Immobilised photosensitisers for continuous flow reactions of singlet oxygen in supercritical carbon dioxide
Xue
Han
,
Richard A.
Bourne
,
Martyn
Poliakoff
* and
Michael W.
George
*
School of Chemistry, University of Nottingham, University Park Nottingham, NG7 2RD, UK. E-mail: mike.george@nottingham.ac.uk; martyn.poliakoff@nottingham.ac.uk; Fax: +44 155 9513508; Tel: +44 115 9513512
First published on 11th March 2011
Abstract
Photosensitisers have been incorporated into both polymer and aerogel supports for the photochemical generation of singlet oxygen, 1O2, in supercritical carbon dioxide, scCO2. These systems showed high activity and an acceptable lifetime for the photo-oxidation of α-terpinene and of citronellol in continuous reactors over a period of at least 5 h. The most effective photosensitiser was a covalently coupled analogue of tetradi(2,6)chloro-phenylporphyrin (TDCPP) on polyvinyl chloride (PVC). This immobilisation removes the need to separate the photosensitiser from the downstream solution.
Introduction
Selective oxidation is an important reaction in many organic syntheses and there is a continuing research effort to develop clean, accessible and industrially viable reaction methodologies.1,2 Singlet Oxygen, 1O2 can be used for a wide range of transformations such as [2 + 2] and [2 + 4] cycloadditions, ene reactions, oxidation of heteroatoms and peroxide synthesis. 1O2 is typically generated from a mixture of sodium hypochlorite and hydrogen peroxide or by using gaseous 3O2 in the presence of light with an appropriate sensitiser.3 Photocatalysis is the most commonly used method for oxidation with 1O2 and common sensitisers include Rose Bengal, methylene blue and tetraphenylporphyrin. However, reactions with photosensitised 1O2 have not been used for large scale industrial applications because of two problems (i) overcoming the decreased photochemical efficiency with the increased concentration of reagents used for industrial scale processes and (ii) since CCl4 is no longer acceptable as a large-scale industrial solvent, there is difficulty in finding a suitable non-flammable solvent which is also slow in relaxing 1O2 to the ground state.We have recently reported4,5 a new approach using supercritical carbon dioxide (scCO2) that addresses some of these issues; scCO2 is neither flammable nor toxic. scCO2 is completely miscible with gaseous O2 and has a lower viscosity and higher diffusivity than more conventional solvents. The complete miscibility of scCO2 with O2 removes mass transport limitations and enables O2 concentration to be controlled accurately and kept safely below explosion limits.6 This enables photocatalytic oxidation reactions to be carried out in continuous flow much more easily than can be done in biphasic gas–liquid systems. In addition, scCO2 can be easily separated from the product mixture by simply depressurising to give solvent-free product. Whilst the high pressures required for performing reactions with scCO2 do limit the applicability of our approach, we believe that the advantages described overcome this drawback and enable photo-oxidised products to be produced in a higher space-time yield, continuously and more cleanly than traditional methods.
Photo-oxidation reactions in scCO2 using molecular O2 and CO2-soluble metal catalysts7–10 have been successfully performed with the advantages described. Photophysical experiments have demonstrated that 1O2 can be effectively generated in scCO2 with an extended lifetime compared to many conventional solvents.11–13 We have demonstrated the quantitative conversion of α-terpinene to ascaridole (Scheme 1) using a highly fluorinated and CO2-soluble homogeneous photosensitiser, 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin (TPFPP). These reactions followed zero-order kinetics in a batch high-pressure spectroscopy cell because the reaction rate largely depends on the intensity of the light.4 We have also used LED-arrays to illuminate a sapphire tube reactor for performing continuous photo-oxidations using TPFPP with a significantly enhanced space-time yield compared to more conventional photo-oxidation reactors.5,14 However there remain the problems of (i) separating the photosensitiser from the usually thermally sensitive product and (ii) the need to use ever increasing amounts of photosensitiser for scale-up.
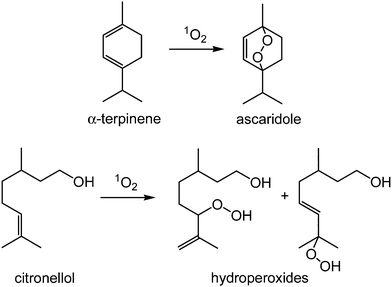 | ||
| Scheme 1 Photo-oxidation of α-terpinene to ascaridole (top) and citronellol to hydroperoxide products (bottom) | ||
These problems could be overcome by the use of an immobilised sensitiser. This would provide several advantages over our original flow experiments with dissolved TPFPP; particularly removing the need to separate the catalyst from product, the reuse of the photo-catalyst, and giving the opportunity to use inexpensive CO2-insoluble sensitisers. Ideally, the photocatalyst and support must possess good stability under both supercritical and irradiative conditions.
The concept of immobilised photosensitisers per se is not new. Porphyrins can generate 1O2 with high stability to decomposition,15 and they have been previously immobilised on supports including polystyrene,16 polyethylene glycol,17 Merrifield resin,18zeolite,19 and silica.20 Immobilised porphyrins have been used for photo-oxidations in batch mode, but limitations due to the long term stability has prevented them from being fully exploited by operating continuously.
However, the high intensity of our LEDs and the properties of scCO2 introduce additional challenges to making a successful immobilised photosensitiser.
Aims and strategy
In this paper we aim to find an immobilised photosensitiser which can give us a performance comparable to that of the homogeneous or surfactant-solubilised photosensitisers that we have used until now for continuous 1O2 reactions in scCO2. For this, we have used the criterion that the immobilised photosensitiser should be able to work in continuous flow for at least five hours without a significant drop in activity, conveniently quantified as the apparent space-time yield, STY.Our strategy has been to test a range of eight photosensitisers with a number of different immobilisation methods for photo-oxidation in scCO2. We began with commercially available heterogenized Rose Bengal, and then used methods which require an increasing numbers of steps to prepare the immobilised photocatalyst.
We have used two test reactions, Scheme 1; the conversion of α-terpinene to ascaridole, and the reaction of citronellol to give a mixture of hydroperoxides, the first step in the synthesis of rose oxide. These reactions were chosen because (i) we already had considerable data for the performance of homogeneous photosensitisers in scCO2 for these reactions and (ii) the compounds involved in the citronellol reaction are considerably more polar than those in the α-terpinene reaction, thereby increasing the chances of leaching the sensitiser from the support.
Initially, each immobilised photosensitiser in Scheme 2 was tested on the reaction of α-terpinene in a batch reactor with sufficient irradiation time to give complete conversion but with process monitoring to establish the STY. If the activity seemed reasonable and if the same sample of photosensitiser could be recycled for several batches, the photosensitiser was then tested for the same reaction in a continuous reactor. Finally, the photosensitiser was tested with the citronellol reaction in the same continuous reactor. In each case, we were looking for good conversion of the reactant and high selectivity to the desired product, together with an acceptably high STY combined with a low rate of bleaching or leaching.
![The photosensitisers used in this work: (RB) Rose Bengal; (TPP) tetraphenylporphyrin; (ZnTPP) Zinc tetraphenylporphyrin; (TDCPP) tetradi(2,6)chloro-phenylporphyrin; (TPP+) 5,10,15,20-tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluenesulfonate); [Ru(bpy)3]2+ tris-bipyridyl Ruthenium; (TDCPP+) [TDCPP-CONH–C2H4–N(CH3)3]+ I− and (TDCPP-COOH) 4-[10,15,20-tris(2,6-dichlorophenyl)-21H,23H-porphin-5-yl]-benzoic acid (see Experimental Section for details of synthesis and immobilisation)](/image/article/2011/SC/c0sc00641f/c0sc00641f-s2.gif) | ||
| Scheme 2 The photosensitisers used in this work: (RB) Rose Bengal; (TPP) tetraphenylporphyrin; (ZnTPP) Zinc tetraphenylporphyrin; (TDCPP) tetradi(2,6)chloro-phenylporphyrin; (TPP+) 5,10,15,20-tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluenesulfonate); [Ru(bpy)3]2+ tris-bipyridyl Ruthenium; (TDCPP+) [TDCPP-CONH–C2H4–N(CH3)3]+ I− and (TDCPP-COOH) 4-[10,15,20-tris(2,6-dichlorophenyl)-21H,23H-porphin-5-yl]-benzoic acid (see Experimental Section for details of synthesis and immobilisation) | ||
The overall results are summarised in Table 1 and are discussed in more detail below. However, to anticipate our conclusions, only one system, namely TDCPP-COOH covalently bound to amine derivatised PVC, gave an acceptable performance with both test reactions.
| Method of Immobilisation | Photosensitiser, see Scheme 1 | Reactions, (See Scheme 2) | |||
|---|---|---|---|---|---|
| α-terpinene → ascaridole | citronellol → hydroperoxides | ||||
| Batcha | Continuous | Continuous | |||
| a STY - space-time yield. | |||||
| 1 | Commercial polymer-bound | RB | Low STY | Initial yield good, 70% but rapid bleaching | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| 2 | Trapping in cast PVC films | RB | Low STY | — | Rapid leaching (<60 min) of all sensitisers by citronellol & products |
|
|||||
| Zn-TPP | Medium STY | — | |||
|
|||||
| TPP | High STY | 88% Yield dropping to 50% after 320 min | |||
|
|||||
| TDCPP | Medium STY | >80% yield for 400 min | |||
|
|||||
| 3 | Ionic sensitisers on SiO2 aerogel support | TDCPP+ | — | — | Low conversion, but High TOF (turnover frequency); only small decrease after 320 min |
|
|||||
| TPP+ | Med STY but Low Selectivity with formation of p-cymene | — | Low conversion and bleached rapidly | ||
|
|||||
| [Ru(bpy)3]2+ | — | — | Very Low STY | ||
|
|||||
| 4 | Covalently bonded to amino PVC | TDCPP-COOH | Very high STY | 85% yield maintained with high STY for 320 min | 88% yield, only dropping to 78% after 330 min |
Testing procedure
At the start of each test, the immobilised photosensitiser was put into the reactor, batch or continuous. A key issue is to minimise the risk of an explosive reaction between O2 and the organic substrate. This was achieved by using the equipment shown schematically in Fig. 1.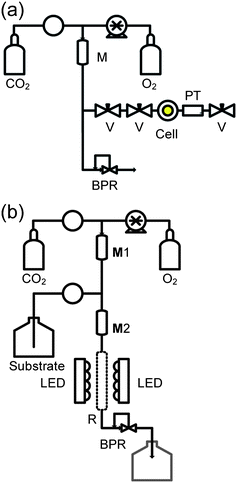 | ||
| Fig. 1 Schematic arrangement of equipment to minimise the risk of explosion by dilution of O2 with CO2 prior to contact with organic substrates. (a) Rig for adding CO2 + O2 to the batch reactor, prefilled with substrate and photosensitiser. (b) Flow equipment for continuous reactions. The components are labelled as follows: V - SSI 1/16 inch valve; BPR - Jasco BP-1580-81 back pressure regulator; M, mixing chamber (5 cm 1/4 inch SS316 tubes at 50 °C); CO2 - delivered from a chilled Jasco™ PU-1580-CO2 pump; O2 - added at a measured rate via a Rheodyne dosage unit; PT - pressure transducer (RDP Electronics); R - photocatalyst packed sapphire tube reactor; Substrate is pumped using a Jasco™ PU-980 HPLC pump. All pipework is constructed from 1/16 inch 316 SS tubing (Swagelok). | ||
In a batch experiment, a spectroscopic cell21 was prefilled with a measured volume of organic substrate, typically (50–150 μL). The cell was then filled with a mixture of CO2 and O2 to a set pressure (typically 140 bar for α-terpinene photo-oxidation and 180 bar for citronellol). Both reactions were initiated using a white light LED (OSTAR; Part Code: LEUWE3B-PZQZ-4C8F, 1000 lumen mounted on an aluminium heat sink) and the progress of batch reactions was monitored via pressure drop. Off-line analysis using 1H-NMR was performed on each sample.
For recycling experiments, the batch cell was opened between each sample and washed with acetone. The whole cell was then flushed with CO2 to remove all traces of solvent prior to running the next experiment. All tasks were run until ca. 100% conversion of substrate had been achieved.
In a typical flow experiment the reactor was loaded with the immobilised photosensitiser and was pressurised with a mixture of CO2 and O2 (2:1 molar ratio of O2: substrate with a total pressure equal to that used in batch experiments). Irradiation was then begun using two arrays, each of four LEDs, and the flow of organic substrate (0.1 mL min−1) was started simultaneously. The composition of the product stream was monitored using on-line GLC for the α-terpinene reaction and off-line 1H-NMR for the citronellol system because GLC was unsuitable due to the thermal instability of the products for this reaction.
Results and discussion
Evaluation of immobilised Rose Bengal photocatalyst
As explained above we began by testing a commercially available Rose Bengal covalently bound on polystyrene support (∼0.3 mmol g−1 resin, 200–400 mesh - Fluka). The immobilised photosensitiser was reused for 6 cycles. An average space-time yield of 14 mmol L−1min−1 was observed with no clear trends of catalyst degradation.However, the results of continuous flow testing were disappointing. 0.3 g of the immobilised photosensitiser was suspended in a glass wool matrix (0.3 g) in the sapphire tube reactor (Fig. 1b). As shown in Fig. 2 the catalyst displays reasonable initial activity with 69% conversion to ascaridole. However, it rapidly degrades due to photobleaching, which leads to a large decrease in conversion.
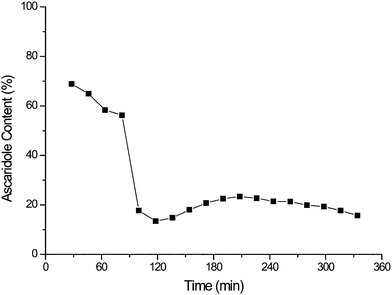 | ||
| Fig. 2 Evidence of rapid bleaching of Rose Bengal on a commercially available support during the continuous oxidation of α-terpinene. | ||
Photo-oxidation with PVC entrapped Photosensitisers in scCO2
We then investigated the trapping of a variety of free photosensitisers into PVC films. Our previous studies using scCO2 for photo-oxidation demonstrated that most of the common 1O2 photosensitisers are insoluble in scCO2 and that either a high degree of fluorination of the photosensitiser or the addition of fluorinated surfactants is required.22 So, we tried to exploit the low solvent power of scCO2 to avoid leaching of the photosensitiser from the film.Preliminary testing with a variety of polymer supports (polyethylene, polystyrene, polyethylene oxide, polyvinyl chloride, PVC) indicated that PVC gave a good combination of facile incorporation of photosensitiser, low solubility in scCO2 and low reactivity with 1O2.23
A series of PVC films were cast by evaporation of a solution of high molecular weight PVC and the appropriate photosensitiser in THF. In all cases the result was a thin, uniform PVC film loaded with photosensitiser (see Experimental Section for full details). A 14 mm disc was cut out from this film and placed against the back window of our photochemical cell.21
Each of the four sensitisers was tested three times for the photochemical oxidation of α-terpinene in batch mode. 50 μL of α-terpinene was injected into the cell which was then filled with a mixture of CO2/O2 (6 mol%) at 140 bar and 40 °C, see Fig. 1a. The cell was then irradiated using a white LED. Fig. 3 shows the STY for the photocatalysts tested, all of which showed significant activity. FTIR monitoring was used to determine the reaction kinetics, which in each case was zero-order. The product distribution was confirmed by 1H-NMR spectroscopy. Only Rose Bengal on PVC displayed low selectivity (ca. 80%), due to oxidative dehydrogenation to para-cymene (4-isopropyltoluene) and the lowest STY (total reaction time 33 min). The other three photocatalysts, Zn-TPP, TDCPP and TPP all showed high selectivity producing ascaridole in at least 96% yield. TPP and TDCPP on PVC were tested further to determine suitability for continuous operation.
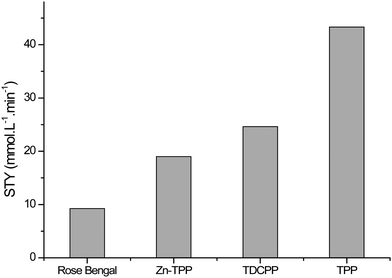 | ||
| Fig. 3 Summary of data from single batch experiments showing the very different space-time yields (STY) for the four photosensitisers suspended in PVC films. | ||
TDCPP is well known as a stable photocatalyst.24,25 We performed a series of batch experiments recycling a single TDCPP/PVC film as shown in Fig. 4. It can be seen that the first cycle gave a lower STY compared to the following experiments, possibly due to progressive swelling which leads to higher reaction rates following treatment in scCO2.
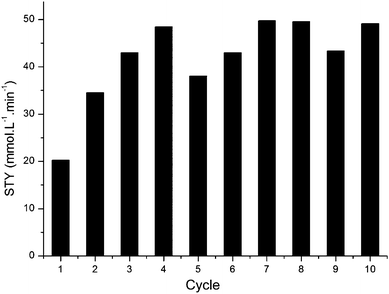 | ||
| Fig. 4 Demonstration of the recyclability of TDCPP in PVC for the batch photo-oxidation of α-terpinene. | ||
Each experiment exhibited zero-order reaction kinetics. The turnover frequency was much lower than that previously observed in homogeneous experiments where TPFPP exhibited a TOF of 960 min−1 compared to the average TOF of 30 min−1 for TDCPP seen in Fig. 4.4 However, due to the higher photosensitiser loadings in the PVC film than the homogeneous experiments, the total time (and hence STY) for complete conversion was comparable to that achieved under homogeneous conditions, taking ca. 3 min and 7 min with homogeneous and supported systems, respectively.
These batch tests demonstrated that the TDCPP/PVC films possess a relatively long lifetime and the ability to produce 1O2 efficiently in scCO2. Following this demonstration, we produced strips of impregnated PVC films for testing in continuous mode and compared the lifetime of TDCPP against TPP for the photo-oxidation of α-terpinene.
Fig. 5 compares the results from TPP in PVC and TDCPP in PVC, both of which exhibit initial high activity. The TPP system unfortunately undergoes photobleaching resulting in a rapid decrease in yield. However, over the duration of the experiment the TDCPP photocatalyst displays a slight increase in the % yield of ascaridole in the colourless liquid emerging from the reactor. No evidence of leaching was observed in the product solution by UV/Vis, suggesting that the ascaridole, α-terpinene and CO2 mixture does not remove photosensitiser from the PVC film.
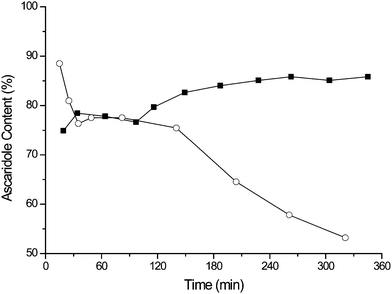 | ||
| Fig. 5 The sustained performance of TDCPP in PVC film (■) compared to the bleaching of TPP in PVC film (○) during the continuous oxidation of α-terpinene. | ||
Following the successful use of TDCPP/PVC film for the photo-oxidation of α-terpinene, we attempted to apply this catalyst to the photo-oxidation of citronellol, see Scheme 1. We have previously performed this photo-oxidation of citronellol using TPFPP with dimethyl carbonate as a co-solvent to enable dissolution of the photosensitiser in the substrate so that it could be delivered by the HPLC pump.5 That approach therefore introduces two additional components, photosensitiser and co-solvent, that need to be removed from the product during purification. An immobilised sensitiser would remove the need for both co-solvent and photosensitiser separation, thereby greatly improving the sustainability characteristics of this reaction.
Again there was a disappointment; citronellol and its derivatives are more polar than α-terpinene and the TDCPP was leached rapidly from the PVC with a coloured product solution emerging from the BPR, with a corresponding loss of conversion. This clearly limits the scope of TDCPP/PVC to non-polar substrates which are unable to leach the photosensitiser from the polymer support.
Aerogel supported sensitisers for photo-oxidation
Aerogels have been previously used to immobilise porphyrins, for example by Baiker as catalysts for the epoxidation of olefins.26 We have successfully prepared a series of new aerogel supported cationic photosensitisers, which were added during the solution sol–gel process. We selected a commercially available ionic porphyrin (5,10,15,20-Tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluenesulfonate) (TPP+), a common ionic 1O2 sensitiser Tris(2,2′-bipyridine)dichlororuthenium(II) hexahydrate ([Ru(bpy)3]2+) and synthesised an ionic TDCPP analogue (TDCPP+) as photosensitisers to immobilise on our SiO2 aerogels.The production of the hybrid photosensitiser-loaded aerogel monoliths (see Experimental Section for details) was performed using a base-catalysed synthesis. The dried aerogel was uniformly coloured. BET characterisation of the three aerogels showed similar values for the surface area (TPP+ 854 m2 g−1, TDCPP+ 866 m2 g−1, [Ru(bpy)3]2+ 860 m2 g−1) and pore width (TPP+ 165 Å, TDCPP+ 164 Å, [Ru(bpy)3]2+ 162 Å). Given the low density of the aerogels, the volumetric sensitiser loadings were much lower than with PVC. In order to load both the high pressure photochemical batch and flow cells, the monoliths were ground up and sieved to give particles in the range of 250–800 μm.
Initial testing was performed using the TPP+ aerogel for the batch photo-oxidation of α-terpinene. The aerogel was recycled 4 times and showed a good STY of ca. 28 mmol L−1min−1. The TPP+ aerogel catalyst exhibited a high TOF (average value of 440 min−1) but a much lower selectivity than the previous catalyst systems due to significant formation of para-cymene. Control experiments under irradiative conditions with undoped silica aerogel showed that para-cymene could be formed in the absence of photosensitiser. Literature experiments27,28 on supported photosensitisers on zeolites have also reported the formation of para-cymene.
Although the TPP+ aerogel was relatively unsuccessful for the photo-oxidation of α-terpinene, batch testing for the selective photo-oxidation of citronellol showed high selectivity for the desired products. All three aerogel catalysts were then used for the continuous oxidation of citronellol. Fig. 6 shows that the [Ru(bpy)3]2+/aerogel system exhibited low activity. However, both of the porphyrin/aerogel catalysts exhibit excellent activity for photo-oxidation of citronellol comparable to the activity observed in previous results with TPFPP dissolved in a dimethyl carbonate co-solvent. Unfortunately TPP+ undergoes relatively rapid photobleaching with a significant drop in yield. By contrast the TDCPP+ system displays both higher activity and low photobleaching. The higher TOF is probably due to the much greater accessibility of the photocatalyst due to the porous structure of the aerogel and the lower loading of photocatalyst of this system. It could be envisaged that a larger sapphire tube could be used to contain more of this low density catalyst to achieve the required conversion.
![Comparison of three cationic photosensitisers on aerogels for the continuous oxidation of citronellol TDCPP+ (■),TPP+ (★) and [Ru(bpy)3]2+ (○).](/image/article/2011/SC/c0sc00641f/c0sc00641f-f6.gif) | ||
| Fig. 6 Comparison of three cationic photosensitisers on aerogels for the continuous oxidation of citronellol TDCPP+ (■),TPP+ (★) and [Ru(bpy)3]2+ (○). | ||
Covalently bound TDCPP based-photosensitisers on Amino-functionalised-PVC
As none of the systems so far tested met our full criteria for success, we synthesised a photosensitiser covalently bound to a PVC support. Amino-functionalised PVC beads were synthesised using a modified literature method,29 see Scheme 3. These were then reacted with TDCPP-COOH to give a chemically bound photocatalyst with 1.4 × 10−5 mol g−1 photosensitiser loading.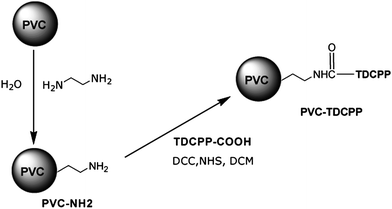 | ||
| Scheme 3 Amino-functionalisation of PVC beads and covalent coupling of TDCPP-COOH to make the most successful of our photosensitisers. | ||
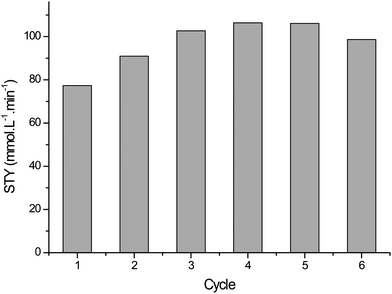
The PVC beads were initially tested for the photo-oxidation of α-terpinene in batch mode with high levels of selectivity and conversion and insignificant loss of activity over 6 cycles, see Fig. 7.
The PVC beads were uniformly distributed in a matrix of glass wool and this mixture was loaded into the continuous reactor. The beads were tested with both the α-terpinene and citronellol reactions. Fig. 8 shows that excellent performance was achieved in both reactions. Only a slight loss in activity was observed over the course of the α-terpinene experiments, presumably due to decomposition of the catalyst.
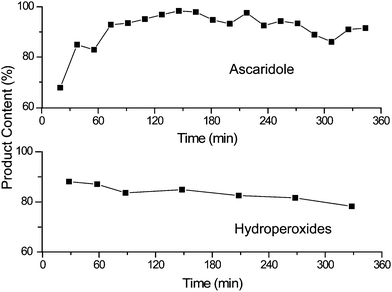 | ||
| Fig. 8 Demonstration of the 6-hour stability of TDCPP-COOH bound to amino PVC (see Scheme 3) for both of our continuous test reactions. None of the other immobilised photosensitisers approached this performance. | ||
The covalently bound nature of the catalyst enabled it to perform well in the photo-oxidation of citronellol by preventing leaching of the catalyst into the product phase. Thus, with this covalently-bound photosensitiser, our criteria had finally been met.
Conclusions
We have demonstrated the potential for using immobilised photosensitisers for the continuous production of peroxides using 1O2 in scCO2. Immobilised photosensitisers enable sensitisers to be used much more efficiently with much greater total turnover and without the need for added co-solvents. This removes the need to separate either the photosensitiser or co-solvent from the product which is therefore cleaner than in previous reactions in scCO2.Initially we showed that a commercially available Rose Bengal catalyst was unsuitable for performing continuous flow reactions due to rapid decomposition of the supported photocatalyst. Following these results we developed three heterogeneous photosensitiser systems. The first focused on a facile system using free non-CO2 philic photosensitisers mixed with PVC. TDCPP was shown to possess a good lifetime and activity for the production of ascaridole. Unfortunately our second model reaction, the photo-oxidation of citronellol, was unsuccessful due to rapid leaching of the TDCPP.
We then explored the use of ionic photosensitisers bound to silica aerogel supports. The photosensitiser in this system showed very high activity, comparable to that of the homogeneous system using TPFPP. The aerogel system was shown to have high activity for the photo-oxidation of citronellol; however moderately low conversions were achieved due to the low photosensitiser content of the aerogel. It also possessed low selectivity in the α-terpinene reaction due to side reactions producing para-cymene.
Finally, we used covalent amide linkages to couple the TDCPP analogue to the amino functionalised PVC beads. These beads combined the advantages of the other catalysts; the beads could produce both ascaridole and the citronellol hydroperoxide products in high yields for a sustained period without leaching of the catalyst occurring. We now have a methodology for producing catalyst-free photo-oxidation products in a continuous manner, enabling facile lab scale production of peroxides with our scCO2 reactor system. This approach is now being applied to a wide range of reactions in our laboratory.
Experimental
Safety Warning: the experiments described involve high pressures and should only be conducted in an apparatus with the appropriate pressure rating and with due regard to the potentially explosive reaction between O 2 and organic compounds.All materials were purchased and used without further purification: Rose Bengal (RB, 95%, Aldrich), 5,10,15,20-Tetraphenyl-21H,23H-porphine zinc (Zn-TPP, 99%, Aldrich), meso-Tetraphenylporphyrin (TPP, 99%, Sigma), 5,10,15,20-Tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluenesulfonate) (TPP+, 95%, Fluka), and Tris(2,2′-bipyridine)dichlororuthenium(II) hexahydrate (technical grade, Fluka), poly(vinyl chloride) (high molecular weight, Aldrich), Dioctyl phthalate (99%, Aldrich), 2,6-Dichlorobenzaldehyde (98%, Fluka), Methyl-4-formyl-benzoate (99%, Aldrich), Pyrrole (98%, Aldrich), Boron trifluoride diethyl etherate (Sigma), Dichloro-dicyano-benzoquinone (98%, Aldrich), Potassium hydroxide (90%, Sigma-Aldrich), Hydrochloric acid (36.5%–38%, Sigma), N,N′-Dicyclohexylcarbodiimide (99%, Aldrich), N-Hydroxysuccinimide (98%, Aldrich), Ethylenediamine (99%, Alfa Aesar), Iodomethane (99%,Sigma-Aldrich), Tetramethoxysilane (TMOS, 98%, Alfa Aesar), Ammonia solution (35%, Fisher Scientific UK Ltd)
PVC film preparation
PVC films for batch experiments were prepared by evaporation of a ca. 5 mL solution of THF with 0.6 g PVC and 0.016 mmol of appropriate photosensitiser in a small laboratory Petri Dish (diameter = 60 mm).For the films used in flow experiments, two solutions composed of 1.8 g PVC (with 17% dioctyl-phalate plasticiser) plus either 50 mg (0.075 mmol) TPP or 72 mg (0.081 mmol) TDCPP in 10 mL of THF were prepared. These solutions were placed in a Petri Dish with a diameter of 110 mm and allowed to dry for 48 h at room temperature covered by a sheet of filter paper.
Aerogel synthesis
The synthesis involved two solutions A and B. Solution A contained 1.9 mL TMOS and 2.3 mL MeOH; solution B contained 2.3 mL MeOH, 0.8 mL H2O, 10 μL NH4.OH (35%) and 2.0 μmol TPP +. Solutions A and B were then fully mixed and poured into a cylindrical Teflon mould (10 cm long by 1.4 cm diameter) and aged for 24 h to form the gel. The gel was then removed from the mould and soaked in EtOH for 24 h, with fresh EtOH being added regularly. Following this the solvent was switched to acetone and the same procedure repeated. The wet gel was then dried by supercritical drying with scCO2 flushed through the gel at 3 mL min−1 at 140 bar, and 40 °C for 1 h. The pressure was then released linearly over the course of 3 h using a programmable back pressure regulator.Covalently bound TDCPP on PVC beads synthesis
10 g PVC was stirred in 100 mL 1,2-diaminoethane solution (80% diamine in H2O) at 80 °C for 6 h. The reaction mixture was then cooled to room temperature. The PVC-NH2 beads thus formed were filtered and washed with 2 L H2O followed by 50 mL MeOH, were dried at 50 °C under vacuum. The beads were characterised by FTIR spectroscopy and elemental microanalysis. These beads (4 g) were then further reacted with TDCPP-COOH (88 mg, 1.0 × 10−4 mol) in CH2Cl2 (50 mL) with peptide coupling reagents N,N′-dicyclohexylcarbodiimide (DCC) (98 mg, 4.7 × 10−4 mol) and N-hydroxysuccinimide (NHS) (62 mg, 5.4 × 10−4 mol) at room temperature for 48 h. The beads were filtered and washed with MeOH, and dried in a vacuum oven. The filtrate was collected and analysed by UV-Visible absorption spectroscopy to determine the amount of free photosensitiser and hence to deduce the loading of the beads (56% coupling efficiency).Batch experiments
Flow experiments
1. Synthesis of TDCPP-COOMe. 1.23 g (7.0 mmol) 2,6-dichlorobenzaldehyde and 0.6 g (3.7 mmol) methyl-4-formyl-benzoate were dissolved in 350 mL CH2Cl2, dried with MgSO4 before use. The solution was stirred and bubbled with N2 at room temperature for 10 min. Then, 0.8 mL (11.5 mmol) pyrrole was added. After stirring for 10 min, 0.2 mL (1.6 mmol) boron trifluoride diethyl etherate (BF3·OEt2) was added. After 20 min, another 0.2 mL of BF3·OEt2 was added and a further 0.5 mL was added 40 min later. The solution was then left stirring at room temperature for 15 h. Then, 3.5 g (15.4 mmol) dichloro-dicyano-benzoquinone (DDQ) was slowly added into the reaction system, and the solution was kept stirring for 5 min. The solvent was then removed by rotary evaporation. The product was purified by column chromatography (silica/CH2Cl2), and characterised by mass spectrometry and 1H-NMR. 240 mg (0.27 mmol) TDCPP-COOMe was obtained, yield: 12%.
2. Hydrolysis of TDCPP-COOMe to TDCPP-COOH. 240 mg of TDCPP-COOMe were added to a solution of KOH in 40 mL H2O + 40 mL THF. The solution was heated to 40 °C and stirred for 24 h. It was cooled to room temperature. HCl (35%) was dropwise added until pH = 2. The product was extracted by CHCl3, and the organic phase was then dried using MgSO4 and then the solvent was removed by rotary evaporation. The product was characterised by mass spectrometry and 1H-NMR. 189 mg (0.22 mmol) TDCPP-COOH was obtained, yield: 81%.
3. Synthesis of TDCPP-CONH-NH2. 189 mg (0.22 mmol) TDCPP-COOH, 66 mg (0.32 mmol) N,N′-Dicyclohexylcarbodiimide (DCC), and 37 mg (0.32 mmol) N-Hydroxysuccinimide (NHS) were dissolved in 50 mL CH2Cl2, dried with MgSO4 before use. 126 mg (2.1 mmol) ethane-1,2-diamine was then added. The solution was stirred at room temperature for 48 h. It was then filtered, dried by rotary evaporation and then purified by column chromatography (silica, CH2Cl2–MeOH = 2
:3). The product was characterised by mass spectrometry and 1H-NMR. 50 mg (0.055 mmol) TDCPP-CONH-NH2 was obtained, yield: 25%.
4. Synthesis of TDCPP-CONH– N(CH3)3+ I−. 50 mg (0.055 mmol) TDCPP-CONH-NH2 were dissolved in a mixture of 10 mL (160 mmol) iodomethane (MeI) and 5 mL DMF, and the solution was stirred at 45 °C for 5 days. Column chromatography proved unsuccessful but the material was used directly with unreacted non-ionic species being subsequently washed out from the silica gel. The composition of the crude product was calculated from 1H-NMR as having 56% of the desired product TDCPP-CONH– N(CH3)3+ I− and 21% being the mono-methylated product TDCPP-CONH-NH2(CH3)+ I−
TDCPP-CONH–C2H4–N(CH3)3+ I− - 1H-NMRδH (CD3OD, 400 MHz): 7.97–8.04 (m, 9H, 2,6-dichlorophenyl 3-H,4-H, and 5-H), 8.48 (m, 2H, Ar 3-H, 5-H), 8.56 (dd, 2H, 2-H, 6-H), 8.92–8.94 (m, 6H, βpyrrole), 9.02–9.04 (d, 2H, βpyrrole), 3.70 (s, 9H, methyl), 3.37–3.38 (m, 2H, methylene), 3.29–3.30 (m, 2H, methylene). MS: m/z: 949.1 (100%), 951.1 (78.5%), 947.1 (56.2%), 950.1 (55.7%), 952.1 (40.0%), 953.1 (34.6%), 948.1 (34.6%). UV (Acetone) λmax 644 nm (ε 1661 mM−1cm−1), 587 (3828), 541 (2979), 511 (9775), 415 (155175).
Acknowledgements
We thank EPSRC (grant no. EP/FO15275/1) and Thomas Swan & Co. Ltd. for support and Prof G. Pattenden, Dr A. Wells and Dr S. K. Ross for helpful discussions. MWG gratefully acknowledges receipt of a Royal Society Wolfson Merit Award. We thank Dr Sihai Yang for assistance with BET analysis. We also thank M. Dellar, M. Guyler, D. Lichfield, R. Wilson and P. Fields for technical support at the University of Nottingham.Notes and references
- A. Maldotti, A. Molinari and R. Amadelli, Chem. Rev., 2002, 102, 3811–3836 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- J. A. Richard, Synlett, 2009, 1187–1188 CrossRef CAS.
- R. A. Bourne, X. Han, A. O. Chapman, N. J. Arrowsmith, H. Kawanami, M. Poliakoff and M. W. George, Chem. Commun., 2008, 4457–4459 RSC.
- R. A. Bourne, X. Han, M. Poliakoff and M. W. George, Angew. Chem., Int. Ed., 2009, 48, 5322–5325 CrossRef CAS.
- M. A. McHugh and V. J. Krukonis, Supercritical Fluid Extraction Principles & Practice, Butterworth-Heinemann, Boston, 1994 Search PubMed.
- G. Jenzer, T. Mallat and A. Baiker, Catal. Lett., 2001, 73, 5–8 CrossRef CAS.
- G. Jenzer, M. S. Schneider, R. Wandeler, T. Mallat and A. Baiker, J. Catal., 2001, 199, 141–148 CrossRef CAS.
- N. Theyssen, Z. S. Hou and W. Leitner, Chem.–Eur. J., 2006, 12, 3401–3409 CrossRef.
- F. Loeker and W. Leitner, Chem.–Eur. J., 2000, 6, 2011–2015 CrossRef.
- A. A. Abdel-Shafi, F. Wilkinson and D. R. Worrall, Chem. Phys. Lett., 2001, 343, 273–280 CrossRef CAS.
- A. A. Abdel-Shafi and D. R. Worrall, J. Photochem. Photobiol., A, 2007, 186, 263–269 CrossRef CAS.
- M. Okamoto, T. Takagi and F. Tanaka, Chem. Lett., 2000, 1396–1397 CrossRef CAS.
- S. Meyer, D. Tietze, S. Rau, B. Schaefer and G. Kreisel, J. Photochem. Photobiol., A, 2007, 186, 248–253 CrossRef CAS.
- K. Gollnick, Advances in Chemistry Series, 1968, 78–101 CrossRef CAS.
- A. G. Griesbeck, T. T. El-Idreesy and A. Bartoschek, Adv. Synth. Catal., 2004, 346, 245–251 CrossRef CAS.
- M. Benaglia, T. Danelli, F. Fabris, D. Sperandio and G. Pozzi, Org. Lett., 2002, 4, 4229–4232 CrossRef CAS.
- S. M. Ribeiro, A. C. Serra and A. Gonsalves, Tetrahedron, 2007, 63, 7885–7891 CrossRef CAS.
- M. Silva, M. E. Azenha, M. M. Pereira, H. D. Burrows, M. Sarakha, M. F. Ribeiro, A. Fernandes, P. Monsanto and F. Castanheira, Pure Appl. Chem., 2009, 81, 2025–2033 CrossRef CAS.
- N. Kitamura, K. Yamada, K. Ueno and S. Iwata, J. Photochem. Photobiol., A, 2006, 184, 170–176 CAS.
- M. Poliakoff, S. M. Howdle and S. G. Kazarian, Angew. Chem., Int. Ed. Engl., 1995, 34, 1275–1295 CrossRef CAS.
- X. Han, R. A. Bourne, M. Poliakoff and M. W. George, Green Chem., 2009, 11, 1787–1792 RSC.
- X. Han, PhD Thesis, The University of Nottingham, 2011.
- E. G. Azenha, A. C. Serra, M. Pineiro, M. M. Pereira, J. S. de Melo, L. G. Arnaut, S. J. Formosinho and A. M. D. R. Gonsalves, Chem. Phys., 2002, 280, 177–190 CrossRef CAS.
- H. Quast, T. Dietz and A. Witzel, Liebigs Ann., 1995, 1495–1501 Search PubMed.
- M. Bonnet, L. Schmid, A. Baiker and F. Diederich, Adv. Funct. Mater., 2002, 12, 39–42 CrossRef CAS.
- M. Stratakis and M. Stavroulakis, Tetrahedron Lett., 2001, 42, 6409–6411 CrossRef.
- M. Stratakis, M. Stavroulakis and N. Sofikiti, J. Phys. Org. Chem., 2003, 16, 16–20 CrossRef CAS.
- B. Balakrishnan, D. S. Kumar, Y. Yoshida and A. Jayakrishnan, Biomaterials, 2005, 26, 3495–3502 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |