Benzoquinone-derived sulfinyl imines as versatile intermediates for alkaloid synthesis: Total synthesis of (–)-3-demethoxyerythratidinone†
Kangway V.
Chuang
,
Raul
Navarro
and
Sarah E.
Reisman
*
The Warren and Katharine Schlinger Laboratory of Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: reisman@caltech.edu
First published on 25th March 2011
Abstract
The preparation and synthetic applications of benzoquinone monoketal-derived N-tert-butanesulfinyl imines is described. These synthetically versatile intermediates undergo highly diastereoselective 1,2-addition reactions with organometallic reagents to provide 4-aminocyclohexadienones in good yields. The utility of this methodology is demonstrated in a six-step enantioselective synthesis of (–)-3-demethoxyerythratidinone.
Introduction
The enantioselective preparation of chiral amines is an area of considerable interest in the discipline of organic chemistry. Chiral amines are prevalent in modern pharmaceutical agents and are the defining functionality of alkaloid natural products.1 As part of studies aimed at the total synthesis of polycyclic alkaloid natural products, we recognized that enantioenriched 4-aminocyclohexadienones could serve as versatile synthetic intermediates for the preparation of erythrina,2hasubanan,3 and acutumine4alkaloids (Fig. 1). Retrosynthetically, it was envisioned that 4-aminocyclohexadienones (4) could derive from the corresponding benzoquinone imine monoketals (5) via nucleophilic 1,2-addition.5 Whereas Swenton and coworkers have previously reported 1,2-addition reactions of organolithium reagents to benzoquinone imine monoketals,6 there are no general methods available to promote these reactions with control over the absolute stereochemistry of the newly formed C–N bond. As a result, these intermediates are underutilized in synthetic efforts. Herein we report a direct, general method to enantioselectively prepare a variety of 4-aminocyclohexadienones, and illustrate their synthetic utility in a six-step synthesis of (–)-3-demethoxyerythratidinone.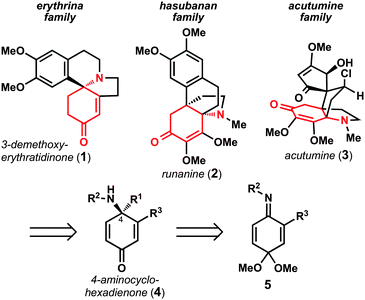 | ||
| Fig. 1 Polycyclic alkaloids envisioned to arise from 4-aminocyclohexadienone 4. | ||
Synthesis plan
In considering general methods to prepare enantioenriched 4-aminocyclohexadienones (4) from benzoquinone imine monoketals (5), we sought to develop a reaction that would: 1) utilize a stable, easy-to-handle benzoquinone imine monoketal; 2) control the absolute configuration at C4; and 3) tolerate a variety of alkyl, aryl, and alkenyl nucleophiles. Moreover, we recognized that 4-aminocyclohexadienones such as 4 are highly susceptible to dienone-phenol rearrangements;7 it was therefore critically important to identify a sufficiently electron-withdrawing nitrogen protecting group in order to disfavor deleterious rearrangements.Despite considerable efforts targeting the preparation of enantioenriched chiral amines,1 the asymmetric synthesis of α,α-disubstituted amines by nucleophilic 1,2-addition to ketimines remains a challenging problem.8–12 Although successful catalytic reactions have emerged for enantioselective allylation10 and Mannich-type reactions,11 there are few reports of catalytic asymmetric addition of aryl or alkyl organometallic reagents to ketimines.12 Hoveyda, Snapper, and coworkers have reported the Zr-catalyzed asymmetric addition of simple alkylzinc reagents to o-anisidine ketimines.12b Alternatively, Shintani, Hayashi, and coworkers recently reported the Rh-catalyzed asymmetric addition of tetra-arylborates to N-tosyl ketimines.12c These two reports represent the state-of-the-art for asymmetric catalytic additions to ketimines; however, the limited scope of nucleophiles and the required use of N-aryl or N-tosyl protecting groups was not expected to be amenable to our desired synthetic applications (vide infra).
With these considerations in mind, it was hypothesized that benzoquinone monoketal-derived N-tert-butanesulfinimines could provide a unique solution. Pioneering studies by Ellman and coworkers have shown that N-tert-butanesulfinyl ketimines undergo highly diastereoselective 1,2-addition reactions with a variety of organometallic reagents.9a,13 In addition, the tert-butanesulfinyl group can be cleaved using mildly acidic conditions. Furthermore, it was envisioned that the electron-withdrawing nature of the sulfinamide product would disfavor the dienone-phenol rearrangement. Taken together, these features of the tert-butanesulfinyl auxiliary were anticipated to prove advantageous in a synthetic context relative to existing asymmetric catalytic approaches.
Results and discussion
Known benzoquinone monoketals 6a–c were prepared in one step from commercially available phenols.14 Our preliminary studies determined that sulfinimine 7a (R = Me) could be prepared directly from 6a by treatment with tert-butanesulfinamide (1.1 equiv) and Ti(OEt)4 at 70 °C for 72 h; however, the desired product was isolated in low yield (Scheme 1). Monitoring the yield of 7a over time revealed that it was decomposing under the reaction conditions. It was hypothesized that use of a quinone monoketal bearing an electron-withdrawing substituent may enhance the rate of sulfinimine formation, allowing for shorter reaction times. Indeed, 2-chloro and 2-bromosulfinimines 7b and 7c were prepared under standard conditions15 in 93% and 85% isolated yield, respectively. Halogenated substrates 7b and 7c possess the advantage of a functional handle for elaboration in natural product synthesis efforts. In both cases, the sulfinimine was formed exclusively as the E-imine isomer (as shown). Sulfinimines 7b and 7c were prepared on gram scale and were stable for months when stored at −20 °C.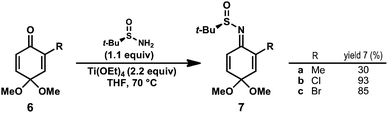 | ||
| Scheme 1 Synthesis of benzoquinone monoketal-derived N-tert-butanesulfinimines. | ||
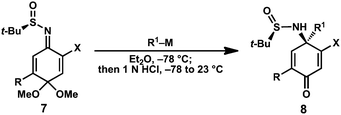
With access to the quinone-derived sulfinimines 7b and 7c, experiments were conducted to evaluate their ability to undergo diastereoselective 1,2-addition reactions with n-butyllithium (Table 1, entries 1 and 3). Treatment of chlorosulfinimine 7b with 1.1 equivalents n-butyllithium at −78 °C followed by quenching with 1N HCl provided 4-aminocyclohexadienone 8a in 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 dr. The major diastereomer was obtained after silica gel chromatography in 90% isolated yield. Notably, hydrolysis of the dimethyl ketal cleanly occurred during the acidic workup; no products of dienone-phenol rearrangement were observed.
:3 dr. The major diastereomer was obtained after silica gel chromatography in 90% isolated yield. Notably, hydrolysis of the dimethyl ketal cleanly occurred during the acidic workup; no products of dienone-phenol rearrangement were observed.
| Entry | X | R | R1-M (equiv) | pdt | dra | Yield (%)b |
|---|---|---|---|---|---|---|
| a Determined by HPLC. b Isolated yield of major diastereomer. c Isolated as a mixture of diastereomers. d Reaction conducted in THF. e Reaction conducted at 0 °C. | ||||||
| 1 | Cl | H (7b) | n-BuLi (1.1) | 8a | 97:3 | 90 |
| 2 | Cl | H | PhLi (1.1) | 8b | 78:22 | 76 |
| 3 | Br | H (7c) | n-BuLi (1.1) | 8c | 98:2 | 88 |
| 4 | Br | H | EtLi (1.1) | 8d | 98:2 | 96 |
| 5 | Br | H | MeLi (1.1) | 8e | 98:2 | 91 |
| 6 | Br | Cl (7d) | MeLi (1.1) | 8f | 97:3 | 92c |
| 7 | Br | Me (7e) | MeLi (1.1) | 8g | 98:2 | 91 |
| 8 | Br | H | PhLi (1.1) | 8h | 80:20 | 74 |
| 9 | Br | H | o-MeC6H4Li (2.0) | 8i | 97:3 | 86 |
| 10 | Br | H | m-MeC6H4Li (2.0) | 8j | 91:9 | 79 |
| 11 | Br | H | p-MeC6H4Li (2.0) | 8k | 91:9 | 78 |
| 12 | Br | H |
 (2.0) (2.0) |
8l | 98:2 | 68d |
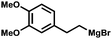
| 13 | Br | H |
 (2.0) (2.0) |
8m | 98:2 | 71d |
| 14 | Br | H |
 (1.1) (1.1) |
8n | 87:13 | 82d |
| 15 | Br | H |
 (1.1) (1.1) |
8o | > 97:3 | 91 |
| 16 | Br | H |
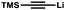 (2.0) (2.0) |
8p | > 98:2 | 99c,d,e |
| 17 | Br | H |
 (1.1) (1.1) |
8q | 96:4 | 82d |
Similar levels of diastereoselectivity were observed for the corresponding bromide 7c.16 A survey of solvents indicated that ethereal solvents were optimal, with Et2O or THF providing the highest levels of diastereoselectivity. A variety of organolithium and organomagnesium reagents were evaluated in order to assess the substrate scope of the reaction. Alkyllithiums provided uniformly high diastereoselectivities (Table 1, entries 1, 3–7); however, addition of phenyllithium proceeded with lower levels of stereocontrol (entries 2 and 8).17 Fortunately, improved diastereoselectivity was observed using substituted aryllithiums (entries 9–11). With less reactive nucleophiles, improved yields were obtained by employing 2 equivalents (entries 9–13, 16). In the case of allyl and propargyl nucleophiles, the readily available Grignard reagents were utilized instead of the corresponding organolithium reagents. Notably, the products shown in Table 1cannot be prepared enantioselectively through any other previously reported methods.
The vinyl halide moiety of the enantioenriched sulfinamide products provides a useful handle for further elaboration. For example, vinyl bromide 8e (R1 = Me) undergoes a variety of palladium-catalyzed cross-coupling reactions, allowing access to the corresponding arene-, alkyne- and allyl-substituted products (Scheme 2, 9, 11, and 12, respectively). On the other hand, vinyl bromide 8n (R1 = allyl) can be coupled with vinyl tributylstannane and treated with Hoveyda-Grubbs second generation catalyst18 to provide bicycle 10 in excellent yield over two steps.
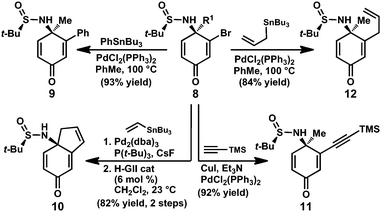 | ||
| Scheme 2 Synthetic transformations of sulfinamide 8. | ||
Having developed a method to prepare enantioenriched 4-aminocyclohexadienones, we sought to highlight this enabling reaction in a total synthesis of the erythrina alkaloid (–)-3-demethoxyerythratidinone (Fig. 1).193-Demethoxyerythratidinone (1) was isolated from Erythrina lithosperma in 1973, and is a representative member of the erythrina family of natural products.20 There have been several total syntheses of 1, the first of which was reported by Tsuda in 1984.21 However, there are only three total syntheses of enantioenriched 1 reported to date;22 thus, the enantioselective synthesis of this natural product remains a significant challenge. The spirocyclic core of (–)-1 was anticipated to derive directly from N-tert-butanesulfinyl imine 7c by a one-pot intermolecular 1,2-addition followed by intramolecular N-alkylation.
In the forward sense, we were pleased to find that addition of aryllithium 1323 (generated in situ at −78 °C from the corresponding aryl bromide by lithium-halogen exchange) to bromosulfinimine 7c provided spirocycle 14 (>98:2 dr), which was isolated in 74% yield as a single diastereomer (Scheme 3). This highly convergent reaction provides direct access to the key spirocyclic amine with excellent levels of stereocontrol.
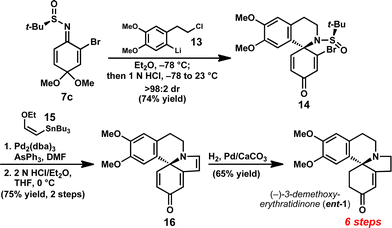 | ||
| Scheme 3 Enantioselective synthesis of (–)-3-demethoxyerythratidinone. | ||
Exposure of sulfinamide 14 to vinyl stannane 15 in the presence of Pd2(dba)3 and AsPh3 at 100 °C furnished the corresponding enol ether in 85% yield.24Sulfinamide deprotection and in situ acid-mediated condensation was effected by treatment of the enol ether with 2N HCl for 2 min at 0 °C to provide tetracycle 16. Notably, use of weaker acids or stirring for longer periods resulted in substantially diminished yields due to competitive decomposition of 16.
Completion of the synthesis was achieved by selective hydrogenation of triene 16 to furnish (–)-1 in 65% yield. At six steps and 26% overall yield, this represents the shortest enantioselective synthesis of 1 reported to date, and illustrates the strategic advantage provided by the ability to promote diastereoselective 1,2-additions to benzoquinone monoketal-derived sulfinimines.
Conclusions
In conclusion, the preparation and synthetic applications of sulfinimines derived from benzoquinone monoketals is reported. This methodology provides the first direct access to a variety of enantioenriched 4-aminocyclohexadienones which possess synthetic handles for further elaboration to alkaloid natural products. The utility of this methodology was demonstrated in a short, enantioselective total synthesis of the natural product 3-demethoxyerythratidinone. The further application of benzoquinone monoketal-derived sulfinimines to the synthesis of alkaloid natural products is the focus of ongoing research in our laboratory.Acknowledgements
We thank Drs. Michael Day and Larry Henling for X-ray crystallographic structural determination, as well as Prof. Brian Stoltz, Dr. Scott Virgil, and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment. We thank the Gates Millennium Scholars Program (R.N.) and the Amgen Scholars Program (K.V.C.) for fellowship support. Andrew Lim is acknowledged for assistance in the preparation of compound 10. HRMS and X-ray crystallographic data were obtained on instruments purchased through awards to the California Institute of Technology by the NSF CRIF program (CHE-0639094, CHE-0541745). Financial support from the California Institute of Technology is gratefully acknowledged.Notes and references
- T. C. Nugent, Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH, Weinheim, 2010 Search PubMed.
- A. F. Parsons, M. J. Palframan, in Alkaloids, Chemistry and Biology, Vol. 68 (Ed: G. A. Cordell), Elsevier, Oxford, 2010, pp 39–81 Search PubMed.
- M. Matsui, in The Alkaloids, Chemistry and Pharmacology, Vol. 33 (Ed: A. Brossi), Academic Press, Inc, San Diego, 1988, pp 307–347 Search PubMed.
- J. Barbosa-Filho, E. V. L. da Cunha, A. I. Gray, in Alkaloids, Chemistry and Biology, Vol. 54 (Ed: G. A. Cordell) Elsevier, Oxford, 2000, pp 1–190 Search PubMed.
- Reviews of nucleophilic 1,2-additions to imines: (a) D. Enders and U. Reinhold, Tetrahedron: Asymmetry, 1997, 8, 1895–1946 CrossRef CAS; (b) R. Bloch, Chem. Rev., 1998, 98, 1407–1438 CrossRef CAS; (c) G. K. Friestad and A. K. Mathies, Tetrahedron, 2007, 63, 2541–2569 CrossRef CAS.
- (a) C.-T. Chou and J. S. Swenton, J. Am. Chem. Soc., 1987, 109, 6898–6899 CrossRef CAS; (b) J. S. Swenton, B. R. Bonke, W. M. Clark, C. P. Chen and K. V. Martin, J. Org. Chem., 1990, 55, 2027–2034 CrossRef CAS.
- A. L. Wilds and C. Djerassi, J. Am. Chem. Soc., 1946, 68, 1715–1719 CrossRef CAS.
- For a review of catalytic asymmetric additions to ketimines and ketones, see: O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873–888 Search PubMed.
- Auxiliary based approaches: (a) J. A. Ellman and D. A. Cogan, J. Am. Chem. Soc., 1999, 121, 268–269 CrossRef CAS; (b) R. Berger, K. Duff and J. L. Leighton, J. Am. Chem. Soc., 2004, 126, 5686–5687 CrossRef CAS; (c) E. Canales, E. Hernandez and J. A. Soderquist, J. Am. Chem. Soc., 2006, 128, 8712–8713 CrossRef CAS.
- Allylation: (a) R. Wada, T. Shibuguchi, M. Sae, M. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 7687–7691 CrossRef CAS; (b) R. Yazaki, T. Nitabaru, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 14477–14479 CrossRef CAS.
- Mannich-Type Reactions: (a) Y. Suto, K. Motomu and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 500–501 CrossRef CAS; (b) Y. Du, L.-W. Xu, Y. Shimizu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 16146–16147 CrossRef CAS; (c) L. C. Wieland, E. M. Vieira, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 570–576 CrossRef CAS.
- (a) C. Lauzon and A. B. Charette, Org. Lett., 2006, 8, 2743–2745 CrossRef CAS; (b) P. Fu, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 5530–5541 CrossRef CAS; (c) R. Shintani, M. Takeda, T. Tsuji and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 13168–13169 CrossRef CAS.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS.
- See Supporting Information.
- G. Liu, D. A. Cogan, T. D. Owens, T. P. Tang and J. A. Ellman, J. Org. Chem., 1999, 64, 1278–1284 CrossRef.
- When n-BuLi is used, the debrominated product is observed in approximately 5% yield.
- The relative stereochemistry of the major diastereomer of 8d was determined by X-ray diffraction and is consistent with addition through a six-membered chair-like transition state (see Ref. 9a). The relative stereochemistry of the remaining products was assigned in analogy based on spectroscopic analysis.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- (–)−1 is the unnatural antipode. Both enantiomers of tert-butanesulfinamide are readily accessible; use of (S)-tert-butanesulfinamide should provide access to the natural antipode.
- D. H. R. Barton, A. A. L. Gunatilaka, R. M. Letcher, A. M. F. T. Lobo and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1, 1973, 874–880 RSC.
- Selected racemic syntheses: (a) Y. Tsuda, A. Nakai, K. Ito, F. Suzuki and M. Haruna, Heterocycles, 1984, 22, 1817–1820 CrossRef CAS; (b) S. J. Danishefsky and J. S. Panek, J. Am. Chem. Soc., 1987, 109, 917–918 CrossRef CAS; (c) H. H. Wasserman and R. M. Amici, J. Org. Chem., 1989, 54, 5843–5844 CrossRef CAS; (d) Q. Wang and A. Padwa, Org. Lett., 2006, 8, 601–604 CrossRef CAS; (e) S. Gao, Y. Q. Tu, X. Hu, S. Wang, R. Hua, Y. Jiang, Y. Zhao, X. Fan and S. Zhang, Org. Lett., 2006, 8, 2373–2376 CrossRef CAS; (f) J. M. Joo, R. A. David, Y. Yuan and C. Lee, Org. Lett., 2010, 12, 5704–5705 CrossRef CAS. For reviews, see Ref. 2 and: (g) E. Reimann, Fortschritte der Chemie organischer Naturstoffe / Progress in the Chemistry of Organic Natural Products, 2007, 1–62 Search PubMed.
- All of the completed syntheses of enantioenriched 1 reported to date utilize chiral auxiliary strategies: (a) Y. Tsuda, S. Hosoi, K. Ishida and M. Sangai, Chem. Pharm. Bull., 1994, 2, 204–213; (b) F. Zhang, N. S. Simpkins and C. Wilson, Tetrahedron Lett., 2007, 48, 5942–5947 CrossRef CAS; (c) S. M. Allin, G. B. Streetley, M. Slater, S. L. James and W. P. Martin, Tetrahedron Lett., 2004, 45, 5493–5496 CrossRef CAS.
- D. J. Jakiela, P. Helquist and L. D. Jones, Org. Synth, 1984, 62, 74–85 CAS . Aryllithium 13 was previously utilized by Swenton and Chou. See Ref. 6a.
- Alternatively, cleavage of the tert-butanesulfinamide group can be effected under acidic conditions to deliver the free amine in excellent yield. See Supporting Information.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data and NMR spectral charts. CCDC reference numbers 780836. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00095k |
| This journal is © The Royal Society of Chemistry 2011 |