Energy versus electron transfer in organic solar cells: a comparison of the photophysics of two indenofluorene: fullerene blend films†
Ying W. Soona,
Tracey M. Clarkea,
Weimin Zhanga,
Tiziano Agostinellib,
James Kirkpatrickb,
Clare Dyer-Smithb,
Iain McCullocha,
Jenny Nelsonb and
James R. Durrant*a
aCentre for Plastic Electronics, Departments of Chemistry, Exhibition Road, London, SW7 2AZ, UK
bCentre for Plastic Electronics, Department of Physics, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: j.durrant@imperial.ac.uk
First published on 18th March 2011
Abstract
In this paper, we compare the photophysics and photovoltaic device performance of two indenofluorene based polymers: poly[2,8-(6,6,12,12-tetraoctylindenofluorene)-co-4,7-(2,1,3-benzothiodiazole] (IF8BT) and poly[2,8-(6,6,12,12-tetraoctylindenofluorene)-co-5,5-(4′,7′-di-2-thienyl-2′,1′,3′-benzothiodiazole] (IF8TBTT) blended with [6,6]-phenyl C61 butyric acid methyl ester (PCBM). Photovoltaic devices made with IF8TBTT exhibit greatly superior photocurrent generation and photovoltaic efficiency compared to those made with IF8BT. The poor device efficiency of IF8BT/PCBM devices is shown to result from efficient, ultrafast singlet Förster energy transfer from IF8BT to PCBM, with the resultant PCBM singlet exciton lacking sufficient energy to drive charge photogeneration. The higher photocurrent generation observed for IF8TBTT/PCBM devices is shown to result from IF8TBTT's relatively weak, red-shifted photoluminescence characteristics, which switches off the polymer to fullerene singlet energy transfer pathway. As a consequence, IF8TBTT singlet excitons are able to drive charge separation at the polymer/fullerene interface, resulting in efficient photocurrent generation. These results are discussed in terms of the impact of donor/acceptor energy transfer upon photophysics and energetics of charge photogeneration in organic photovoltaic devices. The relevance of these results to the design of polymers for organic photovoltaic applications is also discussed, particularly with regard to explaining why highly luminescent polymers developed for organic light emitting diode applications often give relatively poor performance in organic photovoltaic devices.
Introduction
Semiconducting polymers are attracting extensive interest for a range of optoelectronic device applications, including organic light emitting diodes (OLEDs), displays, organic field effect transistors (OFETs) and solar cells.1–4 A key consideration of the design of semiconducting polymers for such applications is the extent to which polymers found to be effective for one application may also be attractive for another. This has particularly been an issue for organic photovoltaics (OPVs), where until recently many of the polymers studied had been originally developed for the more mature OLED and OFET fields.5,6An important polymer design consideration for OLED applications is the photoluminescence (PL) emission yield, with a high PL yield being a pre-requisite for efficient OLED function. In general, a high PL yield has also been thought to be a positive indicator of a polymer's suitability for OPV applications, as a high PL yield implies a long exciton lifetime in the neat polymer, increasing the likely exciton diffusion length and therefore the probability of photogenerated excitons surviving until they reach the donor acceptor interface where charge pair generation can occur.7 Herein we report a study of two polyindenofluorene polymers blended with the electron acceptor[6,6]-phenyl C61butyric acid methyl ester (PCBM) to investigate the influence of photophysical processes on charge generation. We show that the lower photovoltaic device performance observed for one of these polymers can be directly attributed to its relatively intense, and blue shifted PL intensity, which results in ultrafast, energetically downhill singlet energy transfer from the polymer to the fullerene. We thus demonstrate that, in contrast to the requirements for OLEDs, a high PL yield is not necessarily a positive attribute for polymers for use in OPV.
Charge photogeneration in polymer/fullerene based organic solar cells is typically considered to be based upon photoinduced electron transfer from the polymer singlet exciton to the fullerene acceptor. However, relatively little attention has been placed upon the potential impact of polymer to fullerene singlet energy transfer upon the overall film photophysics.8–11 Such singlet, resonance or ‘Förster’ energy transfer will be dependent upon the photoluminescence intensity of the neat polymer, and the spectral overlap of this photoluminescence with the fullerene absorption. In principle, singlet fullerene excitons generated by such energy transfer (or by direct optical excitation) may also drive charge separation at the polymer/fullerene interface (in this case corresponding to the HOMO to HOMO hole transfer rather than the more generally considered LUMO to LUMO electron transfer). Such ‘reverse’ charge separation has been reported in some recent studies,12,13 although its importance in determining overall device performance and the relevance of such energy and electron transfer processes to polymer design are at present only poorly understood.
Studies of charge photogeneration in organic solar cells are increasingly focusing upon the potential role of interfacial charge transfer states (sometimes referred to as radical ion pair or exciplex states)14–18 in mediating the charge separation process. Several studies have addressed the impact of the relative energetics of singlet and triplet exciton states versus charge transfer and charge separated states19,20 in determining the efficiency of this charge photogeneration, and related such dependencies to photophysical models of film function. A further consideration has been the potential role of charge transfer states in mediating recombination losses in such devices, including charge recombination from triplet charge transfer states to yield neutral triplet excitons. In this regard, the potential presence of energy transfer processes, and therefore consideration of both forward (LUMO to LUMO) and reverse (HOMO to HOMO) charge separation may be an important consideration in understanding differences in behaviour observed between different materials series.
Polyfluorenes have attracted extensive interest for application in light emitting diodes due to their good transport properties, high photoluminescence (PL) yields, stability and synthetic tunability.21,22 However, polyfluorenes found to yield efficient OLEDs, such as poly(9,9-dioctylfluorene-alt-benzothiadiazole) (F8BT), poly(9,9-dioctylfluorene) (PFO) and poly(9,9-dioctylfluorene-co-bithiophene) (F8T2) have typically been found to give relatively poor photovoltaic device efficiencies when blended with PCBM.23–25 In contrast, other polyfluorenes, such as the APFO series of fluorene-based co-polymers, usually exhibit comparatively lower PL yields26,27 (thus being relatively inefficient in OLED applications) and promising OPV performances.28,29
For the case of F8BT, the poor OPV performance in blend films with PCBM has been correlated with its high ionisation potential (5.9 eV). This raises the energy of the F8BT+/PCBM− charge-separated state such that its formation is a thermodynamically less favourable process.20 In addition, F8BT's photoluminescence shows good spectral overlap with PCBM absorption, likely to result in efficient singlet energy transfer from F8BT to PCBM followed by intersystem crossing to yield PCBM triplet states.25 This absence of charge pair generation has also been observed in spectroscopic studies of blends of other polyfluorenes with high PL efficiencies with PCBM. Benson-Smith et al.17,20 conducted transient absorption spectroscopy on a range of polyfluorenes and reported that photoexcitation of blends of PCBM with polyfluorenes with ionisation potentials greater than 5.5 eV produced evidence of PCBM triplets formation rather than polarons, whilst polymers with ionisation potentials of 5.2 eV or less showed evidence of polaron generation. We note that the energetic dependence of polaron photogeneration upon polymer ionisation potential observed for this polyfluorene series was found to be distinct from the energetic dependence observed for polythiophene/PCBM blend films,19,30 where charge photogeneration efficiency was observed to correlate not with polymer ionisation potential but rather with the energy offset between the polymer singlet exciton and the charge separated polarons (corresponding effectively to a dependence upon the polymer LUMO energy). As we show herein, this difference in behaviour can be understood in terms of differences in energy transfer dynamics between these two materials systems.
In contrast to the aforementioned F8BT and related polymers, a number of other polyfluorene copolymers with donor–acceptor intrachain character and reduced band gap have shown good potential for use in photovoltaics. For example, blends of APFO-3 with PCBM have produced reasonably efficient solar cells with power conversion efficiencies of up to 4.2%.31,32 Furthermore, ultrafast spectroscopic studies of APFO-3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PCBM blend films have revealed evidence of polymer polarons, despite the high ionisation potential (IP) of the polymer (5.8 eV).33 A statistical copolymer, known as Red F, containing the same dithiophene-benzothiadiazole unit as APFO-3 also showed promising OPV performance in blends with PCBM.34 The APFO class of polyfluorenes therefore do show clear evidence of charge separation and thus behave very differently from polyfluorenes such as F8BT. We note that such APFOpolymers typically show weaker, red-shifted photoluminescence, and relatively poor OLED performance.35
:PCBM blend films have revealed evidence of polymer polarons, despite the high ionisation potential (IP) of the polymer (5.8 eV).33 A statistical copolymer, known as Red F, containing the same dithiophene-benzothiadiazole unit as APFO-3 also showed promising OPV performance in blends with PCBM.34 The APFO class of polyfluorenes therefore do show clear evidence of charge separation and thus behave very differently from polyfluorenes such as F8BT. We note that such APFOpolymers typically show weaker, red-shifted photoluminescence, and relatively poor OLED performance.35
This paper focuses upon a comparative study of the photophysics of two poly-indenofluorenes, poly[2,8-(6,6,12,12-tetraoctylindenofluorene)-co-4,7-(2,1,3-benzothiodiazole] (IF8BT) and poly[2,8-(6,6,12,12-tetraoctylindenofluorene)-co-5,5-(4′,7′-di-2-thienyl-2′,1′,3′-benzothiodiazole] (IF8TBTT) (see Fig. 1) both in neat films and blended with PCBM. The indenofluorene moiety is composed of three benzene rings fused into a rigid structure by tetravalent bridging carbons. This structure results in a planar aryl geometry with extended conjugation and has been shown to favour chain stacking and charge transport36,37—indeed this potential for high charge transport was one of the initial motivations for this study. IF8BT and IF8TBTT are donor acceptor copolymers that are analogous to F8BT and APFO-3, with the indenofluorene unit replacing fluorene. In spite of high charge mobilities38 these indenofluorenes did not lead to superior photovoltaic device performance compared to the fluorene analogues. Here we focus rather on a comparative study of the photophysics of these two indenofluorenepolymers when blended with PCBM and address the impact of energy versuselectron transfer on the performance of organic solar cells.
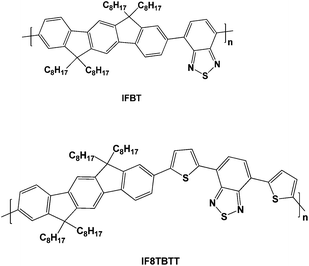 | ||
| Fig. 1 Molecular structures of the polymers discussed in this paper. | ||
Results
Photovoltaic devices fabricated with IF8BT/PCBM blend films exhibited negligible photocurrent generation, consistent with previous reports on F8BT-based devices.24 In contrast, IF8TBTT/PCBM based devices exhibited reasonably good photovoltaic performance, with an 80 wt% blend device achieving an average power conversion efficiency (PCE) of 1.9% (short circuit current, JSC = 4.5 mA cm−2; open circuit voltage, VOC = 0.96 V; fill factor, FF = 0.42), as shown in Fig. 2 and Table 1. Similar device performance was obtained for the analogous fluorenecopolymerAPFO-3 (data not shown), in agreement with previous literature.28 We note that this photovoltaic performance for IF8TBTT, and particularly the reasonably efficient generation of photocurrent, required a high PCBM loading in the blend film, as shown in Table 1. Reducing the PCBM loading to 50 wt% results in a three-fold reduction of short circuit photocurrent relative to the 80 wt% blend. We will return to this composition dependence later.| PCBM(wt)/% | Thickness/nm | Voc/V | Jsc/mAcm−2 | FF | PCE/% |
|---|---|---|---|---|---|
| 50 | 140 | 0.83 | 1.4 | 0.25 | 0.28 |
| 66 | 130 | 0.87 | 2.7 | 0.27 | 0.64 |
| 75 | 120 | 0.92 | 4.4 | 0.40 | 1.6 |
| 80 | 120 | 0.96 | 4.5 | 0.42 | 1.9 |
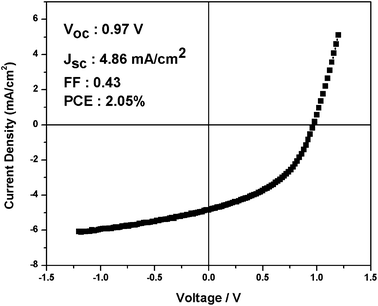 | ||
| Fig. 2 The J V−1 curve of an IF8TBTT/PCBM (80 wt% PCBM) device under simulated AM1.5 sunlight, exhibiting a power conversion efficiency of 2.05%. | ||
The primary aim of this paper is to show how the difference in photocurrent generation between these two polymers can be understood in terms of the photophysics of the blend films. In this regard, we start with a comparison of the properties of the neat polymer films.
Neat IF8BT films possess a broad low energy absorption at 467 nm (Fig. 4a), similar to that of neat F8BT (460 nm) and F8T2 (470 nm), as well as higher energy absorption bands at 320 and 360 nm. In contrast, neat IF8TBTT films exhibit an absorption maximum at 540 nm (Fig. 4a), similar to that observed for APFO-3 and red-shifted by over 70 nm compared to IF8BT, as well as a higher energy maximum at 397 nm. For both IF8BT and IF8TBTT, their absorption bands do not significantly shift in wavelength with varying PCBM concentration (Fig. S1, supporting information†). This lack of composition dependence and the lack of vibronic structure (compared to P3HT) suggest that the PCBM does not influence the packing of the polymer chains, consistent with their relatively amorphous nature.30,39,40
In order to investigate these absorption spectra further, quantum chemical calculations were performed on dimer41 models of IF8BT and IF8TBTT. Time dependent formalism (TDDFT) calculations on IF8BT reveal two photo-active bands: one at 520 nm and one at 360 nm. The lower energy band is predominantly HOMO–LUMO in character, whereas the higher band is predominantly HOMO–LUMO + 2. These molecular orbitals are illustrated in Figure 5. IF8TBTT also has two bright excited states: one at 640 nm, and one at 430 nm. We assign these to the two visible bands observed for this polymer, even though the energy of the lowest band is somewhat underestimated due to the failure of DFT in treating donor acceptor excitations.42 The 640 nm excitation has predominantly HOMO–LUMO character, whereas the 430 nm excitation is predominantly HOMO–LUMO + 1. Since the HOMO of IF8TBTT is predominantly localised across the thiophene and benzothiadiazole unit, the low energy absorption is mainly unaffected by the indenofluorene unit and is rather similar to absorption in APFO-3. These results are in agreement to those obtained from semi-empirical calculations previously performed on APFO-3.43 The second absorption band differs in wavelength slightly between the IF8TBTT and APFO-3 due to the significant contribution of the fluorene/indenofluorene unit to this transition. A significant difference between IF8BT and IF8TBTT is that in IF8BT the HOMO is delocalised over the entire backbone and is deeper by 0.27 eV than the HOMO of IF8TBTT. DFT calculations suggest that the reduction in band gap is mainly due to the strong electron donating character of the thiophene units.
Photoluminescence data for neat films of IF8BT and IF8TBTT are shown in Fig. 4b. It is apparent that IF8TBTT's PL is red-shifted and much weaker (∼14 fold) than that of IF8BT. As discussed above, IF8TBTT has a more hybridised molecular orbital character due to the introduction of the electron rich thiophene units which results in the red shift. The lower emission intensity for IF8TBTT may be due to the solid state intermolecular aggregating effect of the planar dithiophene-benzothiadiazole units along the backbone, or result from HOMO orbital delocalisation over its consistuent thiophene rings (thiophene based polymers typically exhibit low photoluminescence yields).
It is apparent that the spectral overlap between polymer photoluminescence and PCBM absorption (also shown in Fig. 4b) is much larger for IF8BT than for IF8TBTT. As a consequence of stronger PL intensity in the neat IF8BT film and hence higher spectral overlap with PCBM absorption, we can expect the rate constant for Förster resonance energy transfer from polymer singlet excitons to PCBM to be ∼50 fold faster in IF8BT/PCBM blend films relative to IF8TBTT/PCBM (see supporting information for details of this calculation†). Note that direct determination of the rate of energy transfer will be addressed in subsequent studies.
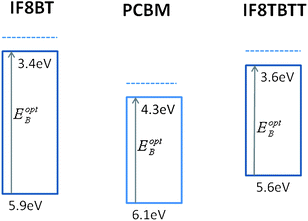 | ||
| Fig. 3 Energy levels diagram of IF8BT, PCBM and IF8TBTT HOMO and LUMO levels, employing ionisation potentials determined by UPSspectroscopy and optical bandgaps determined from optical absorption and emission spectra. The dotted lines are the electron affinity estimated assuming an exciton binding energy of ∼300 meV. We note there is significant uncertainty in the literature over the absolute electron affinity of PCBM, with values ranging between 3.7 and 4.3 eV.33,69,70 | ||
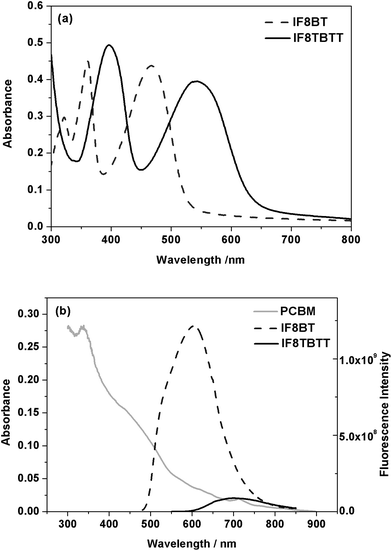 | ||
| Fig. 4 (a) Absorption spectra of IF8BT and IF8TBTT in neat polymer films. (b) Emission spectra of IF8BT and IF8TBTT films normalised for differences in film absorption at the excitation wavelength of 468 nm, overlaid with the PCBM absorption spectrum (gray line). | ||
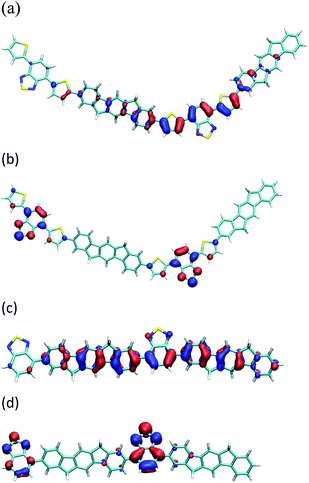 | ||
| Fig. 5 The (a) HOMO and (b) LUMO of a IF8TBTT dimer models, (c) HOMO and (d) LUMO of a IF8BT dimer models, calculated using the B3LYP/6-31G(d) method and basis set. | ||
We turn now to consideration of the band energies in polymer/fullerene blend films. Ionisation energies for IF8BT and IF8TBTT were determined by an ambient ultraviolet photoelectron spectroscopy (PESA) technique44 to be 5.9 ± 0.05 and 5.6 ± 0.05 eV, respectively, an energy difference consistent in sign and magnitude with our DFT calculations. We employ the optical data above to calculate optical bandgaps and therefore LUMO levels, as illustrated in Fig. 3. It is apparent that in terms of LUMO level energies, IF8BT shows the largest energy offset relative to the PCBM LUMO, and therefore might be expected to favour electron transfer compared to IF8TBTT. However, in terms of HOMO level energies, IF8TBTT shows the larger energy level offset relative to the PCBM HOMO level. As we discuss below, this HOMO level offset is likely to influence the rate of hole transfer from PCBM singlet excitons to the polymer, sometimes referred to as reverse electron transfer.
Having considered the properties of neat polymer films, we now consider the behaviour of blend films with PCBM. Fig. 6 shows photoluminescence (PL) spectra for IF8BT/PCBM and IF8TBTT/PCBM blend films as a function of PCBM composition, exciting the polymer absorption bands at 468 nm and 540 nm, respectively. Both blend films series showed strong PL quenching in the presence of only 5 wt% PCBM, with IF8BT showing even stronger quenching than IF8TBTT (quantitatively the addition of 5 wt% PCBM resulted in quenching of the polymerPL down to 0.5% and 8% of the neat IF8BT and IF8TBTT film PL respectively). We note the presence of PCBM in IF8BT blend films also causes a blue shift in the PL spectra, indicative of a change in polymer morphology. In addition, the IF8BT/PCBM blend films all showed the clear appearance of a new emission band at 720 nm, characteristic of PCBM singlet exciton emission.45 This PCBM emission was observed even at only 5 wt% PCBM, where PCBM absorption at the excitation wavelength is negligible. As such, this emission cannot derive from direct optical excitation of the PCBM, but rather must originate from singlet energy transfer from IF8BT to PCBM. In contrast, no PCBM singlet emission could be observed in IF8TBTT/PCBM blend films, apart from a very weak band observed at 75 wt% PCBM content, which could be due to direct optical excitation of large PCBM domains at this high PCBM composition. As such, we conclude that for both polymers the addition of only 5 wt% PCBM results in strong (>90%) quenching of polymer singlet excitons. For IF8BT/PCBM blend films, we observe clear evidence of singlet energy transfer from IF8BT to PCBM, similar to that observed previously for F8BT/PCBM blend films.25 However, for IF8TBTT/PCBM blend films we observe no evidence for such singlet energy transfer.
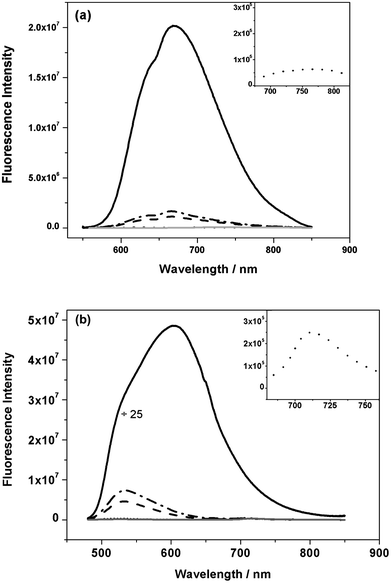 | ||
Fig. 6 The photoluminescence spectra of (a) IF8TBTT and (b) IF8BT in neat polymer films and in blends with PCBM of varying composition with insets showing the magnified spectra for 50 wt% PCBM. The inset for IF8BT blend film shows an additional emission band at 720 nm. (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) neat, neat, ![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) 5% PCBM, 5% PCBM, ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) 10% PCBM, ⋯ 50% PCBM, 75% PCBM). 10% PCBM, ⋯ 50% PCBM, 75% PCBM). | ||
Transient absorption spectroscopy was then utilised in order to identify the transient species formed as a result of photoexcitation for both IF8BT/PCBM and IF8TBTT/PCBM. The transient spectrum of a neat IF8BT film reveals a broad oxygen-sensitive band centred around 900 nm, which can be assigned to the polymer triplet state with a monoexponential lifetime in the absence of oxygen of 3 μs (Fig. 7). In blend films with PCBM, however, a transient oxygen-sensitive band at 710 nm is observed with a monoexponential lifetime in the absence of oxygen of 30 μs, consistent with previously reported data for the PCBM triplet state.20,46 These results, in conjunction with the PCBM singlet peak observed in the PL spectra, support the occurrence of energy transfer in this IF8BT/PCBM blend system. The initial photoexcitation of the polymer generates the polymer singlet state, which is subsequently quenched by energy transfer to the PCBM singlet state, followed by intersystem crossing to form the long-lived PCBM triplet observed in the transient spectroscopic data. This is likely to be the dominant mechanism, although we note that the possibility of some triplet generation via a charge transfer state cannot be entirely ruled out.20
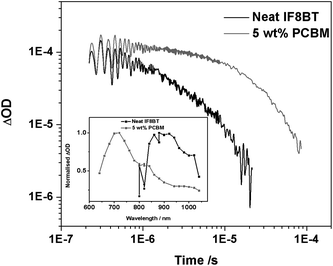 | ||
| Fig. 7 The transient absorption data for an IF8BT neat film and an IF8BT/PCBM (5 wt% PCBM) blend film, measured using 80 μJ cm−2 excitation at 485 nm and a probe wavelength of 950 nm. The inset shows the transient absorption spectrum of each film at 10 μs (for the neat film) and 1 μs (for the blend). | ||
We now turn to the transient absorption data for IF8TBTT/PCBM (1:1) blend films (data for other compositions and neat IF8TBTT are considered below). Photoexcitation resulted in the initial (1 μs) appearance of a broad absorption with a maximum at 770 nm, which at later times (>25 μs) partially splits into two overlapping absorption bands at ∼740 nm and 950 nm (Fig. 8a insert). Typical decay dynamics, probed at 950 nm, are also shown in Fig. 8a; similar dynamics were observed at all wavelengths between 700 and 1000 nm. These dynamics are clearly biphasic, where the relatively large, oxygen dependent, monoexponential fast phase is assigned to IF8TBTT triplet states with a lifetime of ∼1 μs (in the absence of the oxygen). We note that the spectrum of this fast phase (approximately corresponding to the 1 μs spectrum shown in Fig. 8a) is clearly distinguishable from that of PCBM triplet absorption, thereby ruling out assignment of this initial, oxygen dependent signal to PCBM triplet states. This is consistent with our lack of observation of PCBM singlet emission in our PL measurements of IF8TBTT/PCBM blend films. This fast phase assigned to IF8TBTT triplet states is followed by a relatively weak, power law decay, apparent as a straight line on the log/log plot in Fig. 8a. Such power law decays are characteristic of bimolecular (non-geminate) recombination of dissociated charge carriers, as we have discussed previously.30,47 This slow phase is unaffected by the presence of pure oxygen, supporting its assignment to bimolecular polaron recombination. All IF8TBTT/PCBM blend films showed such power law decays with an exponent, α, in the range 0.2 to 0.6. The sub-unity value of α is indicative of bimolecular recombination in the presence of an exponential distribution of localised (trap) states.47 On the basis of these dynamics, we therefore assign the 740 nm and 950 nm absorption bands observed for time delays >25 μs to IF8TBTT polaron absorption. The assignment of the 950 nm band to IF8TBTT polarons was confirmed by chemical oxidation experiments in solution, which resulted in the appearance of an analogous absorption maximum at 985 nm in the absorption spectrum of the radical cation (Fig. S2, supporting information†).48 Further evidence for the assignment of the fast and slow phases to IF8TBTT triplets and dissociated polarons respectively comes from excitation density dependence data (Fig. 8b). The amplitude of the fast phase increases approximately linearly with excitation density, consistent with its assignment to IF8TBTT triplets. In contrast, the amplitude of the power law decay phase saturates at higher excitation densities, consistent with its assignment to trap-limited bimolecular recombination49–51 In summary, we conclude that in IF8BT/PCBM films we observe PCBM triplets, whereas in IF8TBTT/PCBM films we observe IF8TBTT triplets as well as dissociated polarons at longer time delays.
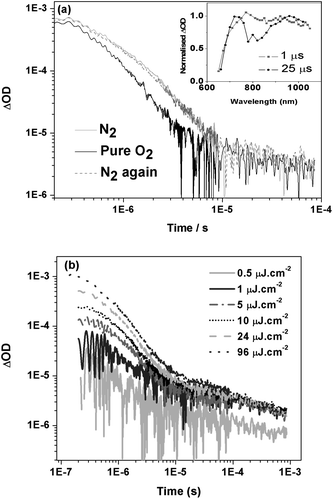 | ||
| Fig. 8 (a) Transient absorption data for an IF8TBTT/PCBM film (50 wt% PCBM) in nitrogen and pure oxygen environments, measured using 90 μJ cm−2 excitation at 520 nm and a probe wavelength of 950 nm. The inset shows the transient absorption spectra of the film at 1 μs and 25 μs. (b) Transient absorption data for an IF8TBTT/PCBM (75 wt% PCBM) blend film as a function of excitation density. | ||
We conclude our results by considering the composition dependence of the transient absorption data for IF8TBTT/PCBM blend films. Fig. 9 shows transient absorption dynamics monitored at 980 nm for neat IF8TBTT, and for blend films up to 75 wt% PCBM. It is apparent that these transient dynamics are relatively insensitive to blend composition. The amplitude of the slow, power law decay increases two-fold between 50 and 75 wt% PCBM content. A similar composition dependency was also observed at ten-fold lower excitation densities, confirming that this composition dependency was not influenced by any saturation effect. This composition dependence is in reasonable agreement with the increase in short circuit photocurrent observed between these two PCBM compositions (increase of factor 3; Table 1), suggesting that this increase in photocurrent derives primarily from an increase in the yield of dissociated charges. Furthermore, the yield of IF8TBTT triplets increases two-fold when going from the neat polymer film to blends with PCBM. This contrasts with the strong PL quenching observed in the blend films, which is indicative of efficient quenching of the IF8TBTT singlet excitons, which would be expected to reduce the yield of IF8TBTT triplet states formed by direct intersystem crossing from IF8TBTT singlet excitons. We note that we have previously observed similar behaviour (strong PL quenching coupled with an increase in polymer triplet yield) for a polythiophene/PCBM blend film.52,53 As previously, we assign the increased triplet yield in the presence of PCBM to triplet formation via radical pair or ‘charge transfer state’ intersystem crossing—whereby the observed triplet states are formed by recombination of triplet charge transfer states.52 Finally we note that we observe a smaller, but non-negligible yield of dissociated polarons in the neat IF8TBTT film, as previously reported for MDMO-PPV and APFO-3 films.50
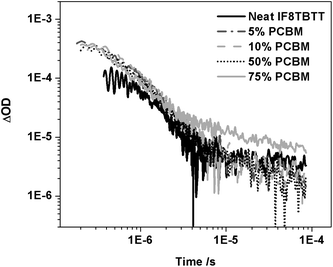 | ||
| Fig. 9 The transient absorption data for an IF8TBTT neat film and IF8TBTT/PCBM blend films with varying PCBM concentration (5, 10, 50 and 75 wt% PCBM), measured using 80 μJ cm−2 excitation at 520 nm and a probe wavelength of 980 nm. | ||
Discussion
It is apparent from the data shown above that the photophysics of IF8BT/PCBM films are strikingly different from those of IF8TBTT/PCBM films. Photoexcitation of IF8BT in IF8BT/PCBM blend films results in singlet energy transfer to PCBM followed by intersystem crossing to yield PCBM triplet states. In contrast, photoexcitation of IF8TBTT/PCBM blend films results in the generation of IF8TBTT triplet states and dissociated polarons. The observation of polarons for the IF8TBTT/PCBM blend films but not for IF8BT/PCBM correlates with our observation of photocurrent generation for the IF8TBTT/PCBM based solar cells but not for IF8BT/PCBM solar cells. Furthermore, the increase in polaron yield at PCBM concentrations greater than 50 wt% in IF8TBTT/PCBM films is in good agreement with the increase in photocurrent observed in the corresponding devices. It can thus be concluded that the differences in photovoltaic performance of these two polymers can be understood directly from differences in the photophysics of the blend films.Fig. 10 summarises the conclusions of our photophysical studies of IF8TBTT/PCBM and IF8BT/PCBM blend films and the energetics of the relevant electronic states. This figure includes consideration of the role of interfacial charge transfer states in mediating charge photogeneration in these blend films. We, and others, have previously reported several studies addressing the importance of such charge transfer states in determining the efficiency of charge photogeneration in organic solar cells, as we have reviewed elsewhere.14
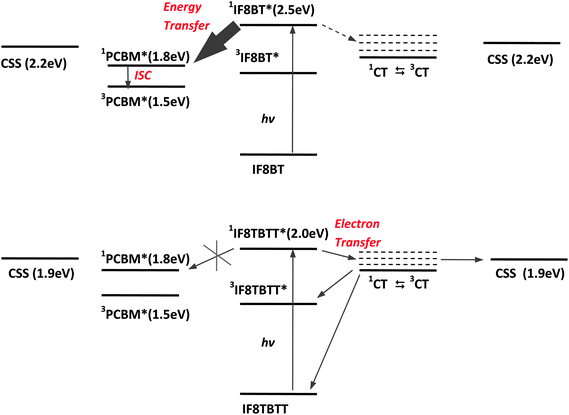 | ||
| Fig. 10 (a) Summary of the photophysics of IF8BT/PCBM blend films. Photogeneration of IF8BT singlet excitons results in rapid energy transfer to PCBM (solid arrow). The resultant PCBM singlet excitons have insufficient free energy to drive charge separation; these singlet excitons therefore undergo either radiative or non-radiative decay to the ground state or intersystem crossing to the PCBM triplet state (3PCBM*). (b) Summary of the photophysics of IF8TBTT/PCBM blend films. Photogeneration of IF8TBTT singlet excitons results in charge separation to generate interfacial charge transfer states. These charge transfer states either dissociate to yield free charges or undergo geminate recombination, primarily via spin mixing to the triplet CT state followed by recombination to the polymer triplet state. Singlet energy transfer from IF8TBTT to PCBM is not observed due to relatively weak, red-shifted IF8TBTT photoluminescence, resulting in a low energy transfer rate constant. Both plots assume a PCBM electron affinity of 3.7 eV, although we note that using a value of 4 eV would not qualitatively change the figure. | ||
It appears likely that the lack of dissociated charge photogeneration in IF8BT/PCBM blend films compared to IF8TBTT/PCBM does not result from slower electron transfer or dissociation kinetics for IF8BT/PCBM. Indeed the energetic driving force for electron transfer from the IF8BT singlet exciton to yield the charge separated state IF8BT+/PCBM− is greater than for IF8TBTT (see Fig. 10 or Fig. 3). We have previously shown for polythiophene/PCBM films that a large energetic driving force can be correlated to the yield of long-lived charges.19 This behaviour was assigned to the need for charges to provide sufficient thermal energy to overcome their Coulomb attraction in the initial geminate pair state, and avoid geminate recombination. Here, the absence of charge photogeneration for IF8BT/PCBM cannot be explained by a lower driving force for electron transfer. Rather, it appears more likely to result from much faster singlet Förster energy transfer from the polymer to PCBM. The blue-shifted and stronger photoluminescence quantum yield observed for IF8BT compared to IF8TBTT predicts a ∼50 fold increase in the Förster energy transfer rate constant (see above). We note that the sixteen-fold stronger PL quenching observed for IF8BT/PCBM relative to IF8TBTT/PCBM (Fig. 6) provides support for the presence of a rapid quenching pathway for IF8BT/PCBM blend films, consistent with much faster Förster energy transfer. Assuming a lifetime for the IF8BT singlet exciton of ∼500 ps (as observed for F8BT25), our observation of 99.5% PL quenching with the addition of only 5% PCBM indicates an energy transfer rate constant of greater than 1011s−1 (see supporting information†). Thus it appears likely that the lack of charge photogeneration observed in IF8BT/PCBM blend films does not result from any inherent inability of IF8BT singlet excitons to drive charge separation, but rather from the rapid quenching of these excitons by energy transfer to PCBM through a process that competes successfully with exciton diffusion to, and separation at, the polymer/fullerene interface. In other words, for these blend films, there exists a kinetic competition between energy and electron transfer from the polymer excitons to the PCBM. For IF8BT, energy transfer dominates; however for IF8TBTT, the lower spectral overlap and emission intensity results in a lower energy transfer rate constant, enabling a higher yield for electron transfer.
Förster singlet energy transfer from the polymer to PCBM does not in itself prevent photoinduced charge separation. Indeed, several studies have previously reported charge photogeneration in organic donor/acceptor heterojunctions originating from acceptor rather than donor singlet excitons.12,13,54,55 Such charge photogeneration corresponds to hole transfer from the acceptor HOMO orbital to the donor HOMO orbital. For the case of IF8BT/PCBM, some 0.7 eV is lost during energy transfer from polymer to PCBM, resulting in a PCBM singlet exciton that has less free energy than the IF8BT+/PCBM− charge separated state, as illustrated in Fig. 10. As such, in these blend films, the PCBM singlet exciton has insufficient energy to drive hole transfer to polymer, so failing to achieve charge photogeneration. This conclusion, based on energetic considerations, is confirmed by our observations of both PCBM singlet exciton photoluminescence and PCBM triplet formation, both indicative of the inability of PCBM singlet excitons to drive efficient charge photogeneration.
We thus conclude that the poor (indeed negligible) photovoltaic performance of IF8BT/PCBM based organic solar cells does not derive from an insufficient energetic driving force between the IF8BT singlet exciton and the charge separated state. Rather, it derives from the presence of a competing energy transfer pathway from IF8BT to PCBM. This energy transfer pathway results in a large loss of exciton free energy (∼0.7 eV), with the resulting PCBM excitons having insufficient free energy to drive charge separation. The efficiency of this competing energy transfer derives from the high photoluminescence efficiency of IF8BT coupled with its relatively blue-shifted emission spectrum, enhancing spectral overlap of this emission with PCBM absorption. Once singlets are generated, PCBM singlet emission, radiationless decay or intersystem crossing are faster than reverse electron transfer, resulting in a negligible photocurrent generation. Thus it appears that a key characteristic of this polyfluorene, namely the high efficiency and spectral position of its PL, which makes such polyfluorenes attractive for OLED applications, is directly the cause of the inefficiency of this polymer in OPV applications.
The studies reported herein have employed only two polyfluorenes. However it appears likely that the key conclusions of this study are directly applicable to other polyfluorenes. It is striking that the fluorene-based polymers that have been shown to be promising for OSC applications, such as APFO-3, typically show either red-shifted or relatively weak luminescence.20,37,56,57 Indeed, our own studies of APFO-3/PCBM blend films exhibited very similar photophysics to the IF8TBTT/PCBM films studied herein, consistent with the reasonably efficient device performance observed with both these polymers. Additionally, one of the reported polyfluorenes, F8T2 which has a similar IP (5.5 eV) to IF8TBTT (5.6 eV), shows only the formation of PCBM triplets via energy transfer when blended with PCBM, with no sign of polaron formation.20 This difference in photophysical behaviour between F8T2 and IF8TBTT can be attributed to the different emission behaviour, with F8T2 having a blue-shifted emission and higher photoluminescence yield, consistent with efficient energy transfer to PCBM. It has furthermore been shown that for fluorene-based polymers which exhibit relatively blue and/or intense luminescence, such as F8BT, PFO, F8T2, RedF and TFMO ((poly[9,9-dioctylfluorene-co-bi-N-(4-methoxy-phenyl) diphenylamine), charge photogeneration and device photocurrent generation depend critically upon the polymer ionisation potential. For example, it has been shown20,24 that blend films employing polyfluorenes with ionisation potentials higher than 5.5 eV produced PCBM triplets while those employing polymers with IPs of 5.2 eV or lower produced polymer polarons. This observation is consistent with the results reported herein, with a lower polymerIP increasing the energetic driving force for HOMO to HOMO charge photogeneration (i.e. hole transfer) from the PCBM singlet exciton. We thus conclude that the clear dependence of charge photogeneration upon polymer ionisation potential for this polyfluorene materials series most probably results from efficient polymer to fullerene energy transfer in such blend films, with charge photogeneration therefore primarily resulting from HOMO to HOMO reverse charge separation. It is also likely that such hole transfer based charge separation plays a role in systems where optical excitation of the acceptor is important.10
We note that this energy transfer/reverse charge separation model contrasts with the organic blends where the donor polymer does not exhibit strong resonance energy transfer to PCBM (e.g.APFO-type co-polymers, polythiophene based polymers and low bandgap charge transfer polymers) where charge separation can be expected to be based on LUMO to LUMO electron transfer. In such blends the charge photogeneration yield has been observed not to correlate with polymer ionisation potential but rather with donor/acceptor electron affinity.19,58 We further note that this reverse charge separation (hole transfer) mechanism, which is preceded by rapid energy transfer from polymer to PCBM, is energetically relatively inefficient due to the energy loss typically associated with the energy transfer step. As such, for OSCs where light absorption is primarily undertaken by the donor moiety, this charge photogeneration mechanism is likely to yield lower optimised OSC photovoltaic efficiencies relative to devices based primarily upon direct charge photogeneration from the donor exciton.
We now turn to charge photogeneration in IF8TBTT/PCBM blend films. It is striking that the yield of dissociated polarons approximately doubles when the PCBM concentration is increased from 50 to 75 wt%. This cannot be assigned to enhanced exciton quenching and CT state formation, as PL quenching already exceeds 99% with only 50 wt% PCBM. It appears more likely that this increase in polaron yield is associated with an increase in PCBM domain size favouring dissociation of interfacial CT states into dissociated charges. Such an influence of nanomorphology upon charge dissociation would be consistent with a wide range of previous studies which have indicated that efficient charge photogeneration is dependent upon the formation of sufficiently large donor and/or acceptor domains such that the polarons can escape their Coulomb capture radius.59–64 It is also notable that even for 75 wt% PCBM blend films, the transient absorption signal assigned to dissociated charges is an order of magnitude lower than that of IF8TBTT triplets. This relatively small polaron signal appears not to be due to a low polaron absorption cross-section; chemical oxidation experiments in solution have indicated that this polaron absorption has an extinction coefficient of 26400 ± 500 L mol−1 cm−1, similar to that reported previously for polyfluorene triplets65,66 and polythiophene polarons.67 As such the relatively low signal amplitude appears more probably indicative of relatively low polaron yields (∼10%) in these blend films. We note that such polaron yields are lower than the external quantum yield measurements of photocurrent generation in the corresponding devices (which exhibit EQE values up to 37%), suggesting that charge photogeneration may be more efficient in IF8TBTT/PCBM photovoltaic devices than in thin films without device electrodes (as employed for the transient optical studies herein), potentially indicative of electric field dependent charge generation for these devices.68 A similar observation for another polyfluorene TFMO in blends with a silole has been previously reported.17 A more extensive comparison of charge photogeneration in blend films and devices will be presented elsewhere.68
We conclude by considering the implications of these results for polymer design for OPV device applications. The key conclusion of this paper is that in bulk heterojunction organic blend films where there exists strong spectral overlap between donor emission and acceptor absorption, and where the donor is highly emissive, energy transfer from donor to acceptor can be very fast, and can compete effectively with electron transfer, even when this electron transfer is energetically favourable. This energy transfer can potentially act as a loss pathway preventing charge photogeneration. This conclusion is important as it may explain, at least in part, why polymers developed for OLED applications, which require high PL efficiencies, often do not show efficient performance in OSC devices. It is also of direct relevance to the design of blue absorbing polymers for tandem OSC applications—implying that a key design criterion for such polymers may be that their singlet excitons should be only weakly emissive.
Acknowledgements
This work was supported by the EPSRC's Nanotechnology Grand Challenge and Supergen programmes with further support from BP Solar Ltd. We are also grateful to Safa Shoaee for helpful discussions and drafting assistance. The Imperial College High Performance Computational centre is acknowledged for the quantum chemical calculations. We thank Scott Watkins at CSIRO for PESA measurements.References
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. De Boer, Polym. Rev., 2008, 48, 531 Search PubMed.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839 CrossRef CAS.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS.
- J. Veres, S. Ogier, G. Lloyd and D. de Leeuw, Chem. Mater., 2004, 16, 4543 CrossRef CAS.
- W. Tang, V. Chellappan, M. Liu, Z.-K. Chen and L. Ke, ACS Appl. Mater. Interfaces, 2009, 1, 1467 CrossRef CAS.
- R. Pacios, J. Nelson, D. D. C. Bradley and C. J. Brabec, Appl. Phys. Lett., 2003, 83, 4764 CrossRef CAS.
- E. W. Snedden, L. A. Cury, K. N. Bourdakos and A. P. Monkman, Chem. Phys. Lett., 2010, 490, 76 CrossRef CAS.
- I. B. Martini, A. D. Smith and B. J. Schwartz, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 035204 CrossRef.
- M. T. Lloyd, Y. F. Lim and G. G. Malliaras, Appl. Phys. Lett., 2008, 92, 143308 CrossRef.
- S. R. Scully, P. B. Armstrong, C. Edder, J. M. J. Fréchet and M. D. McGehee, Adv. Mater., 2007, 19, 2961 CrossRef CAS.
- G. F. Burkhard, E. T. Hoke, S. R. Scully and M. D. McGehee, Nano Lett., 2009, 9, 4037 CrossRef CAS.
- D. C. Coffey, A. J. Ferguson, N. Kopidakis and G. Rumbles, ACS Nano, 2010, 4, 5437 CrossRef CAS.
- A. A. Bakulin, J. C. Hummelen, M. S. Pshenichnikov and P. H. M. van Loosdrecht, Adv. Funct. Mater., 2010, 20, 1653 CrossRef CAS.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS.
- A. C. Morteani, P. Sreearunothai, L. M. Herz, R. H. Friend and C. Silva, Phys. Rev. Lett., 2004, 92, 247402 CrossRef.
- A. Ruseckas, M. Theander, M. R. Andersson, M. Svensson, M. Prato, O. Inganas and V. Sundstrom, Chem. Phys. Lett., 2000, 322, 136 CrossRef CAS.
- C. Dyer-Smith, J. J. Benson-Smith, D. D. C. Bradley, H. Murata, W. J. Mitchell, S. E. Shaheen, S. A. Haque and J. Nelson, J. Phys. Chem. C, 2009, 113, 14533 CrossRef CAS.
- D. Veldman, Ö. Ipek, S. C. J. Meskers, J. Sweelssen, M. M. Koetse, S. C. Veenstra, J. M. Kron, S. S. Van Bavel, J. Loos and R. A. J. Janssen, J. Am. Chem. Soc., 2008, 130, 7721 CrossRef CAS.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, S. Tierney, W. Zhang, M. Heeney, I. McCulloch, J. Nelson, D. D. C. Bradley and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 3030 CrossRef CAS.
- J. J. Benson-Smith, H. Ohkita, S. Cook, J. R. Durrant, D. D. C. Bradley and J. Nelson, Dalton Trans., 2009, 10000 RSC.
- M. T. Bernius, M. Inbasekaran, J. O'Brien and W. Wu, Adv. Mater., 2000, 12, 1737 CrossRef CAS.
- C. I. Wilkinson, D. G. Lidzey, L. C. Palilis, R. B. Fletcher, S. J. Martin, X. H. Wang and D. D. C. Bradley, Appl. Phys. Lett., 2001, 79, 171 CrossRef CAS.
- P. A. Levermore, R. Jin, X. Wang, J. C. de Mello and D. D. C. Bradley, Adv. Funct. Mater., 2009, 19, 950 CrossRef CAS.
- J. J. Benson-Smith, L. Goris, K. Vandewal, K. Haenen, J. V. Manca, D. Vanderzande, D. D. C. Bradley and J. Nelson, Adv. Funct. Mater., 2007, 17, 451 CrossRef CAS.
- S. Cook, H. Ohkita, J. R. Durrant, Y. Kim, J. J. Benson-Smith, J. Nelson and D. D. C. Bradley, Appl. Phys. Lett., 2006, 89, 101128 CrossRef.
- F. L. Zhang, E. Perzon, X. J. Wang, W. Mammo, M. R. Andersson and O. Inganas, Adv. Funct. Mater., 2005, 15, 745 CrossRef CAS.
- H. M. P. Wong, P. Wang, A. Abrusci, M. Svensson, M. R. Andersson and N. C. Greenham, J. Phys. Chem. C, 2007, 111, 5244 CrossRef CAS.
- M. Svensson, F. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersson, Adv. Mater., 2003, 15, 988 CrossRef CAS.
- C. M. B. Svanstrom, J. Rysz, A. Bernasik, A. Budkowski, F. Zhang, O. Inganas, M. R. Andersson, K. O. Magnusson, J. J. Benson-Smith, J. Nelson and E. Moons, Adv. Mater., 2009, 21, 4398 CrossRef.
- T. M. Clarke, F. C. Jamieson and J. R. Durrant, J. Phys. Chem. C, 2009, 113, 20934 CrossRef CAS.
- L. H. Slooff, S. C. Veenstra, J. M. Kroon, D. J. D. Moet, J. Sweelssen and M. M. Koetse, Appl. Phys. Lett., 2007, 90, 143506 CrossRef.
- O. Inganäs, M. Svensson, F. Zhang, A. Gadisa, N. K. Persson, X. Wang and M. R. Andersson, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 31 CrossRef.
- L. M. Andersson and O. Inganas, Appl. Phys. Lett., 2006, 88, 082103 CrossRef.
- R. Pacios, D. D. C. Bradley, J. Nelson and C. J. Brabec, Synth. Met., 2003, 137, 1469 CrossRef CAS.
- A. Gadisa, E. Perzon, M. R. Andersson and O. Inganas, Appl. Phys. Lett., 2007, 90, 113510 CrossRef.
- P. Sonar, L. Oldridge, A. C. Grimsdale, K. Mullen, M. Surin, R. Lazzaroni, P. Leclere, J. Pinto, L. L. Chua, H. Sirringhaus and R. H. Friend, Synth. Met., 2010, 160, 468 CrossRef CAS.
- Q. Zheng, B. J. Jung, J. Sun and H. E. Katz, J. Am. Chem. Soc., 2010, 132, 5394 CrossRef CAS.
- W. Zhang, J. Smith, R. Hamilton, M. Heeney, J. Kirkpatrick, K. Song, S. E. Watkins, T. Anthopoulos and I. McCulloch, J. Am. Chem. Soc., 2009, 131, 10814 CrossRef CAS.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. Mcculloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197 CrossRef CAS.
- W. H. Baek, T. S. Yoon, H. H. Lee and Y. S. Kim, Org. Electron., 2010, 11, 933 CrossRef CAS.
- Calculations on trimers were carried out and revealed a change in HOMO of less than approximately 50 meV.
- D. Jacquemin, E. A. Perpete, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123 CrossRef CAS.
- K. G. Jespersen, W. J. D. Beenken, Y. Zaushitsyn, A. Yartsev, M. Andersson, T. Pullerits and V. Sundstrom, J. Chem. Phys., 2004, 121, 12613 CrossRef CAS.
- M. Uda, Jpn. J. Appl. Phys., 1985 Search PubMed , Supplement 24-4, 284.
- J. K. J. van Duren, X. Yang, J. Loos, C. W. T. Bulle-Lieuwma, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Adv. Funct. Mater., 2004, 14, 425 CrossRef CAS.
- S. Cook, H. Ohkita, Y. Kim, J. J. Benson-Smith, D. D. C. Bradley and J. R. Durrant, Chem. Phys. Lett., 2007, 445, 276 CrossRef CAS.
- J. Nelson, Phys. Rev. B: Condens. Matter, 2003, 67, 155209 CrossRef.
- We note that the long-lived 740 nm band observed in the transient spectrum was not observed in the solution spectrum of the radical cation, suggesting that this band may result from intermolecular delocalised polymer polarons that form only in the solid film.
- At higher excitation densities, the trap are all filled, resulting in fast bimolecular recombination of free (untrapped) polarons on a timescale faster than the time resolution of these experiments (∼100 ns).
- A. F. Nogueira, I. Montanari, J. Nelson, J. R. Durrant, C. Winder, N. S. Sariciftci and C. Brabec, J. Phys. Chem. B, 2003, 107, 1567 CrossRef CAS.
- I. Montanari, A. F. Nogueira, J. Nelson, J. R. Durrant, C. Winder, M. A. Loi, N. S. Sariciftci and C. Brabec, Appl. Phys. Lett., 2002, 81, 3001 CrossRef CAS.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, M. Heeney, S. Tierney, I. McCulloch, D. D. Bradley and J. R. Durrant, Chem. Commun., 2006, 3939 RSC.
- C. Dyer-Smith, L. X. Reynolds, A. Bruno, D. D. C. Bradley, S. A. Haque and J. Nelson, Adv. Funct. Mater., 2010, 20, 2701 CrossRef CAS.
- T. I. Hukka, T. Toivonen, E. Hennebicq, J. L. Brédas, R. A. J. Janssen and D. Beljonne, Adv. Mater., 2006, 18, 1301 CrossRef CAS.
- P. A. van Hal, R. A. J. Janssen, G. Lanzani, G. Cerullo, M. Zavelani-Rossi and S. De Silvestri, Phys. Rev. B: Condens. Matter, 2001, 64, 075206 CrossRef.
- M. Svensson, F. Zhang, O. Inganäs and M. R. Andersson, Synth. Met., 2003, 135–136, 137 CrossRef CAS.
- W. Mammo, S. Admassie, A. Gadisa, F. Zhang, O. Inganäs and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2007, 91, 1010 CrossRef CAS.
- S. Shoaee, T. M. Clarke, C. Huang, S. Barlow, S. R. Marder, M. Heeney, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2010, 132, 12919 CrossRef CAS.
- D. Veldman, O. Opek, S. C. J. Meskers, J. Sweelssen, M. M. Koetse, S. C. Veenstra, J. M. Kroon, S. S. v. Bavel, J. Loos and R. A. J. Janssen, J. Am. Chem. Soc., 2008, 130, 7721 CrossRef CAS.
- P. E. Keivanidis, T. M. Clarke, S. Lilliu, T. Agostinelli, J. E. Macdonald, J. R. Durrant, D. D. C. Bradley and J. Nelson, J. Phys. Chem. Lett., 2010, 1, 734 Search PubMed.
- T. M. Clarke, A. M. Ballantyne, S. Tierney, M. Heeney, W. Duffy, I. McCulloch, J. Nelson and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 8068 CrossRef CAS.
- J. M. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2010, 132, 6154 CrossRef CAS.
- C. R. McNeill, S. Westenhoff, C. Groves, R. H. Friend and N. C. Greenham, J. Phys. Chem. C, 2007, 111, 19153 CrossRef CAS.
- C. R. McNeill, J. J. M. Halls, R. Wilson, G. L. Whiting, S. Berkebile, M. G. Ramsey, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2008, 18, 2309 CrossRef CAS.
- S. M. Fonseca, J. Pina, L. G. Arnaut, J. Seixas de Melo, H. D. Burrows, N. Chattopadhyay, L. Alcácer, A. Charas, J. Morgado, A. P. Monkman, U. Asawapirom, U. Scherf, R. Edge and S. Navaratnam, J. Phys. Chem. B, 2006, 110, 8278 CrossRef CAS.
- T. A. Ford, I. Avilov, D. Beljonne and N. C. Greenham, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125212 CrossRef.
- C. G. Shuttle, B. O. Regan, A. M. Ballantyne, J. Nelson, D. D. C. Bradley and J. R. Durrant, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 113201 CrossRef.
- T. M. Clarke, A. Ballantyne, S. Shoaee, Y. W. Soon, W. Duffy, M. Heeney, I. McCulloch, J. Nelson and J. R. Durrant, Adv. Mater., 2010, 22, 5287–5291 CrossRef CAS.
- S. Cook, R. Katoh and A. Furube, J. Phys. Chem. C, 2009, 113, 2547 CrossRef CAS.
- Y. Kim, S. Choulis, J. Nelson, D. Bradley, S. Cook and J. Durrant, J. Mater. Sci., 2005, 40, 1371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0sc00606h |
| This journal is © The Royal Society of Chemistry 2011 |