.
- (a) O. Eidelman, M. Zangvill, M. Razin, H. Ginsburg and Z. I. Cabantchik, Biochem. J., 1981, 195, 503–513 CAS; (b) M. L. Jennings, Annu. Rev. Biophys. Biophys. Chem., 1989, 18, 397–430 CrossRef CAS.
- Note that following mixing of the RBC suspension with 1, the supernatant is replaced with fresh buffer before starting incubation (also see the Methods section).
- (a) B. Y. Feng, A. Shelat, T. N. Doman, R. K. Guy and B. K. Shoichet, Nat. Chem. Biol., 2005, 1, 146–148 CrossRef CAS; (b) K. E. D. Coan and B. K. Shoichet, Mol. BioSyst., 2007, 3, 208–213 RSC; (c) B. Y. Feng, B. H. Toyama, H. Wille, D. Y. Colby, S. R. Collins, B. C. H. May, S. B. Prusiner, J. Weissman and B. K. Shoichet, Nat. Chem. Biol., 2008, 4, 197–199 CrossRef CAS; (d) K. E. D. Coan and B. K. Shoichet, J. Am. Chem. Soc., 2008, 130, 9606–9612 CrossRef CAS.
- Y. V. Frenkel, A. D. J. Clark, K. Das, Y.-H. Wang, P. J. Lewi, P. A. J. Janssen and E. Arnold, J. Med. Chem., 2005, 48, 1974–1983 CrossRef CAS.
- C. Chazalette, B. Masereel, S. Rolin, A. Thiry, A. Scozzafava, A. Innocenti and C. T. Supuran, Bioorg. Med. Chem. Lett., 2004, 14, 5781–5786 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: DLS analyses, optical densities, representative HPLC chromatograms, 19F NMR spectra and SDS-PAGE analyses. Figs. S1–S5. See DOI: 10.1039/c0sc00513d |
| ‡ Present address: Top Runner Incubation Center for Academia-Industry Fusion, Nagaoka University of Technology, 1603-1 Kamitomioka, Nagaoka, Niigata 940-2188, Japan. |
| This journal is © The Royal Society of Chemistry 2011 |