Amyloidogenesis highlighted by designed peptides forming supramolecular self-assemblies†
Myriam
Ouberai
,
Gunnar T.
Dolphin
,
Pascal
Dumy
and
Julian
Garcia
*
Département de Chimie Moléculaire (DCM), UMR 5250, ICMG-FR, Université Joseph Fourier, BP 53, 38041, Grenoble cedex 9, France. E-mail: julian.garcia@ujf-grenoble.fr; Fax: +33 4 56 52 08 05; Tel: +33 4 56 52 08 31
First published on 21st April 2011
Abstract
Amyloid peptides and proteins are associated with a class of pathologies named amyloidoses such as Alzheimer′s and Parkinson′s diseases. These peptides and proteins, in conditions that are still unclear, fold into a cross-β-sheet structure and form fibrils. To aid the search for therapeutic strategies, detailed knowledge of the mechanisms of fibril formation as well as structural information of toxic intermediates is of current interest. In order to produce a comprehensive model of amyloidogenesis, we have synthesized and characterized designed supramolecular edifices. All edifices fold into cross-β-sheet structure, self-assemble into fibrils and present a neuronal toxicity. The presented results show that fibrillation occurs via the formation of a common key intermediate composed of at least four peptide fragments forming β-strands and stabilized by a hydrogen bonding network and hydrophobic interactions. The cell toxicity study shows that early stage oligomers formed from this minimal structure are related to the toxic species. These edifices are promising tools to decipher in detail the driving forces and factors underlining the aggregation of peptide and proteins into amyloid fibrils.
Introduction
Amyloidogenesis is an incompletely understood process involved in severe pathologies named amyloidoses, in which different proteins cause distinct diseases such as Alzheimer′s disease (AD), Parkinson′s disease, and systemic non-neurological diseases like type II diabetes.1–3 In the case of neurodegenerative disorders, the loss of cells can be considerable and progressive, and the brain of a person can be dramatically reduced. In systemic non-neurological diseases, accumulation of large quantities of aggregated proteins in the tissues causes inflammation and loss of the organ functionality.4 Despite the lack of relevant similarities for the primary sequence of amyloidogenic proteins, the fibrillar structures that are being formed share common characteristics such as unbranched fibrillar morphology, cross-β-sheet structure and exhibit common kinetic features, cytotoxicity and mechanical properties. This suggests that the amyloid state is a generic, widely accessible, stable structure of peptides and proteins.2,5–7 The low solubility of fibrils and the high tendency of amyloid proteins to aggregate make difficult to get an atomic resolution structure. However a variety of experimental methods including magnetic resonance spectroscopy, microscopies, X ray and solid state NMR (SS-NMR) have provided progress in the understanding of the organization of peptides that form fibrils. For instance, Tycko and co-workers have proposed by SS-NMR a structural model for the β-amyloid peptide (Aβ), in which peptides are associated in cross-β structure with the β-strands oriented perpendicular to the long axis of the fibril, while the direction of the interchain hydrogen-bonds that stabilize the β-strands is parallel to the same axis.8,9It is of significant importance for the design of effective therapeutic strategies to understand how aggregation occurs and which are the driving forces involved in this process. A particular attention has to be paid to the initial stages of fibrillogenesis since many studies have shown that the toxicity of the β-amyloid peptide is tightly linked to the formation of oligomers rather than to mature fibres.10,11 The main biophysical problems to study the aggregation in detail are the difficulty to start with monomeric species, the lack of comprehensive techniques and methods to follow individual aggregates with time, and the reproducibility of the experiments attributed to a stochastic nature of the formation of seeds. Hence, peptide models that enable to start the aggregation with monomeric entities and that enable to manipulate to a certain degree the nucleation event by chemical modification are of prime interest.12–17 These models are based for example on empirical approaches with the use of amyloid fragments and mutagenesis12–14 or on rational approaches with de novo designed peptides.15–17 Nonetheless, early oligomers remain poorly characterized and consequently it is not clear which species are actually present in the different studies.18 In this work, our strategy consists in attaching designed peptide fragments to a cyclic decapeptide scaffold to control peptide secondary-structure in well-defined and controlled spatial orientations. Furthermore, the creation of a high local concentration will improve the folding via intramolecular interactions and induce the formation of fibres. Previously, we reported the synthesis of a water-soluble β-amyloid (Aβ) fibril model composed of four identical linear sequences formed by residues 16–37 of full length Aβ40 covalently fixed to a cyclic decapeptide scaffold.19–21 To control amyloid folding kinetics the sequence was mutated at three positions leading to a large net positive charge that creates charge repulsions between fibrils.
In this study we have designed three other peptides that can form amyloid fibrils based on the same approach (Fig. 1). The aim of this research is by using different peptide edifices to demonstrate that the formation of amyloid fibril occurs via the folding into a common fibrillation key intermediate. In addition, we show that they all display amyloid fibril structure and toxicity related to early stage oligomers similar to the precursor wild type amyloid peptide.
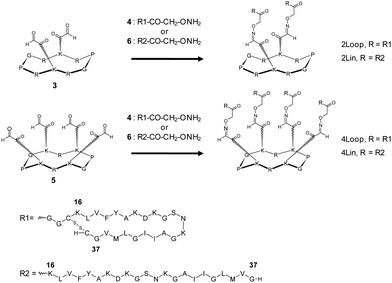 | ||
| Fig. 1 Chemical structures of peptide edifices. | ||
Results
Design and synthesis of amyloid models
The first edifice designed and named 2Lin is composed of two linear amyloid fragments Aβ16-37Y20K22K24 attached on the scaffold (Fig. 1). The two other edifices designed, named 2Loop and 4Loop, are composed respectively of two and four constrained cyclic amyloid fragments Aβ16-37Y20K22K24 attached on the scaffold. We expect that cyclization will promote the folding in cross-β-sheet structure, a hallmark of amyloids, as described by Shivaprasad and Wetzel.22 Synthesis strategy is based in part on that described earlier for compound (Aβ16-37Y20K22K24)419 which is called 4Lin in this paper for simplicity and homogeneity with other discussed molecules. In brief, cyclic decapeptide scaffolds presenting two or four glyoxilic aldehydes (peptides 3 and 5, respectively) and amyloid fragments functionalized at the N-terminal position with an aminooxy (peptides 4 and 6) were first prepared (Fig. 1). The synthesis of these precursor peptides (peptides 3, 4, 5 and 6) is described in the ESI.†To obtain cyclic peptide 4 a disulfide bond was used by introduction of two cysteine residues to C- and N-terminal positions of the Aβ16-37Y20K22K24 fragment. Moreover, as a spacer at the N-terminal position, two glycine residues were added between the cysteine and the aminoxy function. Since synthesis of amyloid sequences is known to be highly challenging, functionalization steps with aminoxy and cyclization by disulfide bond were realized on resin before purification (see Figure S3 in the ESI†). The coupling reactions by oxime bond formation between scaffolds and amyloid peptides were performed in solution, purified by RP-HPLC and characterized by mass ESI. All supramolecular edifices with masses ranging from 6000 to 12000 Da were successfully obtained in satisfying yield and purity.
Fibrillization is triggered by addition of phosphate buffer
Folding and self-assembly into fibrils were studied by far-UV circular dichroism and binding to dyes Thioflavin T (ThT) and Congo Red (CR) in the presence of 20 mM phosphate buffer, pH 7.1 at 21 °C. We previously showed that formation of fibrils is induced by dianions such as HPO42−, through charge-neutralization of the net positively charged assembly.19,20 Folding of compounds from random coil to β-sheet structure was first monitored by CD spectroscopy. In water all edifices were soluble and displayed CD spectra typical of a random coil structure with a minimum at 195–200 nm (Fig. 2). Upon addition of phosphate buffer, all edifices suffered secondary conformational changes, which gave rise to a characteristic β-sheet structure with a minimum around 214–217 nm. However dramatic discrepancies were observed between the folding kinetics of these molecules. For instance, compound 4Loop that exhibited similar behavior to compound 4Lin was the fastest with formation of a β-sheet conformation after less than 15 min, whereas compound 2Loop needed one hour (Fig. 2). On the opposite side the slowest kinetic was displayed by compound 2Lin, for which a mixture of random coil and β-sheet conformations was visible at 96 h, the amount of β-sheet increasing until 17 days (Fig. 2). | ||
| Fig. 2 CD spectra evolution of designed peptides. CD of compounds in water, i.e.t = 0 (…) and after addition of 20 mM phosphate buffer pH 7.1 21 °C at t = 15 min (---), t = 1 h (—) for 4Loop, 2Loop and 4Lin at 10 μM and t = 96 h (---), t = 17 days (—) for 2Lin at 25 μM. | ||
Amyloid structures were identified by the binding to the fluorescent dye Thioflavin T, which is highly specific for the cross-β-sheet structure.23,24ThT bound to each edifice as enhanced fluorescence intensity was seen at 480 nm compared to ThT alone (data not shown). In order to check if the scaffold is able to self aggregate and so to affect the aggregation we have also incubated cyclodecapeptides presenting acetylated lysine residues instead of lysinyl-amyloid fragments with ThT. Neither of these cyclodecapeptides showed ThT binding after incubation for 8 days (data not shown). The fibrillization kinetics were quantified by means of ThT fluorescence study that fitted well to a Finke-Watzky (F-W) two-step model (nucleation and autocatalytic growth).25,26 Reproducible rate constants were obtained for all the models comparable to that described in literature for other amyloid peptides and proteins (Table S1 in the ESI†)). Finally, amyloid structures were also confirmed by their binding to the dye Congo Red, another specific indicator of fibrils formation.27,28 CR binding to our assemblies was demonstrated spectroscopically by hyperchromic effect and bathochrome shift at 530 nm of the absorbance in the UV spectra of CR compared to CR alone (data not shown).
Morphological features of designed peptides
Electron microscopy has been used to probe the morphology evolution of all the assemblies at different periods of incubation. At short incubation time (i.e. 40 h) in 20 mM phosphate buffer short protofilaments with around 50 nm in length and 5 to 6 nm in diameter are seen for compounds 4Lin, 4Loop and 2Loop (Fig. 3a,band c, respectively), while no fibrils are seen for compounds 2Lin and Aβ40 (data not shown). After 71 h of incubation, short protofilaments of 2Lin and Aβ40 were formed similar to those previously observed for the others assemblies at a shorter incubation time (i.e. 40 h) with a length of less than 50 nm and with a diameter of 5 nm (Fig. 3d, not shown for Aβ40). At this incubation time, the other assemblies displayed well formed fibrils that are composed of protofilaments of 5 to 6 nm in diameter but with different global morphologies. For edifices 4Lin and 4Loop, fibrils with an average diameter of 10–12 nm were observed resulting of the interaction of two protofilaments of 5–6 nm in diameter (Fig. 4a, b, respectively). For compound 2Loop protofilaments with length of about 1 μm were organized in bundle of 80 nm in width (Fig. 4c). For compound 2Lin and Aβ40, 7 days of incubation were necessary to observe twisted and longer fibrils, less than 1 μm in length and with 30 nm in diameter composed of an average of 6 protofilaments of 5 to 6 nm in diameter (Fig. 4d, not shown for Aβ40). | ||
| Fig. 3 Morphology of designed peptides after incubation in phosphate buffer. TEM image collected after 40 h for 4Lin 6 μM (a), 4Loop 25 μM (b), 2Loop 25 μM (c), and after 71 h for 2Lin 25 μM (d). | ||
 | ||
| Fig. 4 Morphology of designed peptides after incubation in phosphate buffer. TEM image collected after 71 h for 4Lin 6 μM (a), 4Loop 25 μM (b), 2Loop 25 μM (c), and after 7 days for 2Lin 25 μM (d). | ||
At least 4 fragments are required for fibril to form
To provide further insights into the mechanism of aggregation, fibril growth was simulated by molecular modelling based on TEM measurements and CD spectroscopy results. Compound 4Lin was previously described.19 We showed that the four peptides linked to the template fold as cross β-strands with the inner strands forming a hydrophobic core inside the fibril and the β-strands laying perpendicular to the main axis of the template (Fig. 5a). For edifice 4Loop a comparable result was obtained (Fig. 5c). Two orientations of the loops were possible, parallel or perpendicular to the main axis of the template. Energy minimization showed that a parallel orientation of the loops resulted in a steric clash of the template when forming fibrils. Stable conformation was obtained with a perpendicular orientation, i.e. similar to that find for compound 4Lin. A close inspection of TEM images for molecules 2Loop and 2Lin revealed that the thinnest protofilaments being observed had diameters of 5 ± 1 nm indicating that two monomers are required to form a protofilament. Indeed, it is not possible to get this dimension with only one monomer, the molecule being no more than 2 nm large. We found by energy minimization that the association of two molecules to form the protofilament is more stable when the sequences Ala-30 to Val-36 are involved in the hydrophobic contacts, as compared to sequence V18-K24, presumably due to intermolecular charge repulsions of K22 and K24. The results obtain for molecule 2Lin are comparable to those obtained for compound 2Loop and are presented in Fig. 5b. As for molecule 4Loop, two orientations of the loops were also possible for compound 2Loop relative to the template. For reasons similar to that previously described (i.e. steric hindrance, data not shown) only the conformation with the loops perpendicular to the main axis of the template appeared to be stable (Fig. 5d). This result pointed out that association of at least four peptide fragments is necessary for the protofilament to form. | ||
| Fig. 5 Molecular modelling of designed peptides. Model representations of (a) 4Lin, (b) 2Lin, (c) 4Loop and (d) 2Loop. Top: stick representation of the monomer, bottom: ribbon representation of the fibril. | ||
All peptide edifices exhibit neuronal toxicity due to early stage oligomers
To explore the ability of these compounds to induce neuron death they were co-incubated with human dopaminergic neuroblastoma SH-SY5Y cells at different stages of aggregation. To trigger fibrillization, compounds were first incubated at 25 μM in phosphate buffer for different times before bringing in contact with neuronal cells (5 μM) during 24 h, as described in the experimental section. At an incubation time of 72 h these constructs displayed toxicity similar to the β-amyloid peptide but at a different extend depending on the type of edifice (Fig. 6a). Peptides undergoing fast fibril formation, 4Lin and 4Loop, cause intense neuron death (5.4% and 6.7% of viable cells, respectively). On the opposite side compounds 2Lin and 2Loop with slower kinetic similar to Aβ40 showed reduced but significant toxicity (77.5% and 64.8% of viable cells, respectively).To further understand which aggregation species are responsible for the toxicity we evaluated the effect on neuronal cells of the fastest (4Lin) and the slowest (2Lin) peptide edifices. These two edifices and Aβ40 as a control were co-incubated with the SH-SY5Y cells at different incubation times in phosphate buffer (Fig. 6b). We observed for compound 4Lin a dramatic cell toxicity that grew until 72 h before decreasing markedly at 7 days. In contrast compound 2Lin displayed medium toxicity that became significant at 7 days (47% of viable cells). Similar behavior was observed for Aβ40 but with lower toxicity after 7 days.
 | ||
| Fig. 6 Neurotoxicity of designed peptide edifices. (A) Viability of SH-SY5Y cells in presence of peptides at 5 μM incubated 72 h in 20 mM phosphate buffer pH 7.1. (B) SH-SY5Y cells viability in presence of 5 μM of the aggregation mixture of edifices 4Lin (■), 2Lin (●) and Aβ40 (▲) incubated in 20 mM phosphate buffer pH 7.1, 21 °C during 0 h, 3 h, 36 h, 72 h and 168 h (7 days). | ||
Discussion
It is of major interest to understand the mechanism by which a stable protein can form amyloid fibrils, especially at the initial stages of nucleus formation. This and a better knowledge of the amyloid fibre structure are crucial for the rational design of drugs that can affect fibrils formation from the precursor proteins or that can disassemble the fibrils once they are formed. Furthermore, it has been shown for neurological diseases that early oligomers and not the mature fibre are the main cause of toxicity. This fact further supports the importance of characterizing these oligomeric precursors in order to develop therapeutic treatments of amyloid diseases. Despite the lack of sequence homology between amyloid proteins, the resulting fibrils are highly ordered structures sharing many common features. Thus, we can assume that the amyloid self-assembly process is more elaborated than a simple non specific aggregation process and peptides and proteins possess an inherent capacity to form amyloid fibrils.Many models have so far been proposed in the literature to identify the events occurring during aggregation. However it is not easy to discriminate between these models which one best describes the experimental or theoretical results. For each model a monomeric soluble peptide or protein undergoes conformational changes leading to the formation of fibrils but these changes can be induced, stabilized or be independent of a pre-existing misfolded intermediate. In a first model named Nucleated Polymerization (NP) the initial step of the aggregation consists in the formation of a nucleus, which results from the equilibrium between protein oligomers and monomers.29,30 Once the nucleus is formed the polymerization can continue on the growing end of the fibril. In this model the formation of the nucleus constitutes the rate-determining step, which is characterized by a slow lag phase that can be shortened by adding seeds, i.e. small aggregates. In a second model proposed by Prusinier,31,32 two different forms of soluble monomers co-exist. In this model, named Monomer-Direct-conversion (MDC), both the folded and unfolded proteins are stable in solution and the conformational conversion is induced by the second on the first. The induced conformation represents the aggregation-prone species that can aggregate once a critical concentration is reached. In the most complex model,33 soluble and conformationally dynamic oligomers undergo a nucleation transition leading to aggregation-prone species that are able to catalyze the polymerization reaction of single monomers. The nucleation event represents the rate-limiting step, and because of the simultaneous transitions that a minimum number of monomers must undergo, explains the existence of a lag phase. In fact, this model named nucleated conformational conversion (NCC) allows the presence of numerous species having different conformation and aggregation states.
Various structural models of amyloid fibrils have been presented, the most widely studied being based on the β-amyloid peptide associated with Alzheimer′s disease.8,9,22,34 A common feature of these models is the folding into a strand–loop–strand conformation, which is stabilized by intermolecular hydrogen bonds between two monomers forming typical cross β-sheets packaging.
Previously, we designed a water soluble fibril model by attaching four identical fragments formed by residues 16–37 of full-length Aβ to a cyclic decapeptide scaffold. It is worth noting that this fragment contains three mutations (Y20K22K24), the aim of which being to introduce repulsive charges to avoid uncontrolled aggregation. We showed by CD, TEM and binding of amyloid specific dyes that this construct could fold and aggregate in the designed way. In this paper this compound is named 4Lin. To clarify the mechanism by which fibrils form, we designed and synthesized three more assemblies based on the same strategy and named 2Lin, 2Loop and 4Loop, which contain two linear Aβ fragments, two or four cyclic Aβ fragments, respectively.
The fibrillization process was followed by molecular modelling using experimental data. In an early study we showed that compound 4Lin folds as shown in Fig. 5a, i.e. with the four linear fragments forming cross β-strands. To improve this model and particularly for facilitating the formation of the loop we designed the compound 4Loop, which carries four pre-formed loops. With this assembly we anticipated a faster kinetic relative to compound 4Lin. In fact we obtained similar results indicating that the folding of the four peptide segments, which is thought to be most likely driven by intramolecular hydrophobic interactions, is a very fast event. It is worth noting that in these two models two hydrophobic areas have emerged. The first one is intrastrand and is due to close contacts inside the loop, the second one is interstrand and is formed by residues Ala-30 to Val-36 of both peptide fragments.
For edifices bearing only two peptide fragments, i.e.2Loop and 2Lin, TEM images revealed that the smallest aggregates being observed have average diameters of 5–6 nm. The molecular modelling study showed that this size can be reached when two molecules are associated by close hydrophobic contacts between sequences Ala-30 to Val-36 with formation of a hydrophobic core in the centre of the fibril similar to that observed for compounds 4Loop and 4Lin. This result pointed out that association of at least four peptide fragments is necessary for the protofilament to form. These finding is in concordance with the kinetic differences noticed with CD and TEM experiments, which can be explained by the different levels of conformational organization introduced in each structure. The molecular modelling shows that creation of four β-strands forming a central hydrophobic core is a prerequisite for fibrillation to occur. These conditions are fulfilled for 4Lin and 4Loop, which experience fastest kinetics. The difference between 2Lin and 2Loop is presumably due to the introduction of a loop instead of a linear peptide, i.e. additional pre-organization that speeds up considerably the reaction rate for 2Loop. Compound 2Lin, which does not meet any of the above requisites, showed the slowest kinetic similar to that of Aβ40 peptide. These data demonstrate that the fibril formation occurs for all edifices via the formation of a common intermediate that is a protofilament of 5 to 6 nm in diameter corresponding to the unit of a mature fibril. According to this finding, we can propose for fibril formation the pathway depicted in Fig. 7. Unstructured amyloidogenic peptides undergo fast nucleation driven mostly by intramolecular hydrophobic interactions. A first hydrophobic core is formed inside the sequence but the resulting structure remains unstable and is not able to trigger polymerization. This intermediate is stabilized by intermolecular interactions with other molecules, at least four monomers, giving rise to a nucleus (Fig. 7a). Additional organization of the structure, in this case formation of loops arranged around a second hydrophobic core, induces appropriate orientation of the NH and CO dipoles and allows setting up cross-β-sheets between loops. Since this event needs intermolecular interactions and complex three dimensional rearrangements it constitutes the rate limiting step of the fibrillation process. This building block (Fig. 7b) represents the minimal dimension of the protofilament and the key intermediate capable to elicit further polymerization (Fig. 7c). After a more or less long elongation phase lateral association of two independent protofilaments can lead to formation of thicker fibrils, as shown in TEM experiment, and that we were able to reproduce by molecular modelling (Fig. 7d). Further evolution affords typical fibre morphology with twisting of filaments around one another (Fig. 7e). The proposed fibrillization process displays good correlation with the NCC model,33 where moderate conformational modifications form an unstable intermediate stabilized by intermolecular interactions with other species. The resulting small oligomers can promote further elongation and generate amyloid fibres.
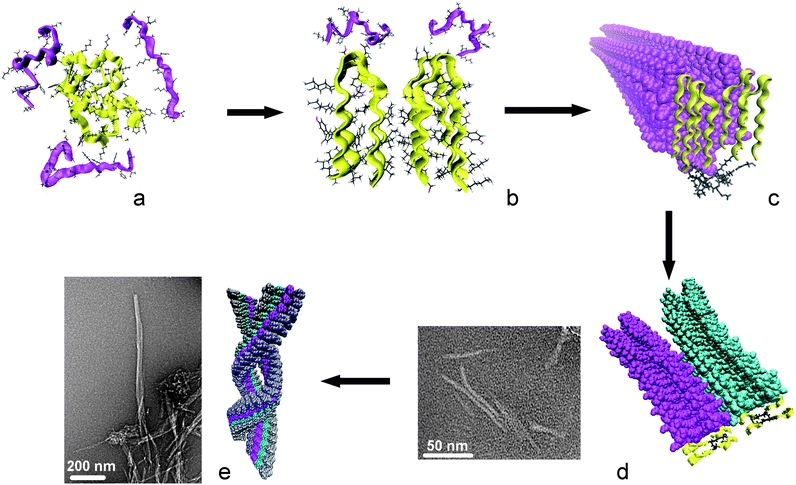 | ||
| Fig. 7 Proposed model for fibril formation, based on compound 4Lin. For clarity the template is not represented in a, b and e, and peptide fragments are represented by sticks and ribbons. The template of the first monomer is highlighted in black in c and d, and the four peptide fragments are ribbon representation in yellow. | ||
The precise mechanisms underlying the toxicity of amyloids remain uncertain and there is still some controversy about what constitutes the neurotoxic species involved in the onset of pathogenesis.35,36 In many diseases37 and particularly for Aβ peptides it has been reported that toxicity resides not in the formation of mature fibres or insoluble aggregates but rather in soluble oligomeric intermediates.38,39 To help in identifying which conformational state is responsible for toxicity we have evaluated the activities of our compounds in inducing neuronal death at different times in vitro. Our experimental procedure relies on TEM and CD results, which give precise fibrillation state of the constructsi.e. oligomers, protofilaments or fibres. Firstly, our results show that all edifices exhibit neuronal toxicity similar or more important than Aβ40 peptide especially in the case of edifices 4Lin and 4Loop that can be related to their high propensity to form oligomers and protofilaments. Moreover, it appears that compounds undergoing fast fibril formation such as 4Lin display intense toxicity during first hours followed by a net attenuation after 72 h. TEM and CD analysis indicated that oligomers and protofilaments are the major constituents present at early times whereas fibres are predominant after 40 h. On the opposite side compounds 2Lin and Aβ40 which experienced slower fibrillization kinetics, showed reduced toxicity but that persists for the entire length of our test. For these two compounds TEM and CD indicated that oligomers and protofilaments are the prevailing species in solution until almost 7 days.
From these results we can correlate neuron toxicity with the presence of species having well-ordered β-sheet structures and corresponding to the first intermediates being formed during the fibrillization process. These oligomers have the minimal structure presented in Fig. 7b, i.e. four peptide fragments forming β-strands and stabilized by hydrophobic interactions. It has been proposed by Chiti and Dobson2 that the toxicity of misfolded proteins could be due to the exposition of residues not normally present at the surface giving rise to abnormal interaction with cellular components. In this study special emphasis has been given to the formation of hydrophobic cores at the early stages of the fibrillization pathway. These cores, involving for instance amino acids Ala-30 to Val-36, are progressively buried inside the fibrils (Fig. 7c) and later inside the fiber (Fig. 7e), resulting in a complete loss of toxicity. More recently Chimon et al.40 have proposed an interesting but hypothetical model based on SS-NMR and ThT experiments, in which the main toxic intermediate is represented by a supramolecular assembly having high parallel β-sheet content. In this study we propose a structure of this intermediate, which will be of valuable interest for the design of drugs targeting β-amyloid or others amyloid-related diseases. Furthermore, it could provide relevant structural basis for the design of synthetic vaccine capable of inducing robust and selective immune response against amyloid oligomers toxicity.
Conclusion
With our supramolecular edifices and protocol we can produce a comprehensive model to describe fibril formation process. These peptides reproduce the characteristics of amyloids as they fold into cross-β-sheet structure, form fibrils and present a neuronal toxicity. For all edifices the fibrillation process occurs via the folding into a complex three-dimensional structure stabilized by hydrogen bonding network and intra-intermolecular hydrophobic interactions. This minimal structure that self-assemble is related in the first stages of fibril formation to the toxic species. This work emphasizes the advantage and usefulness of comparing the behaviour of rational designed peptide edifices, which avoid the stochastic and complex nature of amyloid fibril formation. These models will be highly interesting to analyze the extrinsic factors that influence the process of aggregation such as ionic strength, temperature and inhibitors.Experimental
Peptide synthesis
The synthesis of peptides 3, 4, 5 and 6 is described in the ESI.† Compound 4Lin = (Aβ16-24Y20K22K24)4 was synthesized as previously described.19 Compound 2Lin: the pure peptides 3 (2 mg, 1.13.10−6 mol) and 6 (7 mg, 2.37.10−6 mol) were dissolved in 150 μL of DMSO. The reaction mixture was stirred for 7 h at 40 °C. The product was purified by RP-HPLC affording compound 2Lin with 42% yield (3.5 mg, 4.73.10−7 mol). ESI-MS calc 6029.2, found 6028.0 (Figure S4 in the ESI†). Compound 2Loop: the pure peptides 3 (0.92 mg, 5.24.10−7 mol) and 4 (3.8 mg, 1.16.10−6 mol) were dissolved in DMSO and stirred for 9 h at 40 °C. The product was purified by RP-HPLC affording compound 2Loop with 39% yield (1.7 mg, 2.06.10−7 mol). ESI-MS calc 6665.9, found 6665.4 (Figure S5 in the ESI†). Compound 4Loop: the pure peptides 5 (0.4 mg, 2.52.10−7 mol) and 4 (8.8 mg, 2.69.10−6 mol) were dissolved in DMSO and stirred for 32 h at 40 °C. The product was purified by RP-HPLC affording compound 4Loop with 28% yield (1 mg, 7.07.10−8 mol). ESI-MS calc 12086.5, found 12084.6 (Figure S6 in the ESI†).Circular dichroism spectroscopy
Circular dichroism (CD) spectra were acquired with signal averaging on a Jasco J-810 Spectropolarimeter. Far-UV spectra were recorded from 260 to 200 nm and ellipticities are reported as mean residue ellipticities. Peptides edifices were first dissolved in water to a concentration of 500 μM and then diluted to a final concentration of 10 to 25 μM. The formation of β-sheet was commenced by the addition of stock phosphate to give a final concentration of 20 mM, pH 7.1.Transmission electron microscopy
Microscopy images were recorded using a 6 Philips CM200 Cryo transmission electron microscope operating at 80 kV and with a magnification of 15000, 38000 and 66000. Peptide edifices and Aβ40 were diluted in water to a final concentration of 6 to 25 μM. The formation of the fibrils was commenced by the addition of stock phosphate to give a final concentration of 20 mM, pH 7.1. The solution was left for 40 h to 7 days at 21 °C before application to a glow-discharged 200-mesh carboncoated grid, for three minutes. Thereafter the grid was washed three times with water, one minute each, and finally stained with 2% uranyl acetate.Molecular modelling
Compounds were constructed with the Insight II/Discover software (Version 2005, Accelrys, San Diego, CA, USA) using the CVFF force field. For all the compounds, a loop constituted of amino acids Ser26 to Gly29 was previously introduced. The loops were positioned close enough to allow hydrogen bond formation (distance less than 5 Å). The resulting molecules were subjected to 2500 iterations of steepest descent minimization, followed by 5000 iterations of conjugate gradient minimization and the convergence of minimization was followed until the RMS derivative was less than 0.01 kcal mol−1.Cell viability assays
SH-SY5Y cells were cultured in a medium containing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture Eagle′s Minimum Essential Medium and Ham′s F12 nutriment mix supplemented with 15% fetal bovine serum (FBS), 1% non essential amino-acid, 100 U/mL penicillin, 100 μg mL−1streptomycin and 20 mM glutamine. Cells were maintained in 95% humidified atmosphere with 5% CO2 at 37 °C. The cells were plated in 100 μL of media on 96-well plate at a density of 7.5 × 104cells/well. Peptides Aβ40, 2Lin, 2Loop, 4Lin and 4Loop were incubated at 25 μM in 20 mM phosphate buffer, pH 7.1, 21 °C. After incubation at different times the aggregation mixture was diluted at 5 μM in reduced-serum medium OPTI-MEM® and added to the cells. The treated cells were incubated for 24 h at 37 °C under 5% CO2. Cell viability assay was determined using the MTT assay. After treatment, medium from 96 wells plates was removed. Cells were washed with PBS and 100 μL of MTT solution (500 μg mL−1 in PBS) was added. After incubation (1 h at 37 °C), 100 μL of DMSO was added to dissolve formazan crystals formed by metabolically active cells. The absorbance of formazan was read at 570 nm. Data were normalized to untreated group assigned at 100%. Results are mean ± standard error of the mean of two independent experiments preformed in triplicate.
:1 mixture Eagle′s Minimum Essential Medium and Ham′s F12 nutriment mix supplemented with 15% fetal bovine serum (FBS), 1% non essential amino-acid, 100 U/mL penicillin, 100 μg mL−1streptomycin and 20 mM glutamine. Cells were maintained in 95% humidified atmosphere with 5% CO2 at 37 °C. The cells were plated in 100 μL of media on 96-well plate at a density of 7.5 × 104cells/well. Peptides Aβ40, 2Lin, 2Loop, 4Lin and 4Loop were incubated at 25 μM in 20 mM phosphate buffer, pH 7.1, 21 °C. After incubation at different times the aggregation mixture was diluted at 5 μM in reduced-serum medium OPTI-MEM® and added to the cells. The treated cells were incubated for 24 h at 37 °C under 5% CO2. Cell viability assay was determined using the MTT assay. After treatment, medium from 96 wells plates was removed. Cells were washed with PBS and 100 μL of MTT solution (500 μg mL−1 in PBS) was added. After incubation (1 h at 37 °C), 100 μL of DMSO was added to dissolve formazan crystals formed by metabolically active cells. The absorbance of formazan was read at 570 nm. Data were normalized to untreated group assigned at 100%. Results are mean ± standard error of the mean of two independent experiments preformed in triplicate.
Acknowledgements
We thank Isabelle Paintrand for performing TEM (CERMAV, Grenoble, France). We thank Dr E. Hermans (FARL, UCL-Belgium) for providing us with SH-SY5Y cells, Pr. M. P. Mingeot-Leclercq (FACM, UCL-Belgium) for cell culture access and M.C. Cambier for technical assistance. We are grateful to Dr Pierre Murat for kinetics analysis. This work was supported by grants from the European FP6 EURAMY project “Systemic amyloidoses in Europe”. The authors are grateful to the NanoBio program for the facilities of the Synthesis and Surface Characterization platforms.Notes and references
- V. Bellotti, P. Mangione and M. Stoppini, Cell. Mol. Life Sci., 1999, 55, 977–991 CrossRef CAS.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- J. W. Kelly, Curr. Opin. Struct. Biol., 1998, 8, 101–106 CrossRef CAS.
- M. B. Pepys, Annu. Rev. Med., 2006, 57, 223–241 CrossRef CAS.
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS.
- T. P. Knowles, A. W. Fitzpatrick, S. Meehan, H. R. Mott, M. Vendruscolo, C. M. Dobson and M. E. Welland, Science, 2007, 318, 1900–1903 CrossRef CAS.
- J. F. Smith, T. P. Knowles, C. M. Dobson, C. E. Macphee and M. E. Welland, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15806–15811 CrossRef CAS.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS.
- A. T. Petkova, W. M. Yau and R. Tycko, Biochemistry, 2006, 45, 498–512 CrossRef CAS.
- D. M. Hartley, D. M. Walsh, C. P. Ye, T. Diehl, S. Vasquez, P. M. Vassilev, D. B. Teplow and D. J. Selkoe, J. Neurosci., 1999, 19, 8876–8884 CAS.
- M. Wogulis, S. Wright, D. Cunningham, T. Chilcote, K. Powell and R. E. Rydel, J. Neurosci., 2005, 25, 1071–1080 CrossRef CAS.
- P. Marek, A. Abedini, B. Song, M. Kanungo, M. E. Johnson, R. Gupta, W. Zaman, S. S. Wong and D. P. Raleigh, Biochemistry, 2007, 46, 3255–3261 CrossRef CAS.
- S. M. Tracz, A. Abedini, M. Driscoll and D. P. Raleigh, Biochemistry, 2004, 43, 15901–15908 CrossRef CAS.
- A. D. Williams, E. Portelius, I. Kheterpal, J. T. Guo, K. D. Cook, Y. Xu and R. Wetzel, J. Mol. Biol., 2004, 335, 833–842 CrossRef CAS.
- S. Deechongkit, E. T. Powers, S. L. You and J. W. Kelly, J. Am. Chem. Soc., 2005, 127, 8562–8570 CrossRef CAS.
- H. A. Lashuel, S. R. LaBrenz, L. Woo, L. C. Serpell and J. W. Kelly, J. Am. Chem. Soc., 2000, 122, 5262–5277 CrossRef CAS.
- M. W. West, W. Wang, J. Patterson, J. D. Mancias, J. R. Beasley and M. H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11211–11216 CrossRef CAS.
- G. Bitan, E. A. Fradinger, S. M. Spring and D. B. Teplow, Amyloid, 2005, 12, 88–95 CrossRef.
- G. T. Dolphin, P. Dumy and J. Garcia, Angew. Chem., Int. Ed., 2006, 45, 2699–2702 CrossRef CAS.
- G. T. Dolphin, M. Ouberai, P. Dumy and J. Garcia, ChemMedChem, 2007, 2, 1613–1623 CrossRef CAS.
- G. T. Dolphin, O. Renaudet, M. Ouberai, P. Dumy, J. Garcia and J. L. Reymond, ChemBioChem, 2009, 10, 1325–1329 CrossRef CAS.
- S. Shivaprasad and R. Wetzel, Biochemistry, 2004, 43, 15310–15317 CrossRef CAS.
- H. LeVine 3rd, Protein Sci., 1993, 2, 404–410 CAS.
- H. LeVine 3rd, Methods Enzymol., 1999, 309, 274–284 CAS.
- A. M. Morris, M. A. Watzky and R. G. Finke, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 375–397 CrossRef CAS.
- M. A. Watzky, A. M. Morris, E. D. Ross and R. G. Finke, Biochemistry, 2008, 47, 10790–10800 CrossRef CAS.
- W. E. Klunk, R. F. Jacob and R. P. Mason, Methods Enzymol., 1999, 309, 285–305 CAS.
- W. E. Klunk, J. W. Pettegrew and D. J. Abraham, J. Histochem. Cytochem., 1989, 37, 1273–1281 CAS.
- J. D. Harper and P. T. Lansbury Jr, Annu. Rev. Biochem., 1997, 66, 385–407 CrossRef CAS.
- D. M. Walsh, D. M. Hartley, Y. Kusumoto, Y. Fezoui, M. M. Condron, A. Lomakin, G. B. Benedek, D. J. Selkoe and D. B. Teplow, J. Biol. Chem., 1999, 274, 25945–25952 CrossRef CAS.
- F. E. Cohen and S. B. Prusiner, Annu. Rev. Biochem., 1998, 67, 793–819 CrossRef CAS.
- S. B. Prusiner, Science, 1982, 216, 136–144 CAS.
- T. R. Serio, A. G. Cashikar, A. S. Kowal, G. J. Sawicki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf and S. L. Lindquist, Science, 2000, 289, 1317–1321 CrossRef CAS.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS.
- D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, Nature, 2002, 416, 535–539 CrossRef CAS.
- S. T. Ferreira, M. N. Vieira and F. G. De Felice, IUBMB Life, 2007, 59, 332–345 Search PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetics data, experimental procedures, HPLC profiles and SM spectra for all new compounds. See DOI: 10.1039/c1sc00016k |
| This journal is © The Royal Society of Chemistry 2011 |